TOPIRAMATE- topiramate tablet
Bryant Ranch Prepack
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use topiramate tablets, USP safely and effectively. See full prescribing information for topiramate tablets, USPTopiramate Tablets, USP for oral use
Initial U.S. Approval: 1996 RECENT MAJOR CHANGESWarnings and Precautions, Visual Field Defects (5.2) 01/2014
INDICATIONS AND USAGETopiramate is indicated for:
DOSAGE AND ADMINISTRATIONSee DOSAGE AND ADMINISTRATION, Epilepsy: Monotherapy and Adjunctive Therapy Use for additional details (2.1).
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONSNone (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSThe most common (≥10% more frequent than placebo or low-dose topiramate in monotherapy) adverse reactions at recommended dosing in adult and pediatric controlled, epilepsy clinical trials were paresthesia, anorexia, weight decrease, speech disorder related speech problem, fatigue, dizziness, somnolence, nervousness, psychomotor slowing, abnormal vision, and fever. (6) To report SUSPECTED ADVERSE REACTIONS, contact Camber Pharmaceuticals, Inc. at 1-866-495-8330 or FDA at 1-800-FDA-1088 ORwww.fda.gov/medwatch. (6) DRUG INTERACTIONSSummary of (AED) interactions with topiramate (7.1)
a= Plasma concentration increased 25% in some patients, generally those on a twice a day dosing regimen of phenytoin. b= Is not administered but is an active metabolite of carbamazepine. NC= Less than 10% change in plasma concentration. NE= Not Evaluated.
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 8/2021 |
FULL PRESCRIBING INFORMATION
1. INDICATIONS AND USAGE
1.1 Monotherapy Epilepsy
Topiramate tablets, USP are indicated as initial monotherapy in patients 2 years of age and older with partial onset or primary generalized tonic-clonic seizures. Safety and effectiveness in patients who were converted to monotherapy from a previous regimen of other anticonvulsant drugs have not been established in controlled trials [see Clinical Studies (14.1)].
1.2 Adjunctive Therapy Epilepsy
Topiramate tablets, USP are indicated as adjunctive therapy for adults and pediatric patients ages 2 to 16 years with partial onset seizures or primary generalized tonic-clonic seizures, and in patients 2 years of age and older with seizures associated with Lennox-Gastaut syndrome [see Clinical Studies (14.2)].
2. DOSAGE AND ADMINISTRATION
2.1 Epilepsy
It is not necessary to monitor topiramate plasma concentrations to optimize topiramate tablets therapy.
On occasion, the addition of topiramate tablets to phenytoin may require an adjustment of the dose of phenytoin to achieve optimal clinical outcome. Addition or withdrawal of phenytoin and/or carbamazepine during adjunctive therapy with topiramate tablets may require adjustment of the dose of topiramate tablets.
Because of the bitter taste, tablets should not be broken.
Topiramate tablets can be taken without regard to meals.
Monotherapy Use
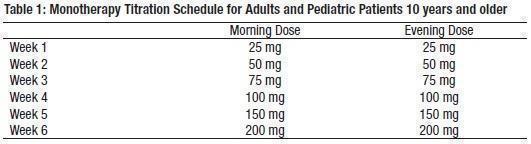
Adults and Pediatric Patients 10 Years and Older
The recommended dose for topiramate tablet monotherapy in adults and pediatric patients 10 years of age and older is 400 mg/day in two divided doses. Approximately 58% of patients randomized to 400 mg/day achieved this maximal dose in the monotherapy controlled trial; the mean dose achieved in the trial was 275 mg/day. The dose should be achieved by titration according to the following schedule (Table 1):
Children Ages 2 to <10 Years
Dosing of topiramate as initial monotherapy in children 2 to < 10 years of age with partial onset or primary generalized tonic-clonic seizures was based on a pharmacometric bridging approach [see Clinical Studies (14.1)].
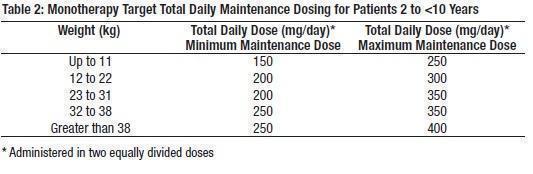
Dosing in patients 2 to <10 years is based on weight. During the titration period, the initial dose of topiramate tablets should be 25 mg/day administered nightly for the first week. Based upon tolerability, the dosage can be increased to 50 mg/day (25 mg twice daily) in the second week. Dosage can be increased by 25 to 50 mg/day each subsequent week as tolerated. Titration to the minimum maintenance dose should be attempted over 5 to 7 weeks of the total titration period. Based upon tolerability and clinical response, additional titration to a higher dose (up to the maximum maintenance dose) can be attempted at 25 to 50 mg/day weekly increments. The total daily dose should not exceed the maximum maintenance dose for each range of body weight (Table 2).
Adjunctive Therapy Use
Adults (17 Years of Age and Over) - Partial Onset Seizures, Primary Generalized Tonic-Clonic Seizures, or Lennox-Gastaut Syndrome
The recommended total daily dose of topiramate tablets as adjunctive therapy in adults with partial onset seizures is 200 to 400 mg/day in two divided doses, and 400 mg/day in two divided doses as adjunctive treatment in adults with primary generalized tonic-clonic seizures. It is recommended that therapy be initiated at 25 to 50 mg/day followed by titration to an effective dose in increments of 25 to 50 mg/day every week. Titrating in increments of 25 mg/day every week may delay the time to reach an effective dose. Doses above 400 mg/day (600 mg, 800 mg or 1,000 mg/day) have not been shown to improve responses in dose-response studies in adults with partial onset seizures. Daily doses above 1,600 mg have not been studied.
In the study of primary generalized tonic-clonic seizures the initial titration rate was slower than in previous studies; the assigned dose was reached at the end of 8 weeks [see Clinical Studies (14.1)].
Pediatric Patients (Ages 2 to 16 Years) – Partial Onset Seizures, Primary Generalized Tonic-Clonic Seizures, or Lennox-Gastaut Syndrome
The recommended total daily dose of topiramate tablets as adjunctive therapy for pediatric patients with partial onset seizures, primary generalized tonic-clonic seizures, or seizures associated with Lennox-Gastaut syndrome is approximately 5 to 9 mg/kg/day in two divided doses. Titration should begin at 25 mg/day (or less, based on a range of 1 to 3 mg/kg/day) nightly for the first week. The dosage should then be increased at 1- or 2-week intervals by increments of 1 to 3 mg/kg/day (administered in two divided doses), to achieve optimal clinical response. Dose titration should be guided by clinical outcome.
In the study of primary generalized tonic-clonic seizures the initial titration rate was slower than in previous studies; the assigned dose of 6 mg/kg/day was reached at the end of 8 weeks [see Clinical Studies (14.1)].
2.4 Patients with Renal Impairment
In renally impaired subjects (creatinine clearance less than 70 mL/min/1.73 m2), one-half of the usual adult dose is recommended. Such patients will require a longer time to reach steady-state at each dose.
2.5 Geriatric Patients (Ages 65 Years and Over)
Dosage adjustment may be indicated in the elderly patient when impaired renal function (creatinine clearance rate <70 mL/min/1.73 m2)) is evident [see Clinical Pharmacology (12.3)].
2.6 Patients Undergoing Hemodialysis
Topiramate is cleared by hemodialysis at a rate that is 4 to 6 times greater than a normal individual. Accordingly, a prolonged period of dialysis may cause topiramate concentration to fall below that required to maintain an anti-seizure effect. To avoid rapid drops in topiramate plasma concentration during hemodialysis, a supplemental dose of topiramate may be required. The actual adjustment should take into account 1) the duration of dialysis period, 2) the clearance rate of the dialysis system being used, and 3) the effective renal clearance of topiramate in the patient being dialyzed.
3. DOSAGE FORMS AND STRENGTHS
Topiramate tablets are available containing 25 mg, 50 mg, 100 mg or 200 mg of topiramate, USP.
The 25 mg tablets are white, film coated, round, biconvex tablets debossed with IG on one side and 278 on other.
The 50 mg tablets are yellow, film coated, round, biconvex tablets debossed with IG on one side and 279 on other.
The 100 mg tablets are light yellow, film coated, round, biconvex tablets debossed with IG on one side and 280 on other.
The 200 mg tablets are pink, film coated, round, biconvex tablets debossed with IG on one side and 281 on other.
5. WARNINGS AND PRECAUTIONS
5.1 Acute Myopia and Secondary Angle Closure Glaucoma
A syndrome consisting of acute myopia associated with secondary angle closure glaucoma has been reported in patients receiving topiramate. Symptoms include acute onset of decreased visual acuity and/or ocular pain. Ophthalmologic findings can include myopia, anterior chamber shallowing, ocular hyperemia (redness) and increased intraocular pressure. Mydriasis may or may not be present. This syndrome may be associated with supraciliary effusion resulting in anterior displacement of the lens and iris, with secondary angle closure glaucoma. Symptoms typically occur within 1 month of initiating topiramate therapy. In contrast to primary narrow angle glaucoma, which is rare under 40 years of age, secondary angle closure glaucoma associated with topiramate has been reported in pediatric patients as well as adults. The primary treatment to reverse symptoms is discontinuation of topiramate tablets as rapidly as possible, according to the judgment of the treating physician. Other measures, in conjunction with discontinuation of topiramate, may be helpful.
Elevated intraocular pressure of any etiology, if left untreated, can lead to serious sequelae including permanent vision loss.
5.2 Visual Field Defects
Visual field defects (independent of elevated intraocular pressure) have been reported in clinical trials and in postmarketing experience in patients receiving topiramate. In clinical trials, most of these events were reversible after topiramate discontinuation. If visual problems occur at any time during topiramate treatment, consideration should be given to discontinuing the drug.
5.3 Oligohidrosis and Hyperthermia
Oligohidrosis (decreased sweating), infrequently resulting in hospitalization, has been reported in association with topiramate use. Decreased sweating and an elevation in body temperature above normal characterized these cases. Some of the cases were reported after exposure to elevated environmental temperatures.
The majority of the reports have been in pediatric patients. Patients, especially pediatric patients, treated with topiramate should be monitored closely for evidence of decreased sweating and increased body temperature, especially in hot weather. Caution should be used when topiramate is prescribed with other drugs that predispose patients to heat-related disorders; these drugs include, but are not limited to, other carbonic anhydrase inhibitors and drugs with anticholinergic activity.
5.4 Metabolic Acidosis
Hyperchloremic, non-anion gap, metabolic acidosis (i.e., decreased serum bicarbonate below the normal reference range in the absence of chronic respiratory alkalosis) is associated with topiramate treatment. This metabolic acidosis is caused by renal bicarbonate loss due to the inhibitory effect of topiramate on carbonic anhydrase. Such electrolyte imbalance has been observed with the use of topiramate in placebo-controlled clinical trials and in the post-marketing period. Generally, topiramate-induced metabolic acidosis occurs early in treatment although cases can occur at any time during treatment. Bicarbonate decrements are usually mild to moderate (average decrease of 4 mEq/L at daily doses of 400 mg in adults and at approximately 6 mg/kg/day in pediatric patients); rarely, patients can experience severe decrements to values below 10 mEq/L. Conditions or therapies that predispose patients to acidosis (such as renal disease, severe respiratory disorders, status epilepticus, diarrhea, ketogenic diet or specific drugs) may be additive to the bicarbonate lowering effects of topiramate.
Some manifestations of acute or chronic metabolic acidosis may include hyperventilation, nonspecific symptoms such as fatigue and anorexia, or more severe sequelae including cardiac arrhythmias or stupor. Chronic, untreated metabolic acidosis may increase the risk for nephrolithiasis or nephrocalcinosis, and may also result in osteomalacia (referred to as rickets in pediatric patients) and/or osteoporosis with an increased risk for fractures. Chronic metabolic acidosis in pediatric patients may also reduce growth rates. A reduction in growth rate may eventually decrease the maximal height achieved. The effect of topiramate on growth and bone-related sequelae has not been systematically investigated in long-term, placebo-controlled trials. Long-term, open-label treatment of infants/toddlers,
with intractable partial epilepsy, for up to 1 year, showed reductions from baseline in Z SCORES for length, weight, and head circumference compared to age and sex-matched normative data, although these patients with epilepsy are likely to have different growth rates than normal infants. Reductions in Z SCORES for length and weight were correlated to the degree of acidosis [see Use in SpecificPopulations (8.4)]. Topiramate treatment that causes metabolic acidosis during pregnancy can possibly produce adverse effects on the fetus and might also cause metabolic acidosis in the neonate from possible transfer of topiramate to the fetus [see Warnings and Precautions (5.7) and Use in Special Populations (8.1)].
Epilepsy
Adult patients
In adults, the incidence of persistent treatment-emergent decreases in serum bicarbonate (levels of <20 mEq/L at two consecutive visits or at the final visit) in controlled clinical trials for adjunctive treatment of epilepsy was 32% for 400 mg/day, and 1% for placebo. Metabolic acidosis has been observed at doses as low as 50 mg/day. The incidence of a markedly abnormally low serum bicarbonate (i.e., absolute value <17 mEq/L and >5 mEq/L decrease from pretreatment) in the adjunctive therapy trials was 3% for 400 mg/day, and 0% for placebo.
The incidence of persistent treatment-emergent decreases in serum bicarbonate in adult patients (≥ 16 years of age) in the epilepsy controlled clinical trial for monotherapy was 14% for 50 mg/day and 25% for 400 mg/day. The incidence of a markedly abnormally low serum bicarbonate (i.e., absolute value < 17 mEq/L and > 5 mEq/L decrease from pretreatment) in this trial for adults was 1% for 50 mg/day and 6% for400 mg/day. Serum bicarbonate levels have not been systematically evaluated at daily doses greater than 400 mg/day.
Pediatric patients
In pediatric patients (2 to 16 years of age), the incidence of persistent treatment-emergent decreases in serum bicarbonate in placebo-controlled trials for adjunctive treatment of Lennox-Gastaut syndrome or refractory partial onset seizures was 67% for topiramate (at approximately 6 mg/kg/day), and 10% for placebo. The incidence of a markedly abnormally low serum bicarbonate (i.e., absolute value < 17 mEq/L and > 5 mEq/L decrease from pretreatment) in these trials was 11% for topiramate and 0% for placebo. Cases of moderately severe metabolic acidosis have been reported in patients as young as 5 months old, especially at daily doses above 5 mg/kg/day.
