BELBUCA- buprenorphine hydrochloride film
Endo Pharmaceuticals
----------
HIGHLIGHTS OF PRESCRIBING INFORMATION
Highlights of Prescribing Information
These highlights do not include all the information needed to use BELBUCA safely and effectively. See full prescribing information for BELBUCA. BELBUCA™ (buprenorphine) buccal film CIII Initial U.S. Approval: 1981 WARNING: ADDICTION, ABUSE, and MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL EXPOSURE; and NEONATAL OPIOID WITHDRAWAL SYNDROMESee full prescribing information for complete boxed warning.
INDICATIONS AND USAGEBELBUCA buccal film contains buprenorphine, a partial opioid agonist. BELBUCA is indicated for the management of pain severe enough to require daily, around-the-clock, long-term opioid treatment and for which alternative treatment options are inadequate. ( 1) Limitations of Use
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSBuccal film available in 75 mcg, 150 mcg, 300 mcg, 450 mcg, 600 mcg, 750 mcg, and 900 mcg dosage strengths. ( 3) CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions >5% include nausea, constipation, headache, vomiting, dizziness, and somnolence. ( 6.1) To report SUSPECTED ADVERSE REACTIONS, contact Endo Pharmaceuticals Inc. at 1-800-462-3636 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 12/2015 |
FULL PRESCRIBING INFORMATION
Warning Information.
These highlights do not include all the information needed to use.
See full prescribing information for complete boxed warning.
Initial U.S. Approval.
Addiction, Abuse, and Misuse
BELBUCA exposes patients and other users to the risks of opioid addiction, abuse, and misuse, which can lead to overdose and death. Assess each patient’s risk prior to prescribing BELBUCA, and monitor patients regularly for the development of these behaviors or conditions [see Warnings and Precautions and Overdosage.
Life-Threatening Respiratory Depression
Serious, life-threatening, or fatal respiratory depression may occur with use of BELBUCA. Monitor for respiratory depression, especially during initiation of BELBUCA or following a dose increase. Misuse or abuse of BELBUCA by chewing, swallowing, snorting or injecting buprenorphine extracted from the buccal film will result in the uncontrolled delivery of buprenorphine and pose a significant risk of overdose and death [see Warnings and Precautions.
Accidental Exposure
Accidental exposure to even one dose of BELBUCA, especially in children, can result in a fatal overdose of buprenorphine [see Warnings and Precautions.
Neonatal Opioid Withdrawal Syndrome
Prolonged use of BELBUCA during pregnancy can result in neonatal opioid withdrawal syndrome, which may be life-threatening if not recognized and treated, and requires management according to protocols developed by neonatology experts. If opioid use is required for a prolonged period in a pregnant woman, advise the patient of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available [see Warnings and Precautions.
1 INDICATIONS AND USAGE
BELBUCA is indicated for the management of pain severe enough to require daily, around-the-clock, long-term opioid treatment and for which alternative treatment options are inadequate.
Limitations of Use
- Because of the risks of addiction, abuse, and misuse with opioids, even at recommended doses, and because of the greater risks of overdose and death with long-acting opioid formulations, reserve BELBUCA for use in patients for whom alternative treatment options (e.g., non-opioid analgesics or immediate-release opioids) are ineffective, not tolerated, or would be otherwise inadequate to provide sufficient management of pain.
- BELBUCA is not indicated as an as-needed (prn) analgesic.
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosage and Administration Instructions
BELBUCA should be prescribed only by healthcare professionals who are knowledgeable in the use of potent opioids for the management of chronic pain.
BELBUCA buccal film is for oral buccal use only and is to be applied to the buccal mucosa every 12 hours.
Instruct patients not to use BELBUCA if the pouch seal is broken or the buccal film is cut, damaged, or changed in any way and to avoid applying BELBUCA to areas of the mouth with any open sores or lesions.
Monitor patients closely for respiratory depression, especially within the first 24-72 hours of initiating therapy and following dosage increases with BELBUCA and adjust the dosage accordingly [see Warnings and Precautions ( 5.2)].
2.2 Initial Dosing
Initiate the dosing regimen for each patient individually, taking into account the patient’s severity of pain, response, prior analgesic treatment experience, and risk factors for addiction, abuse, and misuse [see Warnings and Precautions ( 5.1)] .
