PHOSLYRA- calcium acetate solution
Fresenius Medical Care North America
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use PHOSLYRA
® safely and effectively. See full prescribing information for PHOSLYRA. PHOSLYRA (calcium acetate oral solution)
Initial U.S. Approval: 1990 INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATIONDOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONSADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact Fresenius Medical Care North America at 1-800-323-5188 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch DRUG INTERACTIONSSee 17 for PATIENT COUNSELING INFORMATION. Revised: 9/2020 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
PHOSLYRA ® is indicated as an adjunct to reduction in dietary intake of phosphate and dialysis to reduce serum phosphorus in patients with kidney failure on dialysis.
2 DOSAGE AND ADMINISTRATION
The recommended initial dose of PHOSLYRA for the adult dialysis patient is 10 mL with each meal. Increase the dose gradually to lower serum phosphorus levels to the target range, as long as hypercalcemia does not develop. Titrate the dose every 2 to 3 weeks until an acceptable serum phosphorus level is reached. Most patients require 15–20 mL with each meal.
5 WARNINGS AND PRECAUTIONS
5.1 Hypercalcemia
Patients with kidney failure on dialysis may develop hypercalcemia when treated with calcium, including calcium acetate (PHOSLYRA). Avoid the concurrent use of calcium supplements, including calcium-based nonprescription antacids, with PHOSLYRA.
An overdose of PHOSLYRA may lead to progressive hypercalcemia , which may require emergency measures. Therefore, early in the treatment phase during the dosage adjustment period, monitor serum calcium levels twice weekly. Should hypercalcemia develop, reduce the PHOSLYRA dosage or discontinue the treatment, depending on the severity of hypercalcemia.
More severe hypercalcemia (Ca >12 mg/dL) is associated with confusion, delirium, stupor and coma. Severe hypercalcemia can be treated by acute hemodialysis and discontinuing PHOSLYRA therapy.
Mild hypercalcemia (10.5 to 11.9 mg/dL) may be asymptomatic or manifest as constipation, anorexia, nausea, and vomiting. Mild hypercalcemia is usually controlled by reducing the PHOSLYRA dose or temporarily discontinuing therapy. Decreasing or discontinuing Vitamin D therapy is recommended as well.
Chronic hypercalcemia may lead to vascular calcification and other soft-tissue calcification. Radiographic evaluation of suspected anatomical regions may be helpful in early detection of soft tissue calcification. The long-term effect of PHOSLYRA on the progression of vascular or soft tissue calcification has not been determined.
Hypercalcemia (>11 mg/dL) was reported in 16% of patients in a 3-month study of a solid dose formulation of calcium acetate; all cases resolved upon lowering the dose or discontinuing treatment.
6 ADVERSE REACTIONS
No clinical trials have been performed with PHOSLYRA in the intended population. Because the dose and active ingredients of PHOSLYRA are equivalent to that of the calcium acetate gelcaps or tablets, the scope of the adverse reactions is anticipated to be similar.
Hypercalcemia is discussed elsewhere [see Warnings and Precautions ( 5.1)] .
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
In clinical studies, calcium acetate has been generally well tolerated.
The solid dose formulation of calcium acetate was studied in two clinical trials: a 3-month, openlabel, non-randomized study with 98 kidney failure patients on hemodialysis (HD); and in a 2week, double-blind, placebo-controlled, cross-over clinical trial with 69 kidney failure patients on HD.
| Preferred Term | Total adverse reactions
reported for calcium acetate n=167 n (%) | 3-mo, openlabel study of
calcium acetate n=98 n (%) | Double-blind, placebo-controlled, cross-over study of calcium
acetate n=69 |
|
| Calcium acetate
n (%) | Placebo
n (%) |
|||
| Nausea | 6 (3.6) | 6 (6.1) | 0 (0.0) | 0 (0.0) |
| Vomiting | 4 (2.4) | 4 (4.1) | 0 (0.0) | 0 (0.0) |
| Hypercalcemia | 21 (12.6) | 16 (16.3) | 5 (7.2) | 0 (0.0) |
Calcium acetate oral solution was studied in a randomized, controlled, 3-arm, open label, crossover, single-dose study comparing calcium acetate oral solution to a solid formulation in healthy volunteers on a controlled diet. Of the observed drug-related adverse reactions, diarrhea (5/38, 13.2%) was more common with the oral solution.