Although not approved for use in patients under 2 years of age with partial onset seizures, a controlled trial that examined this population revealed that topiramate produced a metabolic acidosis that is notably greater in magnitude than that observed in controlled trials in older children and adults. The mean treatment difference (25 mg/kg/day topiramate-placebo) was -5.9 mEq/L for bicarbonate. The incidence of metabolic acidosis (defined by a serum bicarbonate < 20 mEq/L) was 0% for placebo, 30% for 5 mg/kg/day, 50% for 15 mg/kg/day, and 45% for 25 mg/kg/day. The incidence of markedly abnormal changes (i.e., < 17 mEq/L and > 5 mEq/L decrease from baseline of ≥ 20 mEq/L) was 0 % for placebo, 4% for 5 mg/kg/day, 5 % for 15 mg/kg/day, and 5 % for 25 mg/kg/day [see Use in Special Populations (8.4)].
In pediatric patients (6 to15 years of age), the incidence of persistent treatment-emergent decreases in serum bicarbonate in the epilepsy controlled clinical trial for monotherapy was 9 % for 50 mg/day and 25 % for 400 mg/day. The incidence of a markedly abnormally low serum bicarbonate (i.e., absolute value <17 mEq/L and >5 mEq/L decrease from pretreatment) in this trial was 1 % for 50 mg/day and 6 % for
400 mg/day.
Measurement of Serum Bicarbonate in Epilepsy Patients
Measurement of baseline and periodic serum bicarbonate during topiramate treatment is recommended. If metabolic acidosis develops and persists, consideration should be given to reducing the dose or discontinuing topiramate (using dose tapering). If the decision is made to continue patients on topiramate in the face of persistent acidosis, alkali treatment should be considered.
5.5 Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including topiramate, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as one week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5 to 100 years) in the clinical trials analyzed.
Table 4 shows absolute and relative risk by indication for all evaluated AEDs.
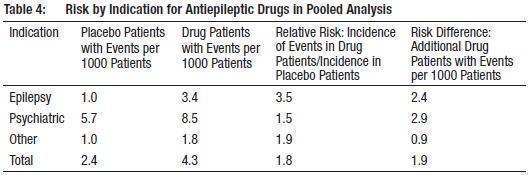
The relative risk for suicidal thoughts or behavior was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing topiramate or any other AED must balance the risk of suicidal thoughts or behavior with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, the prescriber needs to consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
Patients, their caregivers, and families should be informed that AEDs increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of the signs and symptoms of depression, any unusual changes in mood or behavior or the emergence of suicidal thoughts, behavior or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers.
5.6 Cognitive/Neuropsychiatric Adverse Reactions
Adverse reactions most often associated with the use of topiramate were related to the central nervous system and were observed in the epilepsy population. In adults, the most frequent of these can be classified into three general categories:
1) Cognitive-related dysfunction (e.g., confusion, psychomotor slowing, difficulty with concentration/attention, difficulty with memory, speech or language problems, particularly word-finding difficulties);
2) Psychiatric/behavioral disturbances (e.g., depression or mood problems); and
3) Somnolence or fatigue.
Adult Patients
Cognitive-Related Dysfunction
The majority of cognitive-related adverse reactions were mild to moderate in severity, and they frequently occurred in isolation. Rapid titration rate and higher initial dose were associated with higher incidences of these reactions. Many of these reactions contributed to withdrawal from treatment [see Adverse Reactions (6)].
In the add-on epilepsy controlled trials (using rapid titration such as 100 to 200 mg/day weekly increments), the proportion of patients who experienced one or more cognitive-related adverse reactions was 42% for 200 mg/day, 41% for 400 mg/day, 52% for 600 mg/day, 56% for 800 and 1,000 mg/day, and 14% for placebo. These dose-related adverse reactions began with a similar frequency in the titration or in the maintenance phase, although in some patients the events began during titration and persisted into the maintenance phase. Some patients who experienced one or more cognitive-related adverse reactions in the titration phase had a dose-related recurrence of these reactions in the maintenance phase.
In the monotherapy epilepsy controlled trial, the proportion of patients who experienced one or more cognitive-related adverse reactions was 19% for topiramate 50 mg/day and 26% for 400 mg/day.
Psychiatric/Behavioral Disturbances
Psychiatric/behavioral disturbances (depression or mood) were dose-related for the epilepsy population [see Warnings and Precautions (5.5)].
Somnolence/Fatigue
Somnolence and fatigue were the adverse reactions most frequently reported during clinical trials of topiramate for adjunctive epilepsy. For the adjunctive epilepsy population, the incidence of somnolence did not differ substantially between 200 mg/day and 1,000 mg/day, but the incidence of fatigue was dose-related and increased at dosages above 400 mg/day. For the monotherapy epilepsy population in the 50 mg/day and 400 mg/day groups, the incidence of somnolence was dose-related (9% for the 50 mg/day group and 15% for the 400 mg/day group) and the incidence of fatigue was comparable in both treatment groups (14% each).
Additional nonspecific CNS events commonly observed with topiramate in the add-on epilepsy population include dizziness or ataxia.
Pediatric Patients
Epilepsy
In double-blind adjunctive therapy and monotherapy epilepsy clinical studies, the incidences of cognitive/neuropsychiatric adverse reactions in pediatric patients were generally lower than observed in adults. These reactions included psychomotor slowing, difficulty with concentration/attention, speech disorders/related speech problems, and language problems. The most frequently reported neuropsychiatric reactions in pediatric patients during adjunctive therapy double-blind studies were somnolence and fatigue. The most frequently reported neuropsychiatric reactions in pediatric patients in the 50 mg/day and 400 mg/day groups during the monotherapy double-blind study were headache, dizziness, anorexia, and somnolence.
No patients discontinued treatment due to any adverse reactions in the adjunctive epilepsy double-blind trials. In the monotherapy epilepsy double-blind trial, 1 pediatric patient (2%) in the 50 mg/day group and 7 pediatric patients (12%) in the 400 mg/day group discontinued treatment due to any adverse reactions. The most common adverse reaction associated with discontinuation of therapy was difficulty with concentration/attention; all occurred in the 400 mg/day group.
5.7 Fetal Toxicity
Topiramate can cause fetal harm when administered to a pregnant woman. Data from pregnancy registries indicate that infants exposed to topiramate in utero have an increased risk for cleft lip and/or cleft palate (oral clefts). When multiple species of pregnant animals received topiramate at clinically relevant doses, structural malformations, including craniofacial defects, and reduced fetal weights occurred in offspring [see Use in Specific Populations (8.1)].
Consider the benefits and the risks of topiramate when administering this drug in women of childbearing potential, particularly when topiramate is considered for a condition not usually associated with permanent injury or death [see Use in Specific Populations (8.9) and Patient Counseling Information (17)].
Topiramate should be used during pregnancy only if the potential benefit outweighs the potential risk. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus [see Use in Specific Populations (8.1) and (8.9)]
5.8 Withdrawal of Antiepileptic Drugs (AEDs)
In patients with or without a history of seizures or epilepsy, antiepileptic drugs including topiramate should be gradually withdrawn to minimize the potential for seizures or increased seizure frequency [see Clinical Studies (14)]. In situations where rapid withdrawal of topiramate is medically required, appropriate monitoring is recommended.
5.9 Sudden Unexplained Death in Epilepsy (SUDEP)
During the course of premarketing development of topiramate tablets, 10 sudden and unexplained deaths were recorded among a cohort of treated patients (2796 subject years of exposure). This represents an incidence of 0.0035 deaths per patient year. Although this rate exceeds that expected in a healthy population matched for age and sex, it is within the range of estimates for the incidence of sudden unexplained deaths in patients with epilepsy not receiving topiramate (ranging from 0.0005 for the general population of patients with epilepsy, to 0.003 for a clinical trial population similar to that in the topiramate program, to 0.005 for patients with refractory epilepsy).
5.10 Hyperammonemia and Encephalopathy (Without and With Concomitant Valproic Acid [VPA] Use)
Hyperammonemia/Encephalopathy Without Concomitant Valproic Acid (VPA)
Topiramate treatment has produced hyperammonemia (in some instances dose-related) in a clinical investigational program in adolescent patients (12 to 17 years) given topiramate. The incidence of hyperammonemia (above the upper limit of normal reference) at any time in the trial was 9% for placebo, 14% for 50 mg, and 26% for 100 mg topiramate daily. In some patients, hyperammonemia was
observed at the end of the trial at the final visit. The incidence of markedly increased hyperammonemia (at least 50% or higher above upper limit of normal) at any time in the trial in adolescent patients was also increased at 100 mg/day (9%) compared to 50 mg topiramate (0%) or placebo (3%). During this trial, markedly increased ammonia levels returned to normal in all but one patient (in whom the ammonia level fell to high instead of markedly abnormal).
Topiramate treatment has produced hyperammonemia in a clinical investigational program in very young pediatric patients (1 to 24 months) who were treated with adjunctive topiramate for partial onset epilepsy (8% for placebo, 10% for 5 mg/kg/day, 0% for 15 mg/kg/day, 9% for 25 mg/kg/day). In some patients, ammonia was markedly increased (≥ 50% above upper limit of normal). The hyperammonemia associated with topiramate treatment occurred with and without encephalopathy in placebo-controlled trials and in an open-label, extension trial of infants with refractory epilepsy. Dose-related hyperammonemia was observed in the extension trial in pediatric patients up to 2 years old. Clinical symptoms of hyperammonemic encephalopathy often include acute alterations in level of consciousness and/or cognitive function with lethargy or vomiting. Topiramate is not approved as adjunctive treatment of partial onset seizures in pediatric patients less than 2 years old.
Hyperammonemia with and without encephalopathy has also been observed in post-marketing reports in patients who were taking topiramate without concomitant valproic acid (VPA).
Hyperammonemia/Encephalopathy With Concomitant Valproic Acid (VPA)
Concomitant administration of topiramate and valproic acid (VPA) has been associated with hyperammonemia with or without encephalopathy in patients who have tolerated either drug alone based upon post-marketing reports. Although hyperammonemia may be asymptomatic, clinical symptoms of hyperammonemic encephalopathy often include acute alterations in level of consciousness and/or cognitive function with lethargy or vomiting. In most cases, symptoms and signs abated with discontinuation of either drug. This adverse reaction is not due to a pharmacokinetic interaction.
Although topiramate is not indicated for use in infants/toddlers (1 to 24 months) topiramate with VPA clearly produced a dose-related to increase in the incidence of treatment-emergent hyperammonemia (above the upper limit of normal, 0% for placebo, 12% for 5 mg/kg/day, 7% for 15 mg/kg/day, 17% for 25 mg/kg/day) in an investigational program. Markedly increased, dose-related hyperammonemia (0% for placebo and 5 mg/kg/day, 7% for 15 mg/kg/day, 8% for 25 mg/kg /day) also occurred in these infants/toddlers. Dose-related hyperammonemia was similarly observed in a long-term, extension trial in these very young, pediatric patients [see Use in Specific Populations (8.4)].
Hyperammonemia with and without encephalopathy has also been observed in post-marketing reports in patients taking topiramate with VPA.
The hyperammonemia associated with topiramate treatment appears to be more common when topiramate is used concomitantly with VPA.
Monitoring for Hyperammonemia
Patients with inborn errors of metabolism or reduced hepatic mitochondrial activity may be at an increased risk for hyperammonemia with or without encephalopathy. Although not studied, topiramate treatment or an interaction of concomitant topiramate and valproic acid treatment may exacerbate existing defects or unmask deficiencies in susceptible persons.
In patients who develop unexplained lethargy, vomiting, or changes in mental status associated with any topiramate treatment, hyperammonemic encephalopathy should be considered and an ammonia level should be measured.
5.11 Kidney Stones
A total of 32/2086 (1.5%) of adults exposed to topiramate during its adjunctive epilepsy therapy development reported the occurrence of kidney stones, an incidence about 2 to 4 times greater than expected in a similar, untreated population. In the double-blind monotherapy epilepsy study, a total of 4/319 (1.3%) of adults exposed to topiramate reported the occurrence of kidney stones. As in the general population, the incidence of stone formation among topiramate-treated patients was higher in men. Kidney stones have also been reported in pediatric patients taking topiramate for epilepsy.
During long-term (up to 1 year) topiramate treatment in an open-label extension study of 284 pediatric patients 1 to 24 months old with epilepsy, 7% developed kidney or bladder stones that were diagnosed clinically or by sonogram. Topiramate is not approved for treatment of epilepsy in pediatric patients less than 2 years old [see Use in Specific Populations (8.4)].
An explanation for the association of topiramate and kidney stones may lie in the fact that topiramate is a carbonic anhydrase inhibitor. Carbonic anhydrase inhibitors (e.g., zonisamide, acetazolamide or dichlorphenamide) can promote stone formation by reducing urinary citrate excretion and by increasing urinary pH [see Warnings and Precautions (5.3)]. The concomitant use of topiramate with any other drug producing metabolic acidosis, or potentially in patients on a ketogenic diet may create a physiological environment that increases the risk of kidney stone formation, and should therefore be avoided.
Increased fluid intake increases the urinary output, lowering the concentration of substances involved in stone formation. Hydration is recommended to reduce new stone formation.
5.12 Hypothermia with Concomitant Valproic Acid (VPA) Use
Hypothermia, defined as an unintentional drop in body core temperature to <35°C (95°F), has been reported in association with topiramate use with concomitant valproic acid (VPA) both in conjunction with hyperammonemia and in the absence of hyperammonemia. This adverse reaction in patients using concomitant topiramate and valproate can occur after starting topiramate treatment or after increasing the daily dose of topiramate [see Drug Interactions (7.1)]. Consideration should be given to stopping topiramate or valproate in patients who develop hypothermia, which may be manifested by a variety of clinical abnormalities including lethargy, confusion, coma, and significant alterations in other major organ systems such as the cardiovascular and respiratory systems. Clinical management and assessment should include examination of blood ammonia levels.
5.13 Paresthesia
Paresthesia (usually tingling of the extremities), an effect associated with the use of other carbonic anhydrase inhibitors, appears to be a common effect of topiramate in adult and pediatric patients. Paresthesia was more frequently reported in the monotherapy epilepsy trials than in the adjunctive therapy epilepsy trials. In the majority of instances, paresthesia did not lead to treatment discontinuation.
5.14 Adjustment of Dose in Renal Failure
The major route of elimination of unchanged topiramate and its metabolites is via the kidney. Dosage adjustment may be required in patients with reduced renal function [see Dosage and Administration (2.4)].
5.15 Decreased Hepatic Function
In hepatically impaired patients, topiramate should be administered with caution as the clearance of topiramate may be decreased. [see Dosage and Administration (2.7)].
5.16 Monitoring: Laboratory Tests
Topiramate treatment was associated with changes in several clinical laboratory analytes in randomized, double-blind, placebo-controlled studies.