Use of BELBUCA as the Initial Opioid Analgesic (Opioid Naïve Patients)
Initiate treatment in opioid naïve patients with a 75 mcg film once daily or, if tolerated, every 12 hours (see Table 1) for at least 4 days, then increase dose to 150 mcg every 12 hours. Individual titration to a dose that provides adequate analgesia and minimizes adverse reactions should proceed in increments of 150 mcg every 12 hours, no more frequently than every 4 days. Doses up to 450 mcg every 12 hours were studied in opioid naïve patients in the clinical trials [see Clinical Studies ( 14)] .
Conversion from Other Opioids to BELBUCA
There is a potential for buprenorphine to precipitate withdrawal in patients who are already on opioids. To reduce the risk of opioid withdrawal, taper patients to no more than 30 mg oral morphine sulfate equivalents (MSE) daily before beginning BELBUCA. Following analgesic taper, base the starting dose on the patient’s daily opioid dose prior to taper, as described in Table 1. Patients may require additional short-acting analgesics during the taper period and during titration.
BELBUCA may not provide adequate analgesia for patients requiring greater than 160 mg oral MSE per day. Consider the use of an alternate analgesic.
|
Prior Daily Dose of Opioid Analgesic Before Taper to 30 mg Oral MSE |
Initial BELBUCA Dose |
|
Less than 30 mg oral MSE |
BELBUCA 75 mcg once daily or every 12 hours |
|
30 mg to 89 mg oral MSE |
BELBUCA 150 mcg every 12 hours |
|
90 mg to 160 mg oral MSE |
BELBUCA 300 mcg every 12 hours |
|
Greater than 160 mg oral MSE |
Consider alternate analgesic |
BELBUCA doses of 600 mcg, 750 mcg, and 900 mcg are only for use following titration from lower doses of BELBUCA. Individual titration should proceed in increments of 150 mcg every 12 hours, no more frequently than every 4 days.
Conversion from Methadone to BELBUCA
Close monitoring is of particular importance when converting from methadone to other opioid agonists, including BELBUCA. The ratio between methadone and other opioid agonists may vary widely as a function of previous dose exposure. Methadone has a long half-life and can accumulate in the plasma.
2.3 Titration and Maintenance of Therapy
Individually titrate BELBUCA to a dose that provides adequate analgesia and minimizes adverse reactions Continually reevaluate patients receiving BELBUCA to assess the maintenance of pain control and the relative incidence of adverse reactions and monitor for the development of addiction, abuse, or misuse [see Warnings and Precautions ( 5.1)] . Frequent communication is important among the prescriber, other members of the healthcare team, the patient, and the caregiver/family during periods of changing analgesic requirements, including initial titration. During long-term therapy, periodically reassess the continued need for opioid analgesics.
The minimum titration interval of BELBUCA is 4 days, based on the pharmacokinetic profile and time to reach steady-state plasma levels [see Clinical Pharmacology ( 12.3)].
Individual titration should proceed in increments of no more than 150 mcg every 12 hours.
The maximum BELBUCA dose is 900 mcg every 12 hours. Do not exceed a dose of BELBUCA 900 mcg every 12 hours due to the potential for QTc interval prolongation.[see Warnings and Precautions ( 5.7), Adverse Reactions ( 6.1), and Clinical Pharmacology ( 12.2)] . If pain is not adequately managed on BELBUCA 900 mcg, consider an alternate analgesic.
Patients who experience breakthrough pain may require dosage adjustment of BELBUCA or may need rescue medication with an appropriate dose of an immediate-release analgesic. If the level of pain increases after dose stabilization, attempt to identify the source of increased pain before increasing the BELBUCA dose.
If unacceptable opioid-related adverse reactions are observed, adjust the dose to obtain an appropriate balance between the management of pain and opioid-related adverse reactions.
2.4 Discontinuation of BELBUCA
When a patient no longer requires therapy with BELBUCA, use a gradual downward titration of the dose to prevent signs and symptoms of withdrawal in the physically-dependent patient. Do not abruptly discontinue BELBUCA.
2.5 Dosage Modifications in Patients with Severe Hepatic Impairment
In patients with severe hepatic impairment (i.e., Child-Pugh C), reduce the starting dose and reduce the titration dose by half that of patients with normal liver function, from 150 mcg to 75 mcg. [see Warnings and Precautions ( 5.11), Use in Special Populations ( 8.6), Clinical Pharmacology ( 12.3)].