6.2 Postmarketing Experience
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate their frequency or to establish a causal relationship to drug exposure.
The following additional adverse reactions have been identified during post-approval of calcium acetate: dizziness, edema, and weakness.
7 DRUG INTERACTIONS
| Oral drugs that have to be separated from Phoslyra
|
|
| Dosing Recommendations | |
| Flouroquinolones | Take at least 2 hours before or 6 hours after Phoslyra |
| Tetracyclines | Take at least 1 hour before Phoslyra |
| Levothyroxine | Take at least 4 hours before or 4 hours after Phoslyra |
Oral medications not listed in the Table
There are no empirical data on avoiding drug interactions between Phoslyra and most concomitant oral drugs. For oral medications where a reduction in the bioavailability of that medication would have a clinically significant effect on its safety or efficacy, consider separation of the timing of the administration of the two drugs. The duration of separation depends upon the absorption characteristics of the medication concomitantly administered, such as the time to reach peak systemic levels and whether the drug is an immediate release or an extended release product. Consider monitoring clinical responses or blood levels of concomitant medications that have a narrow therapeutic range.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Administration of the recommended dose of PHOSLYRA is not expected to cause major birth defects, miscarriage, or adverse maternal or fetal outcomes. Patients with kidney failure on dialysis may develop hypercalcemia with calcium acetate treatment [see Contraindications ( 4)] and [Warnings and Precautions ( 5.1)]. Maintenance of normal serum calcium levels is important for maternal and fetal wellbeing. There are adverse effects on maternal and fetal outcomes associated with kidney failure on dialysis in pregnancy (see Clinical Considerations). Animal reproduction studies have not been conducted with PHOSLYRA.
Clinical Consideration
Disease-Associated Maternal and/or Embryo/Fetal Risk
Pregnancies that occur in women with kidney failure on dialysis are associated with a high rate of complications, including increased maternal mortality, hypertension, preeclampsia, anemia, intrauterine growth restriction, preterm delivery, and still birth.
8.4 Pediatric Use
Safety and effectiveness of PHOSLYRA in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of calcium acetate did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
10 OVERDOSAGE
Administration of PHOSLYRA in excess of the appropriate daily dosage may result in hypercalcemia [see Warnings and Precautions ( 5.1)] .
11 DESCRIPTION
PHOSLYRA acts as a phosphate binder. Its chemical name is calcium acetate. Its molecular formula is C 4H 6CaO 4, and its molecular weight is 158.17. Its structural formula is:
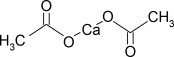
PHOSLYRA for oral administration is provided as pale to light greenish-yellow clear liquid. Each 5 mL of PHOSLYRA contains 667 mg calcium acetate, USP equal to 169 mg (8.45 mEq) calcium. PHOSLYRA also contains the following inactive ingredients: maltitol NF, glycerin USP, Magnasweet 110, propylene glycol USP, povidone K25 USP, sucralose NF, methylparaben NF, artificial black cherry flavor, menthol flavor, purified water USP.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Calcium acetate, when taken with meals, combines with dietary phosphate to form an insoluble calcium-phosphate complex, which is excreted in the feces, resulting in decreased serum phosphorus concentrations.
12.2 Pharmacodynamics
Orally administered calcium acetate from pharmaceutical dosage forms is systemically absorbed up to approximately 40% under fasting conditions and up to approximately 30% under nonfasting conditions. This range represents data from both healthy subjects and renal dialysis patients under various conditions.
A randomized, 3-arm, open-label, cross-over study in healthy volunteers evaluated the bioavailability of PHOSLYRA compared to calcium acetate gelcaps. Each subject received ~1000 mg elemental calcium from each dose of the following study medications: 30 mL PHOSLYRA (test), 6 calcium acetate gelcaps (reference), or 5 calcium citrate caplets (positive control) in three periods. The study medications were administered three times per day with meals from Day 0 through Day 2 and one morning dose on Day 3 of each period.
Treatment (baseline-subtracted) related changes (AUC and C max) in serum calcium and phosphorus assessed over the 6 hours following dosing were similar for PHOSLYRA and calcium acetate gelcaps. Urinary excretion of calcium and phosphorus were not significantly increased with PHOSLYRA compared to calcium acetate gelcaps.