Topiramate treatment causes non-anion gap, hyperchloremic metabolic acidosis manifested by a decrease in serum bicarbonate and an increase in serum chloride. Measurement of baseline and periodic serum bicarbonate during topiramate treatment is recommended [see Warnings and Precautions (5.4)].
Topiramate treatment with or without concomitant valproic acid (VPA) can cause hyperammonemia with or without encephalopathy [see Warnings and Precautions (5.10)].
The clinical significance of decreased serum bicarbonate and associated increased serum chloride reflecting metabolic acidosis and of increased ammonia reflecting hyperammonemia which may be associated with encephalopathy is described [see Warnings and Precautions (5.4 and 5.10)]. However, the clinical significance of these other various abnormalities in other clinical laboratory analytes described here has not been clearly established.
Epilepsy
Controlled trials of adjunctive topiramate treatment of adults for partial onset seizures showed an increased incidence of markedly decreased serum phosphorus (6% topiramate, 2% placebo), markedly increased serum alkaline phosphatase (3% topiramate, 1% placebo), and decreased serum potassium (0.4% topiramate, 0.1% placebo).
Changes in several clinical laboratory analytes (i.e., increased creatinine, BUN, alkaline phosphatase, total protein, total eosinophil count, and decreased potassium) have been observed in a clinical investigational program in very young (<2 years) pediatric patients who were treated with adjunctive topiramate for partial onset seizures [see Use in Specific Populations (8.4)].
Other Use
In pooled double-blind studies in pediatric patients (6 to 17 years), an increased risk for certain abnormalities (value outside normal reference range) in selected clinical laboratory analytes measured in blood has been observed during topiramate treatment of pediatric patients compared to placebo-treated patients. In some instances, abnormalities were also observed at the end of the trial at the final visit and the changes were considered markedly abnormal.
For patients 12 to 17 years, the following were noted to be abnormally increased more frequently with topiramate than with placebo: BUN, creatinine, uric acid, chloride [see Warnings and Precautions (5.4)], ammonia [see Warnings and Precautions (5.10)], total protein, and platelets. The following were abnormally decreased in some subjects: phosphorus, and bicarbonate [see Warnings and Precautions (5.4)].
For patients 6 to 11 years, the following were noted to be abnormally increased more frequently with topiramate than with placebo: alkaline phosphatase, creatinine and eosinophils. Analytes abnormally decreased were: total white count and neutrophils. There was no testing for serum bicarbonate, chloride, ammonia, or phosphorus in these younger patients.
6. ADVERSE REACTIONS
The following adverse reactions are discussed in more detail in other sections of the labeling:
- Acute Myopia and Secondary Angle Closure [see Warnings and Precautions (5.1)]
- Visual Field Defects [see Warnings and Precautions (5.2)]
- Oligohidrosis and Hyperthermia [see Warnings and Precautions (5.3)]
- Metabolic Acidosis [see Warnings and Precautions (5.4)]
- Suicidal Behavior and Ideation [see Warnings and Precautions (5.5)]
- Cognitive/Neuropsychiatric Adverse Reactions[see Warnings and Precautions (5.6)]
- Fetal Toxicity [see Warnings and Precautions (5.7) and Use in Specific Populations (8.1)]
- Sudden Unexplained Death in Epilepsy (SUDEP) [see Warnings and Precautions (5.9)]
- Hyperammonemia and Encephalopathy (Without and With Concomitant Valproic Acid [VPA] Use) [see Warnings and Precautions (5.10)]
- Kidney Stones [see Warnings and Precautions (5.11)]
- Hypothermia with Concomitant Valproic Acid (VPA) Use [see Warnings and Precautions (5.12)]
- Paresthesia [see Warnings and Precautions (5.13)]
The data described in the following section were obtained using topiramate tablets.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the incidence of adverse reactions observed in the clinical trials of a drug cannot be directly compared to the incidence of adverse reactions in the clinical trials of another drug, and may not reflect the incidence of adverse reactions observed in practice.
Increased Risk for Bleeding Topiramate treatment is associated with an increased risk for bleeding. In a pooled analysis of placebo-controlled studies of approved and unapproved indications, bleeding was more frequently reported as an adverse event for topiramatethan for placebo (4.5% versus 3.0% in adult patients, and 4.4% versus 2.3% in pediatric patients). In this analysis, the incidence of serious bleeding events for topiramateand placebo was 0.3% versus 0.2% for adult patients, and 0.4% versus 0% for pediatric patients.
Adverse bleeding reactions reported with topiramateranged from mild epistaxis, ecchymosis, and increased menstrual bleeding to life-threatening hemorrhages. In patients with serious bleeding events, conditions that increased the risk for bleeding were often present, or patients were often taking drugs that cause thrombocytopenia (other antiepileptic drugs) or affect platelet function or coagulation (e.g., aspirin, nonsteroidal anti-inflammatory drugs, selective serotonin reuptake inhibitors, or warfarin or other anticoagulants).
Monotherapy Epilepsy
Adults ≥16 Years
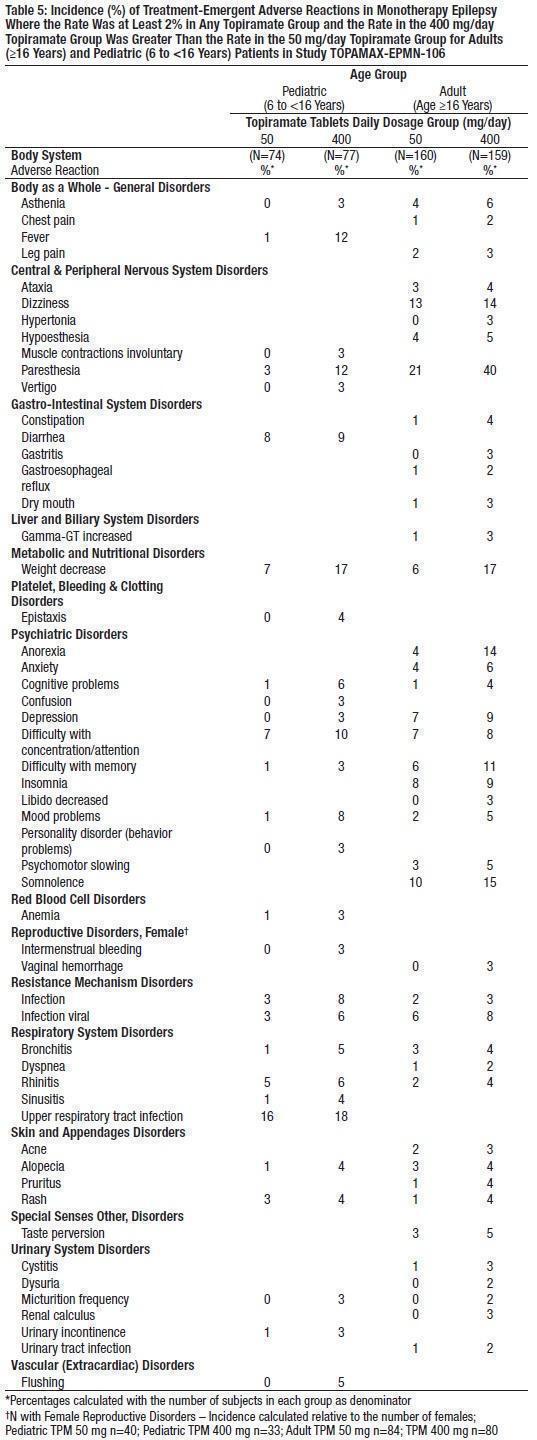
The adverse reactions in the controlled trial that occurred most commonly in adults in the 400 mg/day topiramate group and at a rate higher (≥5 %) than in the 50 mg/day group were: paresthesia, weight decrease, anorexia, somnolence, and difficulty with memory (see Table 5).
Approximately 21% of the 159 adult patients in the 400 mg/day group who received topiramate as monotherapy in the controlled clinical trial discontinued therapy due to adverse reactions. The most common (≥ 2% more frequent than low-dose 50 mg/day topiramate) adverse reactions causing discontinuation in this trial were difficulty with memory, fatigue, asthenia, insomnia, somnolence, and paresthesia.
Pediatric Patients 6 to <16 Years of Age
The adverse reactions in the controlled trial that occurred most commonly in pediatric patients in the 400 mg/day topiramate group and at a rate higher (≥ 5%) than in the 50 mg/day group were fever, weight decrease, mood problems, cognitive problems, infection, flushing, and paresthesia (see Table 5). Table 5 also presents the incidence of adverse reactions occurring in at least 2% of adult and pediatric patients treated with 400 mg/day topiramate and occurring with greater incidence than 50 mg/day topiramate.
Approximately 14 % of the 77 pediatric patients in the 400 mg/day group who received topiramate as monotherapy in the controlled clinical trial discontinued therapy due to adverse reactions. The most common (≥ 2% more frequent than low-dose 50 mg/day topiramate) adverse reactions resulting in discontinuation in this trial were difficulty with concentration/attention, fever, flushing, and confusion.
Adjunctive Therapy Epilepsy
The most commonly observed adverse reactions associated with the use of topiramate at dosages of 200 to 400 mg/day (recommended dose range) in controlled trials in adults with partial onset seizures, primary generalized tonic-clonic seizures, or Lennox-Gastaut syndrome, that were seen at an incidence higher (≥ 5%) than in the placebo group were : somnolence, weight decrease, anorexia, dizziness, ataxia, speech disorders and related speech problems, language problems, psychomotor slowing, confusion, abnormal vision, difficulty with memory, paresthesia, diplopia, nervousness, and asthenia (see Table 6). Dose-related adverse reactions at dosages of 200 to 1,000 mg/day are shown in Table 8.
The most commonly observed adverse reactions associated with the use of topiramate at dosages of 5 to 9 mg/kg/day in controlled trials in pediatric patients with partial onset seizures, primary generalized tonic-clonic seizures, or Lennox-Gastaut syndrome, that were seen at an incidence higher (≥ 5%) than in the placebo group were: fatigue, somnolence, anorexia, nervousness, difficulty with concentration/attention, difficulty with memory, aggressive reaction, and weight decrease (see Table 9). Table 9 also presents the incidence of adverse reactions occurring in at least 1% of pediatric patients treated with topiramate and occurring with greater incidence than placebo.
In controlled clinical trials in adults, 11% of patients receiving topiramate 200 to 400 mg/day as adjunctive therapy discontinued due to adverse reactions. This rate appeared to increase at dosages above 400 mg/day. Adverse reactions associated with discontinuing therapy included somnolence, dizziness, anxiety, difficulty with concentration or attention, fatigue, and paresthesia and increased at dosages above 400 mg/day. None of the pediatric patients who received topiramate adjunctive therapy at 5 to 9 mg/kg/day in controlled clinical trials discontinued due to adverse reactions.
Approximately 28% of the 1757 adults with epilepsy who received topiramate at dosages of 200 to 1,600 mg/day in clinical studies discontinued treatment because of adverse reactions; an individual patient could have reported more than one adverse reaction. These adverse reactions were psychomotor slowing (4%), difficulty with memory (3.2%), fatigue (3.2%), confusion (3.1%), somnolence (3.2%), difficulty with concentration/attention (2.9%), anorexia (2.7%), depression (2.6%), dizziness (2.5%), weight decrease (2.5%), nervousness (2.3%), ataxia (2.1%), and paresthesia (2%). Approximately 11% of the 310 pediatric patients who received topiramate at dosages up to 30 mg/kg/day discontinued due to adverse reactions. Adverse reactions associated with discontinuing therapy included aggravated convulsions (2.3%), difficulty with concentration/attention (1.6%), language problems (1.3%), personality disorder (1.3%), and somnolence (1.3%). Incidence in Epilepsy Controlled Clinical Trials – Adjunctive Therapy – Partial Onset Seizures, Primary Generalized Tonic-Clonic Seizures, and Lennox-Gastaut Syndrome
Table 6 lists the incidence of adverse reactions that occurred in at least 1% of adults treated with 200 to 400 mg/day topiramate (and also higher daily dosing of 600 mg to 1000 mg) in controlled trials that was numerically greater with topiramate than with placebo. In general, most patients who experienced adverse reactions during the first eight weeks of these trials no longer experienced them by their last visit. Table 9 lists the incidence of treatment-emergent adverse reactions that occurred in at least 1% of pediatric patients treated with 5 to 9 mg/kg topiramate in controlled trials and that was numerically greater than the incidence in patients treated with placebo.
The prescriber should be aware that these data were obtained when topiramate was added to concurrent antiepileptic drug therapy and cannot be used to predict the frequency of adverse reactions in the course of usual medical practice where patient characteristics and other factors may differ from those prevailing during clinical studies. Similarly, the cited frequencies cannot be directly compared with data obtained from other clinical investigations involving different treatments, uses, or investigators. Inspection of these frequencies, however, does provide the prescribing physician with a basis to estimate the relative contribution of drug and non-drug factors to the adverse reaction incidences in the population studied.
Other Adverse Reactions Observed During Double-Blind Epilepsy Adjunctive Therapy Trials
Other adverse reactions that occurred in more than 1% of adults treated with 200 to 400 mg of topiramate in placebo-controlled epilepsy trials but with equal or greater frequency in the placebo group were headache, injury, anxiety, rash, pain, convulsions aggravated, coughing, fever, diarrhea, vomiting, muscle weakness, insomnia, personality disorder, dysmenorrhea, upper respiratory tract infection, and eye pain.