2.6 Dosage Modifications in Patients with Oral Mucositis
In patients with known or suspected mucositis, reduce the starting dosage and titration incremental dosage by half compared to patients without mucositis. [see Warnings and Precautions ( 5.15), Clinical Pharmacology ( 12.3)].
2.7 Administration of BELBUCA
BELBUCA should not be used if the package seal is broken or the film is cut, damaged, or changed in any way.
First, the patient must use the tongue to wet the inside of the cheek or rinse the mouth with water to wet the area for placement of BELBUCA. BELBUCA is then applied immediately after removal from the individually sealed package. . The yellow side of the BELBUCA film is placed against the inside of the cheek. The entire BELBUCA film is held in place with clean, dry fingers for 5 seconds and then left BELBUCA in place on the inside of the cheek until fully dissolved.
BELBUCA adheres to the moist buccal mucosa and will completely dissolve after application, usually within 30 minutes. The film should not be manipulated with the tongue or finger(s) and eating food and drinking liquids should be avoided until the film has dissolved.
A BELBUCA film, if chewed or swallowed, may result in lower peak concentrations and lower bioavailability than when used as directed .
Demonstrate proper administration technique to the patient[see Patient Counseling Information ( 17)] .
2.8 Disposal Instructions
Dispose of unused BELBUCA as soon as it is no longer needed.
To dispose of unused BELBUCA film:
- Remove all BELBUCA films from their foil packages.
- Drop the BELBUCA films into toilet and flush.
- Discard foil packaging in trash.
Do not flush BELBUCA down the toilet in the foil packages or cartons [s ee Patient Counseling Information ( 17)] .
3 DOSAGE FORMS AND STRENGTHS
BELBUCA is a buccal film available in 7 dosage strengths: 75 mcg, 150 mcg, 300 mcg, 450 mcg, 600 mcg, 750 mcg, and 900 mcg buprenorphine per film.
4 CONTRAINDICATIONS
BELBUCA is contraindicated in patients with:
- Significant respiratory depression [see Warnings and Precautions ( 5.2)]
- Acute or severe bronchial asthma in an unmonitored setting or in the absence of resuscitative equipment [see Warnings and Precautions ( 5.6)]
- Known or suspected gastrointestinal obstruction, including paralytic ileus [see Warnings and Precautions ( 5.13)]
- Hypersensitivity (e.g., anaphylaxis) to buprenorphine [see Warnings and Precautions ( 5.12), and Adverse Reactions ( 6)]
5 WARNINGS AND PRECAUTIONS
5.1 Addiction, Abuse, and Misuse
BELBUCA contains buprenorphine, a Schedule III controlled substance. As an opioid, BELBUCA exposes users to the risks of addiction, abuse, and misuse.
Assess each patient’s risk for opioid addiction, abuse, or misuse prior to prescribing BELBUCA and monitor all patients receiving BELBUCA for the development of these behaviors or conditions. Risks are increased in patients with a personal or family history of substance abuse (including drug or alcohol abuse or addiction) or mental illness (e.g., major depression). The potential for these risks should not, however, prevent the proper management of pain in any given patient. Patients at increased risk may be prescribed BELBUCA, but use in such patients necessitates intensive counseling about the risks and proper use of BELBUCA, along with intensive monitoring for signs of addiction, abuse, or misuse. Although the risk of addiction in any individual is unknown, it can occur in patients appropriately prescribed BELBUCA. Addiction can occur at recommended dosages and if the drug is misused or abused [see Drug Abuse and Dependence ( 9)].
Abuse or misuse of BELBUCA by swallowing may cause choking, overdose, and death [see Overdosage ( 10)].
Opioid agonists such as buprenorphine are sought by drug abusers and people with addiction disorders and are subject to criminal diversion. Consider these risks when prescribing or dispensing BELBUCA. Strategies to reduce the risk include prescribing the drug in the smallest appropriate quantity and advising the patient on the proper disposal of unused drug [see Patient Counseling Information ( 17)] . Contact local state professional licensing board or state controlled substances authority for information on how to prevent and detect abuse or diversion of this product.
5.2 Life-Threatening Respiratory Depression
Serious, life-threatening, or fatal respiratory depression has been reported with the use of buprenorphine, even when used as recommended. Respiratory depression, from opioid use, if not immediately recognized and treated, may lead to respiratory arrest and death. Management of respiratory depression may include close observation, supportive measures, and use of opioid antagonists, depending on the patient’s clinical status [see Overdosage ( 10)] . Carbon dioxide (CO 2) retention from opioid-induced respiratory depression can exacerbate the sedating effects of opioids.