14 CLINICAL STUDIES
Effectiveness of calcium acetate in decreasing serum phosphorus has been demonstrated in two studies of the solid dosage form.
Ninety-one patients with kidney failure who were undergoing hemodialysis and were hyperphosphatemic (serum phosphorus >5.5 mg/dL) following a 1-week phosphate binder washout period contributed efficacy data to an open-label, non-randomized study.
The patients received calcium acetate 667 mg tablets at each meal for a period of 12 weeks. The initial starting dose was 2 tablets per meal for 3 meals a day, and the dose was adjusted as necessary to control serum phosphorus levels. The average final dose after 12 weeks of treatment was 3.4 tablets per meal. Although there was a decrease in serum phosphorus, in the absence of a control group the true magnitude of effect is uncertain.
The data presented in Table 2 demonstrate the efficacy of calcium acetate in the treatment of hyperphosphatemia in kidney failure on patients on Hemodialysis. The effects on serum calcium levels are also presented.
|
a Values expressed as mean ± SE. |
|||||
|
b Ninety-one patients completed at least 6 weeks of the study. |
|||||
|
c ANOVA of difference in values at pre-study and study completion. |
|||||
| Parameter | Pre-Study | Week 4 b | Week 8 | Week 12 | p-valuec |
| Phosphorus (mg/dL) a | 7.4 ± 0.17 | 5.9 ± 0.16 | 5.6 ± 0.17 | 5.2 ± 0.17 | ≤0.01 |
| Calcium (mg/dL) a | 8.9 ± 0.09 | 9.5 ± 0.10 | 9.7 ± 0.10 | 9.7 ± 0.10 | ≤0.01 |
There was a 30% decrease in serum phosphorus levels during the 12 week study period (p <0.01). Two-thirds of the decline occurred in the first month of the study. Serum calcium increased 9% during the study mostly in the first month of the study.
Treatment with the phosphate binder was discontinued for patients from the open-label study, and those patients whose serum phosphorus exceeded 5.5 mg/dL were eligible for entry into a double-blind, placebo-controlled, cross-over study. Patients were randomized to receive calcium acetate or placebo, and each continued to receive the same number of tablets as had been individually established during the previous study. Following 2 weeks of treatment, patients switched to the alternative therapy for an additional 2 weeks.
The phosphate binding effect of calcium acetate is shown in Table 3.
|
a Values expressed as mean ± SEM |
||||
|
b ANOVA of calcium acetate vs. placebo after 2 weeks of treatment. |
||||
| Parameter | Pre-Study | Post-Treatment |
p-valueb |
|
| Calcium Acetate | Placebo | |||
| Phosphorus (mg/dL) a | 7.3 ± 0.18 | 5.9 ± 0.24 | 7.8 ± 0.22 | <0.01 |
| Calcium (mg/dL) a | 8.9 ± 0.11 | 9.5 ± 0.13 | 8.8 ± 0.12 | <0.01 |
Overall, 2 weeks of treatment with calcium acetate statistically significantly (p <0.01) decreased serum phosphorus by a mean of 19% and increased serum calcium by a statistically significant (p <0.01) but clinically unimportant mean of 7%.
16 HOW SUPPLIED/STORAGE AND HANDLING
PHOSLYRA for oral administration is a clear solution containing 667 mg calcium acetate per 5 mL. PHOSLYRA is supplied in amber-colored, multiple-dose bottles, packaged with a marked dosing cup in the following size:
473 mL (16 fl. oz) bottle ................................................................................. (NDC 49230-643-31)
17 PATIENT COUNSELING INFORMATION
Inform patients to take PHOSLYRA with meals, adhere to their prescribed diets, and avoid the use of calcium supplements including nonprescription antacids. Inform patients about the symptoms of hypercalcemia [see Warnings and Precautions ( 5.1) and Adverse Reactions ( 6.1)].
Advise patients who are taking an oral medication where a reduction in the bioavailability of that medication would have a clinically significant effect on its safety or efficacy to take the drug one hour before or three hours after PHOSLYRA.
Manufactured for:
Fresenius Medical Care North America
Waltham, MA 02451
1-800-323-5188
Manufactured by: Lyne Laboratories
Brockton, MA 02301
1-800-525-0450
© 2011-2020 Fresenius Medical Care North America. All Rights Reserved.


| PHOSLYRA
calcium acetate solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Fresenius Medical Care North America (958291411) |