Table 6:Incidence of Treatment-Emergent Adverse Reactions in Placebo-Controlled, Add-On Epilepsy Trials in Adultsa, bWhere Incidence Was ≥1% in Any Topiramate Group and Greater Than the Rate in Placebo-Treated Patients
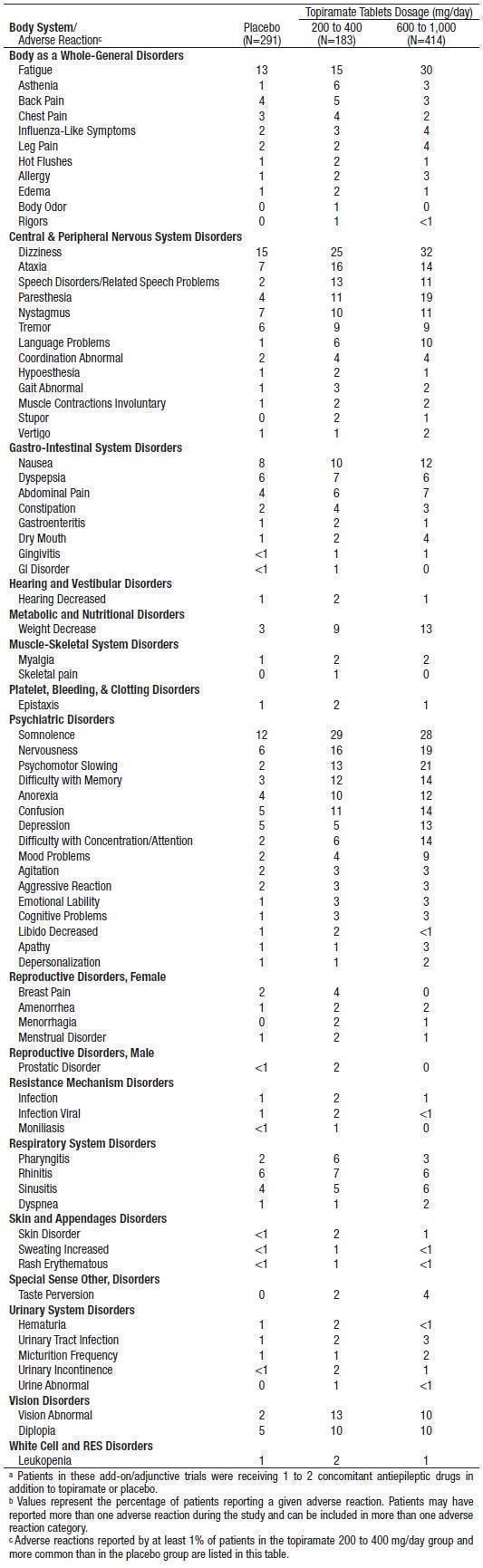
Incidence in Study 119 – Add-On Therapy– Adults with Partial Onset Seizures
Study 119 was a randomized, double-blind, add-on/adjunctive, placebo-controlled, parallel group study with 3 treatment arms: 1) placebo; 2) topiramate 200 mg/day with a 25 mg/day starting dose, increased by 25 mg/day each week for 8 weeks until the 200 mg/day maintenance dose was reached; and 3) topiramate 200 mg/day with a 50 mg/day starting dose, increased by 50 mg/day each week for 4 weeks until the 200 mg/day maintenance dose was reached. All patients were maintained on concomitant carbamazepine with or without another concomitant antiepileptic drug.
The most commonly observed adverse reactions associated with the use of topiramate that were seen at an incidence higher (≥ 5%) than in the placebo group were: paresthesia, nervousness, somnolence, difficulty with concentration/attention, and fatigue (see Table 7). Because these topiramate treatment difference incidence (topiramate % - Placebo %) of many adverse reactions reported in this study were markedly lower than those reported in the previous epilepsy studies, they cannot be directly compared with data obtained in other studies.

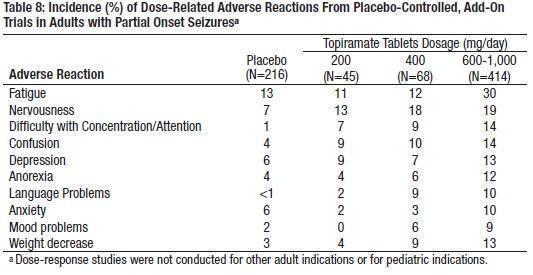
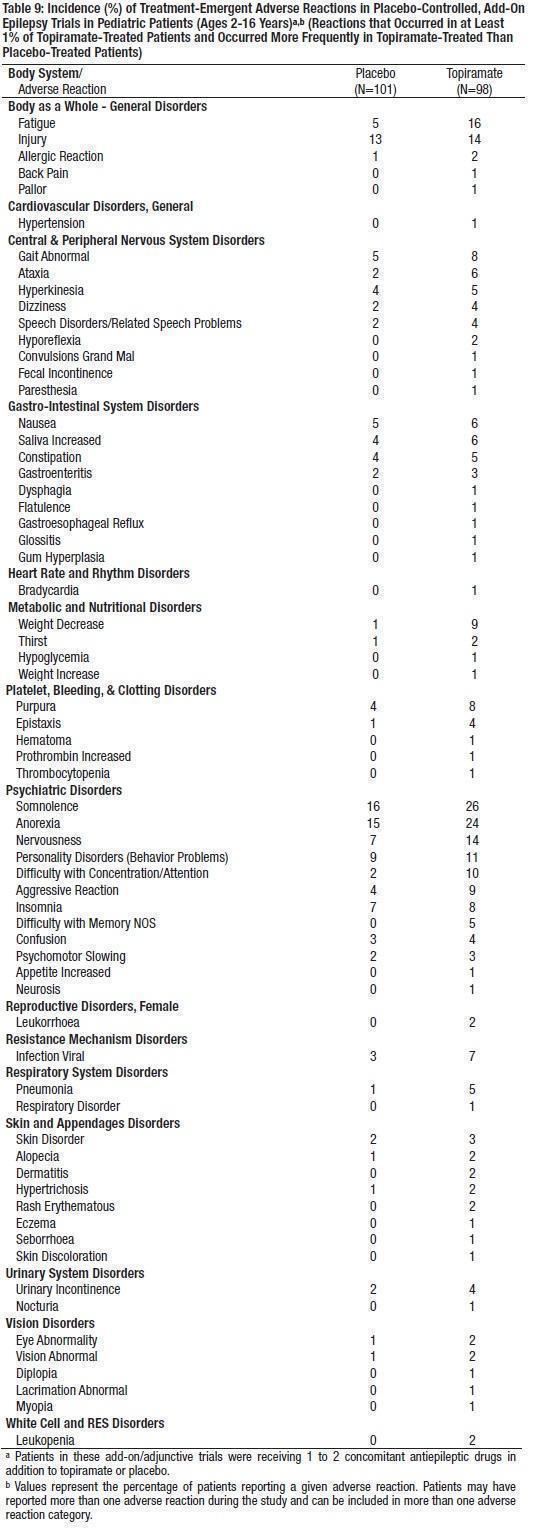
Other Adverse Reactions Observed During All Epilepsy Clinical Trials
Topiramate has been administered to 2246 adults and 427 pediatric patients with epilepsy during all clinical studies, only some of which were placebo-controlled. During these studies, all adverse reactions were recorded by the clinical investigators using terminology of their own choosing. To provide a meaningful estimate of the proportion of individuals having adverse reactions, similar types of reactions were grouped into a smaller number of standardized categories using modified WHOART dictionary terminology. The frequencies presented represent the proportion of patients who experienced a reaction of the type cited on at least one occasion while receiving topiramate. Reported reactions are included except those already listed in the previous tables or text, those too general to be informative, and those not reasonably associated with the use of the drug.
Reactions are classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent occurring in at least 1/100 patients; infrequent occurring in 1/100 to 1/1000 patients; rare occurring in fewer than 1/1000 patients.
Autonomic Nervous System Disorders: Infrequent: vasodilation.
Body as a Whole: Frequent: syncope. Infrequent: abdomen enlarged. Rare: alcohol intolerance.
Cardiovascular Disorders, General: Infrequent: hypotension, postural hypotension, angina pectoris.
Central & Peripheral Nervous System Disorders: Infrequent: neuropathy, apraxia, hyperesthesia, dyskinesia, dysphonia, scotoma, ptosis, dystonia, visual field defect, encephalopathy, EEG abnormal. Rare: upper motor neuron lesion, cerebellar syndrome, tongue paralysis.
Gastrointestinal System Disorders: Infrequent: hemorrhoids, stomatitis, melena, gastritis, esophagitis. Rare: tongue edema.
Heart Rate and Rhythm Disorders: Infrequent: AV block.
Liver and Biliary System Disorders: Infrequent: SGPT increased, SGOT increased.
Metabolic and Nutritional Disorders: Infrequent: dehydration, hypocalcemia, hyperlipemia, hyperglycemia, xerophthalmia, diabetes mellitus. Rare: hypernatremia, hyponatremia, hypocholesterolemia, creatinine increased.
Musculoskeletal System Disorders: Frequent: arthralgia. Infrequent: arthrosis.
Neoplasms: Infrequent: thrombocythemia. Rare: polycythemia.
Platelet, Bleeding, and Clotting Disorders: Infrequent: gingival bleeding, pulmonary embolism.
Psychiatric Disorders: Frequent: impotence, hallucination, psychosis, suicide attempt. Infrequent: euphoria, paranoid reaction, delusion, paranoia, delirium, abnormal dreaming. Rare: libido increased, manic reaction.
Red Blood Cell Disorders: Frequent: anemia. Rare: marrow depression, pancytopenia.
Reproductive Disorders, Male: Infrequent: ejaculation disorder, breast discharge.
Skin and Appendages Disorders: Infrequent: urticaria, photosensitivity reaction, abnormal hair texture. Rare: chloasma.
Special Senses Other, Disorders: Infrequent: taste loss, parosmia.
Urinary System Disorders: Infrequent: urinary retention, face edema, renal pain, albuminuria, polyuria, oliguria.
Vascular (Extracardiac) Disorders: Infrequent: flushing, deep vein thrombosis, phlebitis. Rare: vasospasm.
Vision Disorders: Frequent: conjunctivitis. Infrequent: abnormal accommodation, photophobia, strabismus. Rare: mydriasis, iritis.
White Cell and Reticuloendothelial System Disorders: Infrequent: lymphadenopathy, eosinophilia, lymphopenia, granulocytopenia. Rare: lymphocytosis.
6.2 Postmarketing and Other Experience
In addition to the adverse experiences reported during clinical testing of topiramate, the following adverse experiences have been reported worldwide in patients receiving topiramate post-approval.
These adverse experiences have not been listed above and data are insufficient to support an estimate of their incidence or to establish causation. The listing is alphabetized: bullous skin reactions (including erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis), hepatic failure (including fatalities), hepatitis, maculopathy, pancreatitis, and pemphigus.
7. DRUG INTERACTIONS
In vitro studies indicate that topiramate does not inhibit enzyme activity for CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2D6, CYP2E1, and CYP3A4/5 isozymes. In vitro studies indicate that topiramate is a mild inhibitor of CYP2C19 and a mild inducer of CYP3A4. Drug interactions with some antiepileptic drugs, CNS depressants and oral contraceptives are described here. For other drug interactions, please refer to Clinical Pharmacology (12.3).
7.1 Antiepileptic Drugs
Potential interactions between topiramate and standard AEDs were assessed in controlled clinical pharmacokinetic studies in patients with epilepsy. Concomitant administration of phenytoin or carbamazepine with topiramate decreased plasma concentrations of topiramate by 48% and 40% respectively when compared to topiramate given alone [see Clinical Pharmacology (12.3)].
Concomitant administration of valproic acid and topiramate has been associated with hyperammonemia with and without encephalopathy. Concomitant administration of topiramate with valproic acid has also been associated with hypothermia (with and without hyperammonemia) in patients who have tolerated either drug alone. It may be prudent to examine blood ammonia levels in patients in whom the onset of hypothermia has been reported [see Warnings and Precautions (5.10) (5.12) or Clinical Pharmacology (12.3)].
7.2 CNS Depressants
Concomitant administration of topiramate and alcohol or other CNS depressant drugs has not been evaluated in clinical studies. Because of the potential of topiramate to cause CNS depression, as well as other cognitive and/or neuropsychiatric adverse reactions, topiramate should be used with extreme caution if used in combination with alcohol and other CNS depressants.
7.3 Oral Contraceptives
Exposure to ethinyl estradiol was statistically significantly decreased at doses of 200 mg, 400 mg, and 800 mg/day (18%, 21%, and 30%, respectively) when topiramate was given as adjunctive therapy in patients taking valproic acid. However, norethindrone exposure was not significantly affected. In another pharmacokinetic interaction study in healthy volunteers with a concomitantly administered combination oral contraceptive product containing 1 mg norethindrone (NET) plus 35 mcg ethinyl estradiol (EE), topiramate, given in the absence of other medications at doses of 50 to 200 mg/day, was not associated with statistically significant changes in mean exposure (AUC) to either component of the oral contraceptive. The possibility of decreased contraceptive efficacy and increased breakthrough bleeding should be considered in patients taking combination oral contraceptive products with topiramate. Patients taking estrogen-containing contraceptives should be asked to report any change in their bleeding patterns. Contraceptive efficacy can be decreased even in the absence of breakthrough bleeding [see Clinical Pharmacology (12.3)].
7.4 Metformin
Topiramate treatment can frequently cause metabolic acidosis, a condition for which the use of metformin is contraindicated [see Clinical Pharmacology (12.3)].
7.5 Lithium
In patients, lithium levels were unaffected during treatment with topiramate at doses of 200 mg/day; however, there was an observed increase in systemic exposure of lithium (27% for Cmax and 26% for AUC) following topiramate doses of up to 600 mg/day. Lithium levels should be monitored when coadministered with high-dose topiramate [see Clinical Pharmacology (12.3)].
7.6 Other Carbonic Anhydrase Inhibitors
Concomitant use of topiramate, a carbonic anhydrase inhibitor, with any other carbonic anhydrase inhibitor (e.g., zonisamide, acetazolamide or dichlorphenamide), may increase the severity of metabolic acidosis and may also increase the risk of kidney stone formation. Therefore, if topiramate is given concomitantly with another carbonic anhydrase inhibitor, the patient should be monitored for the appearance or worsening of metabolic acidosis [see Clinical Pharmacology (12.3)].
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category D. [see Warnings and Precautions (5.7)]
Topiramate can cause fetal harm when administered to a pregnant woman. Data from pregnancy registries indicate that infants exposed to topiramate in utero have an increased risk for cleft lip and/or cleft palate (oral clefts). When multiple species of pregnant animals received topiramate at clinically relevant doses, structural malformations, including craniofacial defects, and reduced fetal weights occurred in offspring. Topiramate should be used during pregnancy only if the potential benefit outweighs the potential risk. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus [see Use in Specific Populations (8.9)].
Pregnancy Registry
Patients should be encouraged to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry if they become pregnant. This registry is collecting information about the safety of antiepileptic drugs during pregnancy. To enroll, patients can call the toll free number 1-888-233-2334. Information about the North American Drug Pregnancy Registry can be found at http://www.massgeneral.org/aed/.
Human Data
Data from the NAAED Pregnancy Registry (425 prospective topiramate monotherapy-exposed pregnancies) indicate an increased risk of oral clefts in infants exposed during the first trimester of pregnancy. The prevalence of oral clefts among topiramate-exposed infants was 1.2% compared to a prevalence of 0.39% for infants exposed to a reference AED. In infants of mothers without epilepsy or treatment with other AEDs, the prevalence was 0.12%. For comparison, the Centers for Disease Control and Prevention (CDC) reviewed available data on oral clefts in the United States and found a background rate of 0.17%.