While serious, life-threatening or fatal respiratory depression can occur at any time during the use of BELBUCA, the risk is greatest during initiation of therapy or following a dose increase. Closely monitor patients for respiratory depression when initiating therapy with BELBUCA and following dose increases.
To reduce the risk of respiratory depression, proper dosing and titration of BELBUCA are essential [see Dosage and Administration ( 2.3)] . Overestimating the dose of BELBUCA when converting patients from another opioid product may result in fatal overdose with the first dose.
Accidental exposure to BELBUCA, especially in children, might result in respiratory depression and death due to an overdose of buprenorphine.
5.3 Neonatal Opioid Withdrawal Syndrome
Prolonged use of BELBUCA during pregnancy can result in withdrawal signs in the neonate. Neonatal opioid withdrawal syndrome, unlike opioid withdrawal syndrome in adults, may be life-threatening if not recognized and treated and requires management according to protocols developed by neonatology experts. If opioid use is required for a prolonged period in a pregnant woman, advise the patient of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available.
Neonatal opioid withdrawal syndrome presents as irritability, hyperactivity and abnormal sleep pattern, high pitched cry, tremor, vomiting, diarrhea, failure to gain weight; and there have been reports of convulsions, apnea, respiratory depression, and bradycardia. The onset, duration, and severity of neonatal opioid withdrawal syndrome vary based on the specific opioid used, duration of use, timing and amount of last maternal use, and rate of elimination of the drug by the newborn [see Use in Specific Populations ( 8.1)].
5.4 Risks due to Interactions with Central Nervous System Depressants
Hypotension, profound sedation, coma or respiratory depression may result if BELBUCA is used concomitantly with alcohol or other central nervous system (CNS) depressants (e.g., sedatives, anxiolytics, hypnotics, neuroleptics, other opioids).
When considering the use of BELBUCA in a patient taking a CNS depressant, assess the duration of use of the CNS depressant and the patient’s response, including the degree of tolerance that has developed to CNS depression. Additionally, evaluate the patient’s use of alcohol or illicit drugs that cause CNS depression. If the decision to begin BELBUCA is made, start with a lower dosage of BELBUCA, monitor patients for signs of sedation, respiratory depression, and hypotension, and consider using a lower dosage of the concomitant CNS depressant [see Drug Interactions ( 7)].
5.5 Risk of Life-Threatening Respiratory Depression in Elderly, Cachectic, and Debilitated Patients
Life-threatening respiratory depression is more likely to occur in elderly, cachectic, or debilitated patients as they may have altered pharmacokinetics or altered clearance compared to younger, healthier patients. Monitor such patients closely, particularly when initiating and titrating BELBUCA and when BELBUCA is given concomitantly with other drugs that depress respiration [see Warnings and Precautions ( 5.2)].
5.6 Risk of Apnea in Patients with Chronic Pulmonary Disease
The use of BELBUCA in patients with acute or severe bronchial asthma in an unmonitored setting or in the absence of resuscitative equipment is contraindicated.
BELBUCA-treated patients with significant chronic obstructive pulmonary disease or cor pulmonale, and those with substantially decreased respiratory reserve, hypoxia, hypercapnia, or pre-existing respiratory depression are at increased risk of decreased respiratory drive, including apnea, even at recommended dosages of BELBUCA [see Warnings and Precautions ( 5.2)]. Therefore, closely monitor these patients, especially when initiating and titrating BELBUCA. Alternatively, consider the use of alternative non-opioid analgesics in these patients.
5.7 QTc Prolongation
BELBUCA has been observed to prolong the QTc interval in some subjects participating in clinical trials. Consider these observations in clinical decisions when prescribing BELBUCA to patients with hypokalemia, hypomagnesemia, or clinically unstable cardiac disease, including unstable atrial fibrillation, symptomatic bradycardia, unstable congestive heart failure, or active myocardial ischemia. Periodic electrocardiographic (ECG) monitoring is recommended in these patients. Avoid the use of BELBUCA in patients with a history of Long QT Syndrome or an immediate family member with this condition or those taking Class IA antiarrhythmic medications (e.g., quinidine, procainamide, disopyramide) or Class III antiarrhythmic medications (e.g., sotalol, amiodarone, dofetilide), or other medications that prolong the QT interval. [ see Dosage and Administration ( 2.3), Adverse Reactions ( 6.1), and Clinical Pharmacology ( 12.2)].