The relative risk of oral clefts in topiramate-exposed pregnancies in the NAAED Pregnancy Registry was 9.6 (95% Confidence Interval[CI] 4 to 23) as compared to the risk in a background population of untreated women. The UK Epilepsy and Pregnancy Register reported a similarly increased prevalence of oral clefts of 3.2% among infants exposed to topiramate monotherapy. The observed rate of oral clefts was 16 times higher than the background rate in the UK, which is approximately 0.2%.
Topiramate treatment can cause metabolic acidosis [see Warnings and Precautions (5.4)]. The effect of topiramate-induced metabolic acidosis has not been studied in pregnancy; however, metabolic acidosis in pregnancy (due to other causes) can cause decreased fetal growth, decreased fetal oxygenation, and fetal death, and may affect the fetus’ ability to tolerate labor. Pregnant patients should be monitored for metabolic acidosis and treated as in the nonpregnant state [see Warnings and Precautions (5.4)].
Newborns of mothers treated with topiramate should be monitored for metabolic acidosis because of transfer of topiramate to the fetus and possible occurrence of transient metabolic acidosis following birth.
Animal Data
Topiramate has demonstrated selective developmental toxicity, including teratogenicity, in multiple animal species at clinically relevant doses. When oral doses of 20 mg, 100 mg or 500 mg/kg were administered to pregnant mice during the period of organogenesis, the incidence of fetal malformations (primarily craniofacial defects) was increased at all doses. The low dose is approximately 0.2 times the recommended human dose (RHD) 400 mg/day on a mg/ m2 basis. Fetal body weights and skeletal ossification were reduced at 500 mg/kg in conjunction with decreased maternal body weight gain.
In rat studies (oral doses of 20 mg, 100 mg, and 500 mg/kg or 0.2 mg, 2.5 mg, 30 mg, and 400 mg/kg), the frequency of limb malformations (ectrodactyly, micromelia, and amelia) was increased among the offspring of dams treated with 400 mg/kg (10 times the RHD on a mg/ m2 basis) or greater during the organogenesis period of pregnancy. Embryotoxicity (reduced fetal body weights, increased incidence of structural variations) was observed at doses as low as 20 mg/kg (0.5 times the RHD on a mg/ m2 basis). Clinical signs of maternal toxicity were seen at 400 mg/kg and above, and maternal body weight gain was reduced during treatment with 100 mg/kg or greater.
In rabbit studies (20 mg, 60 mg, or 180 mg/kg or 10 mg, 35 mg, and 120 mg/kg orally during organogenesis), embryo/fetal mortality was increased at 35 mg/kg (2 times the RHD on a mg/ m2 basis) or greater, and teratogenic effects (primarily rib and vertebral malformations) were observed at 120 mg/kg (6 times the RHD on a mg/ m2 basis). Evidence of maternal toxicity (decreased body weight gain, clinical signs, and/or mortality) was seen at 35 mg/kg and above.
When female rats were treated during the latter part of gestation and throughout lactation (0.2 mg, 4 mg, 20 mg, and 100 mg/kg or 2 mg, 20 mg, and 200 mg/kg), offspring exhibited decreased viability and delayed physical development at 200 mg/kg (5 times the RHD on a mg/ m2 basis) and reductions in pre-and/or postweaning body weight gain at 2 mg/kg (0.05 times the RHD on a mg/ m2 basis) and above. Maternal toxicity (decreased body weight gain, clinical signs) was evident at 100 mg/kg or greater.
In a rat embryo/fetal development study with a postnatal component (0.2 mg, 2.5 mg, 30 mg or 400 mg/kg during organogenesis; noted above), pups exhibited delayed physical development at 400 mg/kg (10 times the RHD on a mg/ m2 basis) and persistent reductions in body weight gain at 30 mg/kg (1 times the RHD on a mg/ m2 basis) and higher.
8.2 Labor and Delivery
Although the effect of topiramate on labor and delivery in humans has not been established, the development of topiramate-induced metabolic acidosis in the mother and/or in the fetus might affect the fetus’ ability to tolerate labor [see Use in Specific Populations (8.1)].
8.3 Nursing Mothers
Limited data on 5 breastfeeding infants exposed to topiramate showed infant plasma topiramate levels equal to 10 to 20% of the maternal plasma level. The effects of this exposure on infants are unknown. Caution should be exercised when administered to a nursing woman.
8.4 Pediatric Use
Adjunctive Treatment for Partial Onset Epilepsy in Infants and Toddlers (1 to 24 months)
Safety and effectiveness in patients below the age of 2 years have not been established for the adjunctive therapy treatment of partial onset seizures, primary generalized tonic-clonic seizures, or seizures associated with Lennox-Gastaut syndrome. In a single randomized, double-blind placebo-controlled investigational trial, the efficacy, safety, and tolerability of topiramate oral liquid and sprinkle formulations as an adjunct to concurrent antiepileptic drug therapy in infants 1 to 24 months of age with refractory partial onset seizures were assessed. After 20 days of double-blind treatment, topiramate (at fixed doses of 5 mg, 15 mg, and 25 mg/kg/day) did not demonstrate efficacy compared with placebo in controlling seizures.
In general, the adverse reaction profile in this population was similar to that of older pediatric patients, although results from the above controlled study and an open-label, long-term extension study in these infants/toddlers (1 to 24 months old) suggested some adverse reactions/toxicities (not previously observed in older pediatric patients and adults; i.e, growth/length retardation, certain clinical laboratory abnormalities, and other adverse reactions/toxicities that occurred with a greater frequency and/or greater severity than had been recognized previously from studies in older pediatric patients or adults for various indications.
These very young pediatric patients appeared to experience an increased risk for infections (any topiramate dose 12%, placebo 0%) and of respiratory disorders (any topiramate dose 40%, placebo 16%). The following adverse reactions were observed in at least 3% of patients on topiramate and were 3% to 7% more frequent then in patients on placebo: viral infection, bronchitis, pharyngitis, rhinitis, otitis media, upper respiratory infection, cough, and bronchospasm. A generally similar profile was observed in older children [see Adverse Reactions (6)].
Topiramate resulted in an increased incidence of patients with increased creatinine (any topiramate dose 5%, placebo 0%), BUN (any topiramate dose 3%, placebo 0%), and protein (any topiramate dose 34%, placebo 6%), and an increased incidence of decreased potassium (any topiramate dose 7%, placebo 0%). This increased frequency of abnormal values was not dose-related. Creatinine was the only analyte showing a noteworthy increased incidence (topiramate 25 mg/kg/day 5%, placebo 0%) of a markedly abnormal increase [see Warnings and Precautions (5.16)]. The significance of these finding is uncertain.
Topiramate treatment also produced a dose-related increase in the percentage of patients who had a shift from normal at baseline to high/increased (above the normal reference range) in total eosinophil count at the end of treatment. The incidence of these abnormal shifts was 6 % for placebo, 10% for 5 mg/kg/day, 9% for 15 mg/kg/day, 14% for 25 mg/kg/day, and 11% for any topiramate dose [see Warnings and Precautions (5.16)]. There was a mean dose-related increase in alkaline phosphatase. The significance of these findings is uncertain.
Topiramate produced a dose-related increased incidence of treatment-emergent hyperammonemia [see Warnings and Precautions (5.10)].
Treatment with topiramate for up to 1 year was associated with reductions in Z SCORES for length, weight, and head circumference [see Warnings and Precautions (5.4) and Adverse Reactions (6)].
In open-label, uncontrolled experience, increasing impairment of adaptive behavior was documented in behavioral testing over time in this population. There was a suggestion that this effect was dose-related. However, because of the absence of an appropriate control group, it is not known if this decrement in function was treatment-related or reflects the patient’s underlying disease (e.g., patients who received higher doses may have more severe underlying disease) [see Warnings and Precautions (5.6)].
In this open-label, uncontrolled study, the mortality was 37 deaths/1000 patient years. It is not possible to know whether this mortality rate is related to topiramate treatment, because the background mortality rate for a similar, significantly refractory, young pediatric population (1 to 24 months) with partial epilepsy is not known.
Monotherapy Treatment in Partial Onset Epilepsy in Patients <2 Years Old
Safety and effectiveness in patients below the age of 2 years have not been established for the monotherapy treatment of epilepsy.
Juvenile Animal Studies
When topiramate (30 mg, 90 mg, or 300 mg/kg/day) was administered orally to rats during the juvenile period of development (postnatal days 12 to 50), bone growth plate thickness was reduced in males at the highest dose, which is approximately 5 to 8 times the maximum recommended pediatric dose (9 mg/kg/day) on a body surface area (mg/ m2) basis.
8.5 Geriatric Use
In clinical trials, 3% of patients were over 60. No age-related difference in effectiveness or adverse effects was evident. However, clinical studies of topiramate did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently than younger subjects. Dosage adjustment may be necessary for elderly with impaired renal function (creatinine clearance rate <70 mL/min/1.73 m2) due to reduced clearance of topiramate [see Clinical Pharmacology (12.3) and Dosage and Administration (2.5)].
8.6 Race and Gender Effects
Evaluation of effectiveness and safety in clinical trials has shown no race- or gender- related effects.
8.7 Renal Impairment
The clearance of topiramate was reduced by 42% in moderately renally impaired (creatinine clearance 30 to 69 mL/min/1.73 m2) and by 54% in severely renally impaired subjects (creatinine clearance <30 mL/min/1.73 m2) compared to normal renal function subjects (creatinine clearance >70 mL/min/1.73 m2). One-half the usual starting and maintenance dose is recommended in patients with moderate or severe renal impairment [see Dosage and Administration (2.6) and Clinical Pharmacology (12.3)].
8.8 Patients Undergoing Hemodialysis
Topiramate is cleared by hemodialysis at a rate that is 4 to 6 times greater than in a normal individual. Accordingly, a prolonged period of dialysis may cause topiramate concentration to fall below that required to maintain an anti-seizure effect. To avoid rapid drops in topiramate plasma concentration during hemodialysis, a supplemental dose of topiramate may be required.
The actual adjustment should take into account the duration of dialysis period, the clearance rate of the dialysis system being used, and the effective renal clearance of topiramate in the patient being dialyzed [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
8.9 Women of Childbearing Potential
Data from pregnancy registries indicate that infants exposed to topiramate in utero have an increased risk for cleft lip and/or cleft palate (oral clefts) [see Warnings and Precautions (5.7) and Use in Specific Populations (8.1)]. Consider the benefits and the risks of topiramate when prescribing this drug to women of childbearing potential, particularly when topiramate is considered for a condition not usually associated with permanent injury or death. Because of the risk of oral clefts to the fetus, which occur in the first trimester of pregnancy before many women know they are pregnant, all women of childbearing potential should be apprised of the potential hazard to the fetus from exposure to topiramate. If the decision is made to use topiramate, women who are not planning a pregnancy should use effective contraception [see Drug Interactions (7.3)]. Women who are planning a pregnancy should be counseled regarding the relative risks and benefits of topiramate use during pregnancy, and alternative therapeutic options should be considered for these patients
10. OVERDOSAGE
Overdoses of topiramate have been reported. Signs and symptoms included convulsions, drowsiness, speech disturbance, blurred vision, diplopia, mentation impaired, lethargy, abnormal coordination, stupor, hypotension, abdominal pain, agitation, dizziness and depression. The clinical consequences were not severe in most cases, but deaths have been reported after poly-drug overdoses involving topiramate.
Topiramate overdose has resulted in severe metabolic acidosis [see Warnings and Precautions (5.4)].
A patient who ingested a dose between 96 and 110 g topiramate was admitted to a hospital with a coma lasting 20 to 24 hours followed by full recovery after 3 to 4 days.
In acute topiramate overdose, if the ingestion is recent, the stomach should be emptied immediately by lavage or by induction of emesis. Activated charcoal has been shown to adsorb topiramate in vitro. Treatment should be appropriately supportive. Hemodialysis is an effective means of removing topiramate from the body.
11. DESCRIPTION
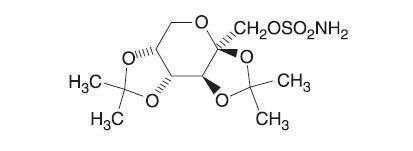
Topiramate is a sulfamate-substituted monosaccharide. Topiramate tablets, USP are available as 25 mg, 50 mg, 100 mg, and 200 mg round tablets for oral administration.
Topiramate, USP is a white crystalline powder with a bitter taste. Topiramate is most soluble in alkaline solutions containing sodium hydroxide or sodium phosphate and having a pH of 9 to 10. It is freely soluble in acetone, chloroform, dimethylsulfoxide, and ethanol. The solubility in water is 9.8 mg/mL. Its saturated solution has a pH of 6.3. Topiramate has the molecular formula C12H21NO8S and a molecular weight of 339.36. Topiramate is designated chemically as 2, 3:4, 5-Di-O-isopropylidene-β-D-fructopyranose sulfamate and has the following structural formula:
Topiramate tablets contain the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, pre-gelatinized starch (maize), sodium starch glycolate, magnesium stearate, opadry white (titanium dioxide, hypromellose 3cp, hypromellose 6cp, PEG 400, polysorbate 80) for 25 mg tablets, opadry yellow (titanium dioxide, hypromellose 3cp, hypromellose 6cp, PEG 400, polysorbate 80, iron oxide yellow) for 50 mg tablets, opadry yellow (hypromellose 3cp, hypromellose 6cp, titanium dioxide, PEG 400, iron oxide yellow, polysorbate 80, iron oxide red) for 100 mg tablets and, opadry pink (titanium dioxide, hypromellose 6cp, PEG 400,iron oxide red) for 200 mg tablets.
12. CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The precise mechanisms by which topiramate exerts its anticonvulsant and other effects are unknown; however, preclinical studies have revealed four properties that may contribute to topiramate's efficacy for epilepsy. Electrophysiological and biochemical evidence suggests that topiramate, at pharmacologically relevant concentrations, blocks voltage-dependent sodium channels, augments the activity of the neurotransmitter gamma-aminobutyrate at some subtypes of the GABA-A receptor, antagonizes the AMPA/kainate subtype of the glutamate receptor, and inhibits the carbonic anhydrase enzyme, particularly isozymes II and IV.
12.2 Pharmacodynamics
Topiramate has anticonvulsant activity in rat and mouse maximal electroshock seizure (MES) tests. Topiramate is only weakly effective in blocking clonic seizures induced by the GABAA receptor antagonist, pentylenetetrazole. Topiramate is also effective in rodent models of epilepsy, which include tonic and absence-like seizures in the spontaneous epileptic rat (SER) and tonic and clonic seizures induced in rats by kindling of the amygdala or by global ischemia.