5.8 Severe Hypotension
BELBUCA may cause severe hypotension including orthostatic hypotension and syncope in ambulatory patients. There is an increased risk in patients whose ability to maintain blood pressure has already been compromised by a reduced blood volume or concurrent administration of certain CNS depressant drugs (e.g., phenothiazines or general anesthetics) [see Drug Interactions ( 7)]. Monitor these patients for signs of hypotension after initiating or titrating the dose of BELBUCA. In patients with circulatory shock, BELBUCA may cause vasodilation that can further reduce cardiac output and blood pressure. Avoid the use of BELBUCA in patients with circulatory shock.
5.9 Risks of Use in Patients with Head Injury or Increased Intracranial Pressure
In patients who may be susceptible to the intracranial effects of CO 2 retention (e.g., those with evidence of increased intracranial pressure or brain tumors), BELBUCA may reduce respiratory drive, and the resultant CO 2 retention can further increase intracranial pressure. Monitor such patients for signs of sedation and respiratory depression, particularly when initiating therapy with BELBUCA.
Opioids may also obscure the clinical course in a patient with a head injury. Avoid the use of BELBUCA in patients with impaired consciousness or coma.
5.10 Hepatotoxicity
Cases of cytolytic hepatitis and hepatitis with jaundice have been observed in individuals receiving sublingual formulations of buprenorphine for the treatment of opioid dependence, both in clinical trials and in post-marketing adverse events reports. The spectrum of abnormalities ranges from transient asymptomatic elevations in hepatic transaminases to case reports of hepatic failure, hepatic necrosis, hepatorenal syndrome, and hepatic encephalopathy. In many cases, the presence of pre-existing liver enzyme abnormalities, infection with hepatitis B or hepatitis C virus, concomitant usage of other potentially hepatotoxic drugs, and ongoing injection drug abuse may have played a causative or contributory role. For patients at increased risk of hepatoxicity (e.g., patients with a history of excessive alcohol intake, intravenous drug abuse or liver disease), obtain baseline liver enzyme levels and monitor periodically during treatment with BELBUCA.
5.11 Risk of Overdose in Patients With Moderate to Severe Hepatic Impairment
In a pharmacokinetic study in subjects dosed with buprenorphine sublingual tablets, buprenorphine plasma levels were found to be higher and the half-life was found to be longer in subjects with moderate and severe hepatic impairment, but not in subjects with mild hepatic impairment. For patients with severe hepatic impairment, a dose adjustment is recommended, and patients with moderate or severe hepatic impairment should be monitored for signs and symptoms of toxicity or overdose caused by increased levels of buprenorphine. [see Dosage and Administration ( 2.5) , Use in Specific Populations ( 8.6) ].
5.12 Anaphylactic/Allergic Reactions
Cases of acute and chronic hypersensitivity to buprenorphine have been reported both in clinical trials and in post-marketing experience. The most common signs and symptoms include rashes, hives, and pruritus. Cases of bronchospasm, angioneurotic edema, and anaphylactic shock have been reported. BELBUCA is contraindicated in patients with a history of hypersensitivity to buprenorphine.
5.13 Risk of Use in Patients with Gastrointestinal Conditions
BELBUCA is contraindicated in patients with known or suspected gastrointestinal obstruction, including paralytic ileus.
BELBUCA may cause spasm of the sphincter of Oddi. Opioids may cause increases in the serum amylase. Monitor patients with biliary tract disease, including acute pancreatitis, for worsening symptoms.
5.14 Increased Risk of Seizures in Patients with Seizure Disorders
Buprenorphine may increase the frequency of seizures in patients with seizure disorders and may increase the risk of seizures occurring in other clinical settings associated with seizures. Monitor patients with a history of seizure disorders for worsened seizure control during BELBUCA therapy.
5.15 Risks of Use in Cancer Patients with Oral Mucositis
Cancer patients with oral mucositis may absorb buprenorphine more rapidly than intended and are likely to experience higher plasma levels of the opioid. For patients with known or suspected mucositis, a dose reduction is recommended. Monitor these patients carefully for signs and symptoms of toxicity or overdose caused by increased levels of buprenorphine. [see Dosage and Administration ( 2.6), Clinical Pharmacology ( 12.3)] .
5.16 Risks of Driving and Operating Machinery
BELBUCA may impair the mental and physical abilities needed to perform potentially hazardous activities such as driving a car or operating machinery. Warn patients not to drive or operate dangerous machinery unless they are tolerant to side effects of BELBUCA and know how they will react to the medication.
6 ADVERSE REACTIONS
The following serious adverse reactions described elsewhere in the labeling include:
- Addiction, Abuse, and Misuse [see Warnings and Precautions ( 5.1)]
- Life-Threatening Respiratory Depression [see Warnings and Precautions ( 5.2)]
- Neonatal Opioid Withdrawal Syndrome [see Warnings and Precautions ( 5.3)]
- Interactions with other CNS Depressants [see Warnings and Precautions ( 5.4)]
- QTc Prolongation [see Warnings and Precautions ( 5.7)]
- Severe Hypotension [see Warnings and Precautions ( 5.8)]
- Anaphylactic/Allergic Reactions [see Warnings and Precautions ( 5.12)]
- Gastrointestinal Adverse Reactions [see Warnings and Precautions ( 5.13)]
- Seizures [see Warnings and Precautions ( 5.14)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 2,127 patients were treated with BELBUCA in controlled and open-label chronic pain trials. There were 504 patients treated for approximately six months and 253 patients treated for approximately one year. The clinical trial population consisted of patients with chronic moderate-to-severe pain.
The most common serious adverse drug reactions (all ≤ 0.2%) occurring during clinical trials with BELBUCA were: cellulitis, pneumonia, ileus, atrial fibrillation, coronary artery disease, cerebrovascular accident, syncope, transient ischemic attack, chest pain, non-cardiac chest pain, ankle fracture, cholecystitis, osteoarthritis, and dehydration.
The most common adverse events (≥ 2%) leading to discontinuation were nausea, vomiting, and liver function test abnormality.
The most common adverse events (≥ 5%) reported by opioid naïve, opioid experienced, and overall patients exposed to BELBUCA in clinical trials and compared against placebo are shown in Tables 2, 3 and 4:
|
Open-Label Titration Phase |
Double-Blind Treatment Phase |
||
|
MedDRA Preferred Term |
BELBUCA (N=749) |
BELBUCA (N=229) |
Placebo (N=232) |
|
Nausea |
50% |
10% |
7% |
|
Constipation |
13% |
4% |
3% |
|
Vomiting |
8% |
4% |
<1% |
|
Headache |
8% |
2% |
3% |
|
Dizziness |
6% |
2% |
<1% |
|
Somnolence |
7% |
1% |
<1% |
|
Fatigue |
5% |
0% |
1% |
|
Open-Label Titration Phase |
Double-Blind Treatment Phase |
||
|
MedDRA Preferred Term |
BELBUCA (N=810) |
BELBUCA (N=254) |
Placebo (N=256) |
|
Nausea |
17% |
7% |
7% |
|
Constipation |
8% |
3% |
1% |
|
Vomiting |
7% |
5% |
2% |
|
Headache |
7% |
2% |
3% |
|
Dizziness |
5% |
2% |
<1% |
|
Somnolence |
5% |
1% |
<1% |
|
Drug Withdrawal Syndrome |
0% |
4% |
10% |
|
Open-Label Titration Phase |
Double-Blind Treatment Phase |
||
|
MedDRA Preferred Term |
BELBUCA (N=1889) |
BELBUCA (N=600) |
Placebo (N=606) |
|
Nausea |
33% |
9% |
8% |
|
Constipation |
11% |
4% |
2% |
|
Vomiting |
7% |
5% |
2% |
|
Headache |
8% |
4% |
3% |
|
Dizziness |
6% |
2% |
<1% |
|
Somnolence |
6% |
<1% |
<1% |
|
Drug Withdrawal Syndrome |
1% |
2% |
5% |
The most common (≥ 5%), common (≥ 1% to < 5%), and least common (< 1%) adverse reactions reported by patients taking BELBUCA in the controlled and open-label clinical studies are presented below:
Most common adverse reactions (≥ 5%): nausea, constipation, headache, vomiting, fatigue, dizziness, somnolence, diarrhea, dry mouth, and upper respiratory tract infection.