Changes (increases and decreases) from baseline in vital signs (systolic blood pressure-SBP, diastolic blood pressure-DBP, pulse) occurred more frequently in pediatric patients (6 to 17 years) treated with various daily doses of topiramate (50 mg, 100 mg, 200 mg, 2 to 3 mg/kg) than in patients treated with placebo in controlled trials for another indication. The most notable changes were SBP < 90 mm Hg, DBP < 50 mm Hg, SBP or DBP increases or decreases ≥ 20 mm Hg, and pulse increases or decreases ≥ 30 beats per minute. These changes were often dose-related, and were most frequently associated with the greatest treatment difference at the 200 mg dose level. When a position was specified for measurement of vital signs in a trial, measurements were made in a sitting position. Systematic collection of orthostatic vital signs has not been conducted. The clinical significance of these various changes in vital signs has not been clearly established.
12.3 Pharmacokinetics
The sprinkle formulation is bioequivalent to the immediate-release tablet formulation and, therefore, may be substituted as a therapeutic equivalent.
Absorption of topiramate is rapid, with peak plasma concentrations occurring at approximately 2 hours following a 400 mg oral dose. The relative bioavailability of topiramate from the tablet formulation is about 80% compared to a solution. The bioavailability of topiramate is not affected by food.
The pharmacokinetics of topiramate are linear with dose proportional increases in plasma concentration over the dose range studied (200 to 800 mg/day). The mean plasma elimination half-life is 21 hours after single or multiple doses. Steady-state is thus reached in about 4 days in patients with normal renal function. Topiramate is 15% to 41% bound to human plasma proteins over the blood concentration range of 0.5 to 250 µg/mL. The fraction bound decreased as blood concentration increased.
Carbamazepine and phenytoin do not alter the binding of topiramate. Sodium valproate, at 500 µg/mL (a concentration 5 to 10 times higher than considered therapeutic for valproate) decreased the protein binding of topiramate from 23% to 13%. Topiramate does not influence the binding of sodium valproate.
Metabolism and Excretion
Topiramate is not extensively metabolized and is primarily eliminated unchanged in the urine (approximately 70% of an administered dose). Six metabolites have been identified in humans, none of which constitutes more than 5% of an administered dose. The metabolites are formed via hydroxylation, hydrolysis, and glucuronidation. There is evidence of renal tubular reabsorption of topiramate. In rats, given probenecid to inhibit tubular reabsorption, along with topiramate, a significant increase in renal clearance of topiramate was observed. This interaction has not been evaluated in humans. Overall, oral plasma clearance (CL/F) is approximately 20 to 30 mL/min in adults following oral administration.
Special Populations
Renal Impairment
The clearance of topiramate was reduced by 42% in moderately renally impaired (creatinine clearance 30 to 69 mL/min/1.73 m2) and by 54% in severely renally impaired subjects (creatinine clearance <30 mL/min/1.73 m2) compared to normal renal function subjects (creatinine clearance >70 mL/min/1.73 m2). Since topiramate is presumed to undergo significant tubular reabsorption, it is uncertain whether this experience can be generalized to all situations of renal impairment. It is conceivable that some forms of renal disease could differentially affect glomerular filtration rate and tubular reabsorption resulting in a clearance of topiramate not predicted by creatinine clearance. In general, however, use of one-half the usual starting and maintenance dose is recommended in patients with moderate or severe renal impairment [see Dosage and Administration (2.4 ) and (2.5) and Warnings and Precautions (5.14)].
Hemodialysis
Topiramate is cleared by hemodialysis. Using a high-efficiency, counterflow, single pass-dialysate hemodialysis procedure, topiramate dialysis clearance was 120 mL/min with blood flow through the dialyzer at 400 mL/min. This high clearance (compared to 20 to 30 mL/min total oral clearance in healthy adults) will remove a clinically significant amount of topiramate from the patient over the hemodialysis treatment period. Therefore, a supplemental dose may be required [see Dosage and Administration (2.6)].
Hepatic Impairment
In hepatically impaired subjects, the clearance of topiramate may be decreased; the mechanism underlying the decrease is not well understood [see Dosage and Administration (2.7)].
Age, Gender, and Race
The pharmacokinetics of topiramate in elderly subjects (65 to 85 years of age, N=16) were evaluated in a controlled clinical study. The elderly subject population had reduced renal function (creatinine clearance
[-20%]) compared to young adults. Following a single oral 100 mg dose, maximum plasma concentration for elderly and young adults was achieved at approximately 1 to 2 hours. Reflecting the primary renal elimination of topiramate, topiramate plasma and renal clearance were reduced 21% and 19%, respectively, in elderly subjects, compared to young adults. Similarly, topiramate half-life was longer (13%) in the elderly. Reduced topiramate clearance resulted in slightly higher maximum plasma concentration (23%) and AUC (25%) in elderly subjects than observed in young adults. Topiramate clearance is decreased in the elderly only to the extent that renal function is reduced. As recommended for all patients, dosage adjustment may be indicated in the elderly patient when impaired renal function (creatinine clearance rate ≤70 mL/min/1.73 m2) is evident. It may be useful to monitor renal function in the elderly patient [see Dosage and Administration (2.4) and Warnings and Precautions (5.14)].
Clearance of topiramate in adults was not affected by gender or race.
Pediatric Pharmacokinetics
Pharmacokinetics of topiramate were evaluated in patients aged 2 to <16 years. Patients received either no or a combination of other antiepileptic drugs. A population pharmacokinetic model was developed on the basis of pharmacokinetic data from relevant topiramate clinical studies. This dataset contained data from 1217 subjects including 258 pediatric patients aged 2 to <16 years (95 pediatric patients <10 years of age).
Pediatric patients on adjunctive treatment exhibited a higher oral clearance (L/h) of topiramate compared to patients on monotherapy, presumably because of increased clearance from concomitant enzyme-inducing antiepileptic drugs. In comparison, topiramate clearance per kg is greater in pediatric patients than in adults and in young pediatric patients (down to 2 years) than in older pediatric patients. Consequently, the plasma drug concentration for the same mg/kg/day dose would be lower in pediatric patients compared to adults and also in younger pediatric patients compared to older pediatric patients. Clearance was independent of dose.
As in adults, hepatic enzyme-inducing antiepileptic drugs decrease the steady state plasma concentrations of topiramate.
Drug-Drug Interactions
Antiepileptic Drugs
Potential interactions between topiramate and standard AEDs were assessed in controlled clinical pharmacokinetic studies in patients with epilepsy. The effects of these interactions on mean plasma AUCs are summarized in Table 13.
In Table 13, the second column (AED concentration) describes what happens to the concentration of the AED listed in the first column when topiramate is added. The third column (topiramate concentration) describes how the co-administration of a drug listed in the first column modifies the concentration of topiramate in experimental settings when topiramate tablets were given alone.
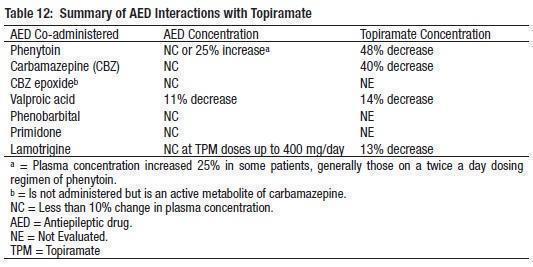
In addition to the pharmacokinetic interaction described in the above table, concomitant administration of valproic acid and topiramate has been associated with hyperammonemia with and without encephalopathy and hypothermia [see Warnings and Precautions (5.10) (5.12) and Drug Interactions (7.1)].
CNS Depressants
Concomitant administration of topiramate and alcohol or other CNS depressant drugs has not been evaluated in clinical studies. Because of the potential of topiramate to cause CNS depression, as well as other cognitive and/or neuropsychiatric adverse reactions, topiramate should be used with extreme caution if used in combination with alcohol and other CNS depressants [see Drug Interactions (7.2)].
Oral Contraceptives
In a pharmacokinetic interaction study in healthy volunteers with a concomitantly administered combination oral contraceptive product containing 1 mg norethindrone (NET) plus 35 mcg ethinyl estradiol (EE), topiramate, given in the absence of other medications at doses of 50 to 200 mg/day, was not associated with statistically significant changes in mean exposure (AUC) to either component of the oral contraceptive. In another study, exposure to EE was statistically significantly decreased at doses of 200 mg, 400 mg, and 800 mg/day (18%, 21%, and 30%, respectively) when given as adjunctive therapy in patients taking valproic acid. In both studies, topiramate (50 mg/day to 800 mg/day) did not significantly affect exposure to NET. Although there was a dose-dependent decrease in EE exposure for doses between 200 and 800 mg/day, there was no significant dose-dependent change in EE exposure for doses of 50 to 200 mg/day. The clinical significance of the changes observed is not known. The possibility of decreased contraceptive efficacy and increased breakthrough bleeding should be considered in patients taking combination oral contraceptive products with topiramate. Patients taking estrogen-containing contraceptives should be asked to report any change in their bleeding patterns. Contraceptive efficacy can be decreased even in the absence of breakthrough bleeding [see Drug Interactions (7.3)].
Digoxin
In a single-dose study, serum digoxin AUC was decreased by 12% with concomitant topiramate administration. The clinical relevance of this observation has not been established.
Hydrochlorothiazide
A drug-drug interaction study conducted in healthy volunteers evaluated the steady-state pharmacokinetics of hydrochlorothiazide (HCTZ) (25 mg q24h) and topiramate (96 mg q12h) when administered alone and concomitantly. The results of this study indicate that topiramate Cmax increased by 27% and AUC increased by 29% when HCTZ was added to topiramate. The clinical significance of this change is unknown. The addition of HCTZ to topiramate therapy may require an adjustment of the topiramate dose. The steady-state pharmacokinetics of HCTZ were not significantly influenced by the concomitant administration of topiramate. Clinical laboratory results indicated decreases in serum potassium after topiramate or HCTZ administration, which were greater when HCTZ and topiramate were administered in combination.
Metformin
Topiramate treatment can frequently cause metabolic acidosis, a condition for which the use of metformin is contraindicated.
A drug-drug interaction study conducted in healthy volunteers evaluated the steady-state pharmacokinetics of metformin (500 mg every 12 hr) and topiramate in plasma when metformin was given alone and when metformin and topiramate (100 mg every 12 hr) were given simultaneously. The results of this study indicated that the mean metformin Cmax and AUC0-12hincreased by 18% and 25%, respectively, when topiramate was added. Topiramate did not affect metformin tmax. The clinical significance of the effect of topiramate on metformin pharmacokinetics is not known. Oral plasma clearance of topiramate appears to be reduced when administered with metformin. The clinical significance of the effect of metformin on topiramate pharmacokinetics is unclear [See Drug Interactions (7.4)].
Pioglitazone
A drug-drug interaction study conducted in healthy volunteers evaluated the steady-state pharmacokinetics of topiramate and pioglitazone when administered alone and concomitantly. A 15% decrease in the AUCτ,ss of pioglitazone with no alteration in Cmax,ss was observed. This finding was not statistically significant. In addition, a 13% and 16% decrease in Cmax,ss and AUCτ,ss respectively, of the active hydroxy-metabolite was noted as well as a 60% decrease in Cmax,ss and AUCτ,ss of the active keto-metabolite. The clinical significance of these findings is not known. When topiramate is added to pioglitazone therapy or pioglitazone is added to topiramate therapy, careful attention should be given to the routine monitoring of patients for adequate control of their diabetic disease state.
Glyburide
A drug-drug interaction study conducted in patients with type 2 diabetes evaluated the steady-state pharmacokinetics of glyburide (5 mg/day) alone and concomitantly with topiramate (150 mg/day). There was a 22% decrease in Cmax and a 25% reduction in AUC24 for glyburide during topiramate administration. Systemic exposure (AUC) of the active metabolites, 4-trans-hydroxy-glyburide (M1) and 3-cis-hydroxyglyburide (M2), was also reduced by 13% and 15%, and Cmax was reduced by 18% and 25%, respectively. The steady-state pharmacokinetics of topiramate were unaffected by concomitant administration of glyburide.
Lithium
In patients, the pharmacokinetics of lithium were unaffected during treatment with topiramate at doses of 200 mg/day; however, there was an observed increase in systemic exposure of lithium (27% for Cmax and 26% for AUC) following topiramate doses up to 600 mg/day. Lithium levels should be monitored when co-administered with high-dose topiramate [See Drug Interactions (7.5)].
Haloperidol
The pharmacokinetics of a single dose of haloperidol (5 mg) were not affected following multiple dosing of topiramate (100 mg every 12 hr) in 13 healthy adults (6 males, 7 females).
Amitriptyline
There was a 12% increase in AUC and Cmax for amitriptyline (25 mg per day) in 18 normal subjects (9 males; 9 females) receiving 200 mg/day of topiramate. Some subjects may experience a large increase in amitriptyline concentration in the presence of topiramate and any adjustments in amitriptyline dose should be made according to the patient's clinical response and not on the basis of plasma levels.
Sumatriptan
Multiple dosing of topiramate (100 mg every 12 hrs) in 24 healthy volunteers (14 males, 10 females) did not affect the pharmacokinetics of single-dose sumatriptan either orally (100 mg) or subcutaneously (6 mg).
Risperidone
When administered concomitantly with topiramate at escalating doses of 100, 250 and 400 mg/day, there was a reduction in risperidone (systemic exposure (16% and 33% for steady-state AUC at the 250 and 400 mg/day doses of topiramate). No alterations of 9-hydroxyrisperidone levels were observed. Co-administration of topiramate 400 mg/day with risperidone resulted in a 14% increase in Cmax and a 12% increase in AUC12 of topiramate. There were no clinically significant changes in the systemic exposure of risperidone plus 9-hydroxyrisperidone or of topiramate; therefore this interaction is not likely to be of clinical significance.
Propranolol
Multiple dosing of topiramate (200 mg/day) in 34 healthy volunteers (17 males, 17 females) did not affect the pharmacokinetics of propranolol following daily 160 mg doses. Propranolol doses of 160 mg/day in 39 volunteers (27 males, 12 females) had no effect on the exposure to topiramate, at a dose of 200 mg/day of topiramate.
Dihydroergotamine
Multiple dosing of topiramate (200 mg/day) in 24 healthy volunteers (12 males, 12 females) did not affect the pharmacokinetics of a 1 mg subcutaneous dose of dihydroergotamine. Similarly, a 1 mg subcutaneous dose of dihydroergotamine did not affect the pharmacokinetics of a 200 mg/day dose of topiramate in the same study.
Diltiazem
Co-administration of diltiazem (240 mg Cardizem CD®) with topiramate (150 mg/day) resulted in a 10% decrease in Cmax and a 25% decrease in diltiazem AUC, a 27% decrease in Cmax and an 18% decrease in des-acetyl diltiazem AUC, and no effect on N-desmethyl diltiazem. Coadministration of topiramate with diltiazem resulted in a 16% increase in Cmax and a 19% increase in AUC12 of topiramate.
Venlafaxine
Multiple dosing of topiramate (150 mg/day) in healthy volunteers did not affect the pharmacokinetics of venlafaxine or O-desmethyl venlafaxine. Multiple dosing of venlafaxine (150 mg Effexor XR®) did not affect the pharmacokinetics of topiramate.
Other Carbonic Anhydrase Inhibitors
Concomitant use of topiramate, a carbonic anhydrase inhibitor, with any other carbonic anhydrase inhibitor (e.g., zonisamide, acetazolamide, or dichlorphenamide) may increase the severity of metabolic acidosis and may also increase the risk of kidney stone formation. Therefore, if topiramate is given concomitantly with another carbonic anhydrase inhibitor, the patient should be monitored for the appearance or worsening of metabolic acidosis [see Drug Interactions (7.6)].
Drug/Laboratory Tests Interactions
There are no known interactions of topiramate with commonly used laboratory tests.
13. NON-CLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenesis
An increase in urinary bladder tumors was observed in mice given topiramate (20 mg, 75 mg, and 300 mg/kg) in the diet for 21 months. The elevated bladder tumor incidence, which was statistically significant in males and females receiving 300 mg/kg, was primarily due to the increased occurrence of a smooth muscle tumor considered histomorphologically unique to mice. Plasma exposures in mice receiving 300 mg/kg were approximately 0.5 to 1 times steady-state exposures measured in patients receiving topiramate monotherapy at the recommended human dose (RHD) of 400 mg, and 1.5 to 2 times steady-state topiramate exposures in patients receiving 400 mg of topiramate plus phenytoin. The relevance of this finding to human carcinogenic risk is uncertain. No evidence of carcinogenicity was seen in rats following oral administration of topiramate for 2 years at doses up to 120 mg/kg (approximately 3 times the RHD on a mg/ m2 basis).
Mutagenesis
Topiramate did not demonstrate genotoxic potential when tested in a battery of in vitro and in vivo assays. Topiramate was not mutagenic in the Ames test or the in vitro mouse lymphoma assay; it did not increase unscheduled DNA synthesis in rat hepatocytes in vitro; and it did not increase chromosomal aberrations in human lymphocytes in vitro or in rat bone marrow in vivo.
Impairment of Fertility
No adverse effects on male or female fertility were observed in rats at doses up to 100 mg/kg (2.5 times the RHD on a mg/ m2 basis).
14. CLINICAL STUDIES
The studies described in the following sections were conducted using topiramate tablets.
14.1 Monotherapy Epilepsy Controlled Trial
Patients with Partial Onset or Primary Generalized Tonic-Clonic Seizures
Adults and Pediatric Patients 10 Years of Age and Older
The effectiveness of topiramate as initial monotherapy in adults and children 10 years of age and older with partial onset or primary generalized tonic-clonic seizures was established in a multicenter, randomized, double-blind, parallel-group trial.
The trial was conducted in 487 patients diagnosed with epilepsy (6 to 83 years of age) who had 1 or 2 well-documented seizures during the 3-month retrospective baseline phase who then entered the study and received topiramate 25 mg/day for 7 days in an open-label fashion. Forty-nine percent of subjects had no prior AED treatment and 17% had a diagnosis of epilepsy for greater than 24 months. Any AED therapy used for temporary or emergency purposes was discontinued prior to randomization. In the double-blind phase, 470 patients were randomized to titrate up to 50 mg/day or 400 mg/day. If the target dose could not be achieved, patients were maintained on the maximum tolerated dose. Fifty-eight percent of patients achieved the maximal dose of 400 mg/day for >2 weeks, and patients who did not tolerate 150 mg/day were discontinued. The primary efficacy assessment was a between-group comparison of time to first seizure during the double-blind phase. Comparison of the Kaplan-Meier survival curves of time to first seizure favored the topiramate 400 mg/day group over the topiramate 50 mg/day group (p=0.0002, log rank test; Figure 1). The treatment effects with respect to time to first seizure were consistent across various patient subgroups defined by age, sex, geographic region, baseline body weight, baseline seizure type, time since diagnosis, and baseline AED use.
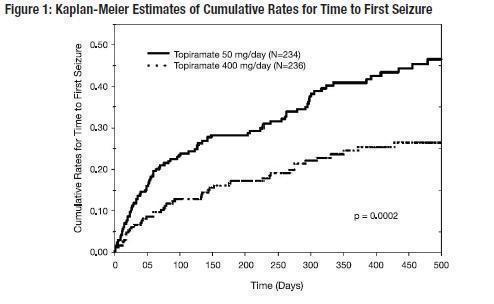
Children 2 to <10 Years of Age
The conclusion that topiramate is effective as initial monotherapy in children 2 to <10 years of age with partial onset or primary generalized tonic-clonic seizures was based on a pharmacometric bridging approach using data from the controlled epilepsy trials described in labeling. This approach consisted of first showing a similar exposure response relationship between pediatric patients down to 2 years of age and adults when topiramate was given as adjunctive therapy. Similarity of exposure-response was also demonstrated in pediatric patients ages 6 to <16 years and adults when topiramate was given as initial monotherapy. Specific dosing in children 2 to <10 years of age was derived from simulations utilizing plasma exposure ranges observed in pediatric and adult patients treated with topiramate initial monotherapy [see Dosage and Administration (2.1)].
14.2 Adjunctive Therapy Epilepsy Controlled Trials
Adult Patients With Partial Onset Seizures
The effectiveness of topiramate as an adjunctive treatment for adults with partial onset seizures was established in six multicenter, randomized, double-blind, placebo-controlled trials, two comparing several dosages of topiramate and placebo and four comparing a single dosage with placebo, in patients with a history of partial onset seizures, with or without secondarily generalized seizures.
Patients in these studies were permitted a maximum of two antiepileptic drugs (AEDs) in addition to topiramate tablets or placebo. In each study, patients were stabilized on optimum dosages of their concomitant AEDs during baseline phase lasting between 4 and 12 weeks. Patients who experienced a prespecified minimum number of partial onset seizures, with or without secondary generalization, during the baseline phase (12 seizures for 12-week baseline, 8 for 8-week baseline or 3 for 4-week baseline) were randomly assigned to placebo or a specified dose of topiramate tablets in addition to their other AEDs.
Following randomization, patients began the double-blind phase of treatment. In five of the six studies, patients received active drug beginning at 100 mg per day; the dose was then increased by 100 mg or 200 mg/day increments weekly or every other week until the assigned dose was reached, unless intolerance prevented increases. In the sixth study (119), the 25 or 50 mg/day initial doses of topiramate were followed by respective weekly increments of 25 or 50 mg/day until the target dose of 200 mg/day was reached. After titration, patients entered a 4, 8 or 12-week stabilization period. The numbers of patients randomized to each dose and the actual mean and median doses in the stabilization period are shown in Table 14.
Pediatric Patients Ages 2 to 16 Years with Partial Onset Seizures
The effectiveness of topiramate as an adjunctive treatment for pediatric patients ages 2 to 16 years with partial onset seizures was established in a multicenter, randomized, double-blind, placebo-controlled trial, comparing topiramate and placebo in patients with a history of partial onset seizures, with or without secondarily generalized seizures(see Table 14)
Patients in this study were permitted a maximum of two antiepileptic drugs (AEDs) in addition to topiramate tablets or placebo. In this study, patients were stabilized on optimum dosages of their concomitant AEDs during an 8-week baseline phase. Patients who experienced at least six partial onset seizures, with or without secondarily generalized seizures, during the baseline phase were randomly assigned to placebo or topiramate tablets in addition to their other AEDs.
Following randomization, patients began the double-blind phase of treatment. Patients received active drug beginning at 25 or 50 mg per day; the dose was then increased by 25 mg to 150 mg/day increments every other week until the assigned dosage of 125 mg, 175 mg, 225 mg, or 400 mg/day based on patients' weight to approximate a dosage of 6 mg/kg per day was reached, unless intolerance prevented increases. After titration, patients entered an 8-week stabilization period.
Patients With Primary Generalized Tonic-Clonic Seizures
The effectiveness of topiramate as an adjunctive treatment for primary generalized tonic-clonic seizures in patients 2 years old and older was established in a multicenter, randomized, double-blind, placebo-controlled trial, comparing a single dosage of topiramate and placebo(see Table 14).
Patients in this study were permitted a maximum of two antiepileptic drugs (AEDs) in addition to topiramate or placebo. Patients were stabilized on optimum dosages of their concomitant AEDs during an 8-week baseline phase. Patients who experienced at least three primary generalized tonic-clonic seizures during the baseline phase were randomly assigned to placebo or topiramate in addition to their other AEDs.
Following randomization, patients began the double-blind phase of treatment. Patients received active drug beginning at 50 mg/day for four weeks; the dose was then increased by 50 mg to 150 mg/day increments every other week until the assigned dose of 175 mg, 225 mg, or 400 mg/day based on patients' body weight to approximate a dosage of 6 mg/kg/day was reached, unless intolerance prevented increases. After titration, patients entered a 12-week stabilization period.
Patients With Lennox-Gastaut Syndrome
The effectiveness of topiramate as an adjunctive treatment for seizures associated with Lennox-Gastaut syndrome was established in a multicenter, randomized, double-blind, placebo-controlled trial comparing a single dosage of topiramate with placebo in patients 2 years of age and older(see Table 14).
Patients in this study were permitted a maximum of two antiepileptic drugs (AEDs) in addition to topiramate or placebo. Patients who were experiencing at least 60 seizures per month before study entry were stabilized on optimum dosages of their concomitant AEDs during a 4-week baseline phase. Following baseline, patients were randomly assigned to placebo or topiramate in addition to their other AEDs. Active drug was titrated beginning at 1 mg/kg/day for a week; the dose was then increased to 3 mg/kg/day for one week, then to 6 mg/kg/day. After titration, patients entered an 8-week stabilization period. The primary measures of effectiveness were the percent reduction in drop attacks and a parental global rating of seizure severity.
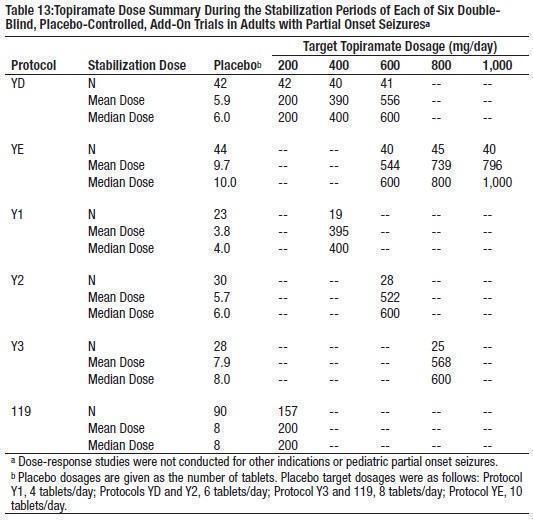
In all add-on trials, the reduction in seizure rate from baseline during the entire double-blind phase was measured. The median percent reductions in seizure rates and the responder rates (fraction of patients with at least a 50% reduction) by treatment group for each study are shown below in Table 10. As described above, a global improvement in seizure severity was also assessed in the Lennox-Gastaut trial.
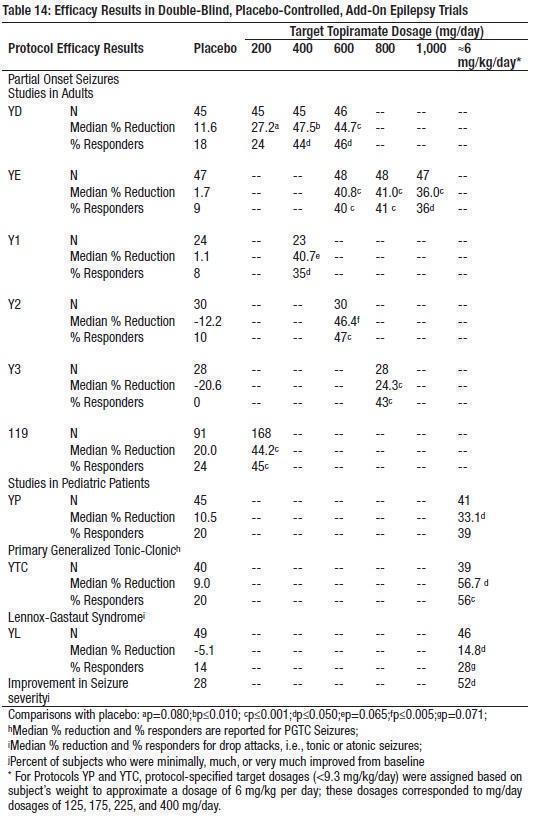
Subset analyses of the antiepileptic efficacy of topiramate tablets in these studies showed no differences as a function of gender, race, age, baseline seizure rate, or concomitant AED.
In clinical trials for epilepsy, daily dosages were decreased in weekly intervals by 50 to 100 mg/day in adults and over a 2- to 8- week period in children; transition was permitted to a new antiepileptic regimen when clinically indicated.
16. HOW SUPPLIED/STORAGE AND HANDLING
Product: 63629-3995
NDC: 63629-3995-1 30 TABLET in a BOTTLE
NDC: 63629-3995-2 60 TABLET in a BOTTLE
NDC: 63629-3995-3 90 TABLET in a BOTTLE
NDC: 63629-3995-4 120 TABLET in a BOTTLE
NDC: 63629-3995-5 7 TABLET in a BOTTLE
NDC: 63629-3995-6 80 TABLET in a BOTTLE
NDC: 63629-3995-7 14 TABLET in a BOTTLE
NDC: 63629-3995-8 45 TABLET in a BOTTLE
NDC: 63629-3995-9 28 TABLET in a BOTTLE
17. PATIENT COUNSELING INFORMATION
Advise the patients to read FDA-approved patient labeling (Medication Guide).
Eye Disorders
Instruct patients taking topiramate to seek immediate medical attention if they experience blurred vision, visual disturbances or periorbital pain [see Warnings and Precautions (5.1), (5.2)].
Oligohydrosis and Hyperthermia
Closely monitor topiramate-treated patients, especially pediatric patients, for evidence of decreased sweating and increased body temperature, especially in hot weather. Counsel patients to contact their healthcare professionals immediately if they develop a high or persistent fever, or decreased sweating [see Warnings and Precautions (5.3)].
Metabolic AcidosisWarn patients about the potential significant risk for metabolic acidosis that may be asymptomatic and may be associated with adverse effects on kidneys (e.g., kidney stones, nephrocalcinosis), bones (e.g., osteoporosis, osteomalacia, and/or rickets in children), and growth (e.g., growth delay/retardation) in pediatric patients, and on the fetus [see Warnings and Precautions (5.4) and Use in Specific Populations (8.1)].
Suicidal Behavior and Ideation
Counsel patients, their caregivers, and families that AEDs, including topiramate, may increase the risk of suicidal thoughts and behavior, and advise of the need to be alert for the emergence or worsening of the signs and symptoms of depression, any unusual changes in mood or behavior or the emergence of suicidal thoughts, or behavior or thoughts about self-harm. Instruct patients to immediately report behaviors of concern to their healthcare providers [see Warnings and Precautions (5.5)].
Interference with Cognitive and Motor Performance
Warn patients about the potential for somnolence, dizziness, confusion, difficulty concentrating, or visual effects, and advise patients not to drive or operate machinery until they have gained sufficient experience on topiramate to gauge whether it adversely affects their mental performance, motor performance, and/or vision [see Warnings and Precautions (5.6)].
Even when taking topiramate or other anticonvulsants, some patients with epilepsy will continue to have unpredictable seizures. Therefore, advise all patients taking topiramate for epilepsy to exercise appropriate caution when engaging in any activities where loss of consciousness could result in serious danger to themselves or those around them (including swimming, driving a car, climbing in high places, etc.). Some patients with refractory epilepsy will need to avoid such activities altogether. Discuss the appropriate level of caution with patients, before patients with epilepsy engage in such activities.
Fetal Toxicity
Inform pregnant women and women of childbearing potential that use of topiramate during pregnancy can cause fetal harm, including an increased risk for cleft lip and/or cleft palate (oral clefts), which occur early in pregnancy before many women know they are pregnant. There may also be risks to the fetus from chronic metabolic acidosis with use of topiramate during pregnancy [see Warnings and Precautions (5.7) and Use in Specific Populations (8.1), (8.9)]. When appropriate, prescribers should counsel pregnant women and women of childbearing potential about alternative therapeutic options. This is particularly important when topiramate use is considered for a condition not usually associated with permanent injury or death.
Advise women of childbearing potential who are not planning a pregnancy to use effective contraception while using topiramate, keeping in mind that there is a potential for decreased contraceptive efficacy when using estrogen-containing birth control with topiramate [see Drug Interactions (7.3)].
Encourage pregnant women using topiramate to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. The registry is collecting information about the safety of antiepileptic drugs during pregnancy. To enroll, patients can call the toll free-number, 1-888-233-2334. Information about the North American Drug Pregnancy Registry can be found at http://www.massgeneral.org/aed/. [see Use in Specific Populations (8.1)].
Hyperammonemia and Encephalopathy
Warn patients about the possible development of hyperammonemia with or without encephalopathy. Although hyperammonemia may be asymptomatic, clinical symptoms of hyperammonemic encephalopathy often include acute alterations in level of consciousness and/or cognitive function with lethargy or vomiting. This hyperammonemia and encephalopathy can develop with topiramate treatment alone or with topiramate treatment with concomitant valproic acid (VPA).
Instruct patients to contact their physician if they develop unexplained lethargy, vomiting, or changes in mental status [see Warnings and Precautions (5.10)].
Kidney Stones
Instruct patients, particularly those with predisposing factors, to maintain an adequate fluid intake in order to minimize the risk of kidney stone formation [see Warnings and Precautions (5.11)].
Instructions for a Missing DoseInstruct patients that if they miss a single dose of topiramate, it should be taken as soon as possible. However, if a patient is within 6 hours of taking the next scheduled dose, tell the patient to wait until then to take the usual dose of topiramate, and to skip the missed dose. Tell patients that they should not take a double dose in the event of a missed dose. Advise patients to contact their healthcare provider if they have missed more than one dose.
Rev:10/15
MEDICATION GUIDE
Topiramate Tablets, USP
(toe pir’a mate).
Read this Medication Guide before you start taking topiramate tablets and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. If you have any questions about topiramate tablets, talk to your healthcare provider or pharmacist.
What is the most important information I should know about topiramate tablets?
Topiramate tablets may cause eye problems. Serious eye problems include:
- any sudden decrease in vision with or without eye pain and redness,
- a blockage of fluid in the eye causing increased pressure in the eye (secondary angle closure glaucoma).
- These eye problems can lead to permanent loss of vision if not treated.
- You should call your healthcare provider right away if you have any new eye symptoms, including any new problems with your vision.
Topiramate tablets may cause decreased sweating and increased body temperature (fever). People, especially children, should be watched for signs of decreased sweating and fever, especially in hot temperatures. Some people may need to be hospitalized for this condition. Call your healthcare provider right away if you have a high fever, a fever that does not go away, or decreased sweating.
Topiramate tablets can increase the level of acid in your blood (metabolic acidosis). If left untreated, metabolic acidosis can cause brittle or soft bones (osteoporosis, osteomalacia, osteopenia), kidney stones, can slow the rate of growth in children, and may possibly harm your baby if you are pregnant. Metabolic acidosis can happen with or without symptoms.
Sometimes people with metabolic acidosis will:
• feel tired
• not feel hungry (loss of appetite)
• feel changes in heartbeat
• have trouble thinking clearly
Your healthcare provider should do a blood test to measure the level of acid in your blood before and during your treatment with topiramate tablets. If you are pregnant, you should talk to your healthcare provider about whether you have metabolic acidosis.
Like other antiepileptic drugs, topiramate tablets may cause suicidal thoughts or actions in a very small number of people, about 1 in 500.
Call a healthcare provider right away if you have any of these symptoms, especially if they are new, worse, or worry you:
- thoughts about suicide or dying
- attempts to commit suicide
- new or worse depression
- new or worse anxiety
- feeling agitated or restless
- panic attacks
- trouble sleeping (insomnia)
- new or worse irritability
- acting aggressive, being angry, or violent
- acting on dangerous impulses
- an extreme increase in activity and talking (mania)
- other unusual changes in behavior or mood
Do not stop topiramate tablets without first talking to a healthcare provider.
- Stopping topiramate tablets suddenly can cause serious problems.
- Suicidal thoughts or actions can be caused by things other than medicines. If you have suicidal thoughts or actions, your healthcare provider may check for other causes.
How can I watch for early symptoms of suicidal thoughts and actions?
- Pay attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings.
- Keep all follow-up visits with your healthcare provider as scheduled.
- Call your healthcare provider between visits as needed, especially if you are worried about symptoms.
Topiramate tablets can harm your unborn baby.
- If you take topiramate tablets during pregnancy, your baby has a higher risk for birth defects called cleft lip and cleft palate. These defects can begin early in pregnancy, even before you know you are pregnant.
- Cleft lip and cleft palate may happen even in children born to women who are not taking any medicines and do not have other risk factors.
- There may be other medicines to treat your condition that have a lower chance of birth defects.
- All women of childbearing age should talk to their healthcare providers about using other possible treatments instead of topiramate tablets. If the decision is made to use topiramate tablets, you should use effective birth control (contraception) unless you are planning to become pregnant. You should talk to your doctor about the best kind of birth control to use while you are taking topiramate tablets.
- Tell your healthcare provider right away if you become pregnant while taking topiramate tablets. You and your healthcare provider should decide if you will continue to take topiramate tablets while you are pregnant.
- Metabolic acidosis may have harmful effects on your baby. Talk to your healthcare provider if topiramate tablets have caused metabolic acidosis during your pregnancy.
- Pregnancy Registry: If you become pregnant while taking topiramate tablets, talk to your healthcare provider about registering with the North American Antiepileptic Drug Pregnancy Registry. You can enroll in this registry by calling 1-888-233-2334. The purpose of this registry is to collect information about the safety of antiepileptic drugs during pregnancy.
What are topiramate tablets?
Topiramate tablets are a prescription medicine used:
- to treat certain types of seizures (partial onset seizures and primary generalized tonic-clonic seizures) in adults and children 2 years and older,
- with other medicines to treat certain types of seizures (partial onset seizures, primary generalized tonic-clonic seizures, and seizures associated with Lennox-Gastaut syndrome) in adults and children 2 years and older
What should I tell my healthcare provider before taking topiramate tablets?
Before taking topiramate tablets, tell your healthcare provider about all your medical conditions, including if you:
- have or have had depression, mood problems, or suicidal thoughts or behavior
- have kidney problems, have kidney stones, or are getting kidney dialysis
- have a history of metabolic acidosis (too much acid in the blood)
- have liver problems
- have weak, brittle, or soft bones (osteomalacia, osteoporosis, osteopenia, or decreased bone density)
- have lung or breathing problems
- have eye problems, especially glaucoma
- have diarrhea
- have a growth problem
- are on a diet high in fat and low in carbohydrates, which is called a ketogenic diet
- are having surgery
- are pregnant or plan to become pregnant
- are breastfeeding. Topiramate passes into breast milk. It is not known if the topiramate that passes into breast milk can harm your baby. Talk to your healthcare provider about the best way to feed your baby if you take topiramate tablets.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. Topiramate tablets and other medicines may affect each other causing side effects.
Especially tell your healthcare provider if you take:
- Valproic acid (such as DEPAKENE® or DEPAKOTE®)
- any medicines that impair or decrease your thinking, concentration, or muscle coordination
- birth control pills. Topiramate tablets may make your birth control pills less effective. Tell your healthcare provider if your menstrual bleeding changes while you are taking birth control pills and topiramate tablets.
Ask your healthcare provider if you are not sure if your medicine is listed above.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist each time you get a new medicine. Do not start a new medicine without talking with your healthcare provider.
How should I take topiramate tablets?
- Take topiramate tablets exactly as prescribed.
- Your healthcare provider may change your dose. Do not change your dose without talking to your healthcare provider.
- Topiramate tablets should be swallowed whole. Do not chew the tablets. They may leave a bitter taste.
- Topiramate tablets can be taken before, during, or after a meal. Drink plenty of fluids during the day. This may help prevent kidney stones while taking topiramate tablets.
- If you take too much topiramate tablets, call your healthcare provider or poison control center right away or go to the nearest emergency room.
- If you miss a single dose of topiramate tablets, take it as soon as you can. However, if you are within 6 hours of taking your next scheduled dose, wait until then to take your usual dose of topiramate tablets, and skip the missed dose. Do not double your dose. If you have missed more than one dose, you should call your healthcare provider for advice.
- Do not stop taking topiramate tablets without talking to your healthcare provider. Stopping topiramate tablets suddenly may cause serious problems. If you have epilepsy and you stop taking topiramate tablets suddenly, you may have seizures that do not stop. Your healthcare provider will tell you how to stop taking topiramate tablets slowly.
- Your healthcare provider may do blood tests while you take topiramate tablets.
What should I avoid while taking topiramate tablets?
- Do not drink alcohol while taking topiramate tablets. Topiramate tablets and alcohol can affect each other causing side effects such as sleepiness and dizziness.
- Do not drive a car or operate heavy machinery until you know how topiramate tablet affects you. Topiramate tablets can slow your thinking and motor skills, and may affect vision.
What are the possible side effects of topiramate tablets?
Topiramate tablets may cause serious side effects including:
See "What is the most important information I should know about topiramate tablets?"
• High blood ammonia levels. High ammonia in the blood can affect your mental activities, slow your alertness, make you feel tired, or cause vomiting. This has happened when topiramate tablets are taken with a medicine called valproic acid (DEPAKENE® and DEPAKOTE®).
• Kidney stones. Drink plenty of fluids when taking topiramate tablets to decrease your chances of getting kidney stones.
- Low body temperature. Taking topiramate tablets when you are also taking valproic acid can cause a drop in body temperature to less then 95oF, feeling tired, confusion, or coma.
• Effects on thinking and alertness. Topiramate tablets may affect how you think and cause confusion, problems with concentration, attention, memory, or speech. Topiramate tablets may cause depression or mood problems, tiredness, and sleepiness.
• Dizziness or loss of muscle coordination.
Call your healthcare provider right away if you have any of the symptoms above.
The most common side effects of topiramate tablets include:
- tingling of the arms and legs (paresthesia)
- not feeling hungry
- nausea
- a change in the way foods taste
- diarrhea
- weight loss
- nervousness
- upper respiratory tract infection
- speech problems
- tiredness
- dizziness
- sleepiness/drowsiness
- slow reactions
- difficulty with memory
- pain in the abdomen
- fever
- abnormal vision
Tell your healthcare provider about any side effect that bothers you or that does not go away.
These are not all the possible side effects of topiramate tablets. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store topiramate tablets?
- Store at 20° to 25°C (68°F to 77°F); [see USP Controlled Room Temperature]. Protect from moisture.
- Keep topiramate tablets in a tightly closed container.
- Keep topiramate tablets dry and away from moisture.
- Keep topiramate tablets and all medicines out of the reach of children.
General information about topiramate tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use topiramate tablets for a condition for which it was not prescribed. Do not give topiramate tablets to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about topiramate tablets. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about topiramate tablets that is written for health professionals.
For more information, go to www.camberpharma.com or call Camber Pharmaceuticals, Inc at 1-866-495-8330.
What are the ingredients in topiramate tablets?
Active ingredient: topiramate, USP
Inactive ingredients: Topiramate tablets contain the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, pre-gelatinized starch (maize), sodium starch glycolate, magnesium stearate, opadry white (titanium dioxide, hypromellose 3cp, hypromellose 6cp, PEG 400, polysorbate 80) for 25 mg tablets, opadry yellow (titanium dioxide, hypromellose 3cp, hypromellose 6cp, PEG 400, polysorbate 80, iron oxide yellow) for 50 mg tablets, opadry yellow (hypromellose 3cp, hypromellose 6cp, titanium dioxide, PEG 400, iron oxide yellow, polysorbate 80, iron oxide red) for 100 mg tablets and, opadry pink (titanium dioxide, hypromellose 6cp, PEG 400,iron oxide red) for 200 mg tablets.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
All brands names listed are the registered trademarks of their respective owners and are not trademarks of Camber Pharmaceuticals, Inc
Manufactured by:
Ascent Pharmaceuticals, Inc.
Central Islip, NY 11722
Manufactured for:
Camber Pharmaceuticals, Inc.
Piscataway, NJ 08854
Rev: 10/15
Barcode: 281-10-2015

| TOPIRAMATE
topiramate tablet |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Labeler - Bryant Ranch Prepack (171714327) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bryant Ranch Prepack | 171714327 | REPACK(63629-3995) , RELABEL(63629-3995) | |