Common (≥ 1% to < 5%) adverse reactions (organized by MedDRA [Medical Dictionary for Regulatory Activities] System Organ Class:
Blood and lymphatic system disorders: anemia
Gastrointestinal disorders: abdominal pain
General disorders and administration site conditions: peripheral edema, pyrexia, drug withdrawal syndrome
Infections and infestations: urinary tract infection, nasopharyngitis, sinusitis, bronchitis, gastroenteritis
Injury, poisoning, and procedural complications: contusion, fall
Metabolism and nutrition disorders: decreased appetite
Musculoskeletal and connective tissue disorders: muscle spasms, back pain
Psychiatric disorders: anxiety, insomnia, depression
Respiratory, thoracic and mediastinal disorders: oropharyngeal pain, sinus congestion
Skin and subcutaneous tissue disorders: hyperhidrosis, pruritus, rash
Vascular disorders: hot flush, hypertension
Least common (<1%) adverse reactions:
Abdominal discomfort, acute sinusitis, dyspepsia, toothache, asthenia, chills, cellulitis, tooth abscess, excoriation, laceration, aspartate aminotransferase increased, blood pressure increased, blood testosterone decreased, electrocardiogram QT prolonged, liver function test abnormal, musculoskeletal pain, neck pain, hypoesthesia, lethargy, migraine, tremor, cough, dyspnea, nasal congestion, rhinorrhea
7 DRUG INTERACTIONS
|
Benzodiazapines |
|
|
Clinical Impact: |
There have been a number of reports regarding coma and death associated with the misuse and abuse of the combination of buprenorphine and benzodiazepines. In many, but not all of these cases, buprenorphine was misused by self-injection of crushed buprenorphine tablets. Preclinical studies have shown that the combination of benzodiazepines and buprenorphine altered the usual ceiling effect on buprenorphine-induced respiratory depression, making the respiratory effects of buprenorphine appear similar to those of full opioid agonists. |
|
Intervention: |
Closely monitor patients with concurrent use of BELBUCA and benzodiazepines. Warn patients that it is extremely dangerous to self-administer benzodiazepines while taking BELBUCA, and warn patients to use benzodiazepines concurrently with BELBUCA only as directed by their physician. |
|
Central Nervous System (CNS) Depressants |
|
|
Clinical Impact: |
Due to additive pharmacologic effects, the concomitant use of CNS depressants can increase the risk of hypotension, respiratory depression, profound sedation, coma, and death. |
|
Intervention: |
Consider dose reduction of one or both drugs. Monitor patients for signs of respiratory depression, sedation, and hypotension [see Warnings and Precautions( 5.4)] . |
|
Examples: |
Alcohol, anxiolytics, general anesthetics, hypnotics, neuroleptics, phenothiazines, sedatives, tranquilizers, other opioids. |
|
Inhibitors of CYP3A4 |
|
|
Clinical Impact: |
The concomitant use of buprenorphine and CYP3A4 inhibitors can increase the plasma concentration of buprenorphine, resulting in increased or prolonged opioid effects, particularly when an inhibitor is added after a stable dose of BELBUCA is achieved. After stopping a CYP3A4 inhibitor, as the effects of the inhibitor decline, the buprenorphine plasma concentration will decrease [see Clinical Pharmacology ( 12.3)] , potentially resulting in decreased opioid efficacy or a withdrawal syndrome in patients who had developed physical dependence to buprenorphine. |
|
Intervention: |
If concomitant use is necessary, consider dosage reduction of BELBUCA until stable drug effects are achieved. Monitor patients for respiratory depression and sedation at frequent intervals. If a CYP3A4 inhibitor is discontinued, consider increasing the BELBUCA dosage until stable drug effects are achieved. Monitor for signs of opioid withdrawal. |
|
CYP3A4 Inducers |
|
|
Clinical Impact: |
The concomitant use of buprenorphine and CYP3A4 inducers can decrease the plasma concentration of buprenorphine [see Clinical Pharmacology ( 12.3)] , potentially resulting in decreased efficacy or onset of a withdrawal syndrome in patients who have developed physical dependence to buprenorphine. After stopping a CYP3A4 inducer, as the effects of the inducer decline, the buprenorphine plasma concentration will increase [see Clinical Pharmacology ( 12.3)] , which could increase or prolong both therapeutic effects and adverse reactions and may cause serious respiratory depression. |
|
Intervention: |
If concomitant use is necessary, consider increasing the BELBUCA dosage until stable drug effects are achieved. Monitor for signs of opioid withdrawal. If a CYP3A4 inducer is discontinued, consider BELBUCA dosage reduction and monitor for signs of respiratory depression. |
|
Mixed Agonist/Antagonist and Partial Agonist Opioid Analgesics |
|
|
Clinical Impact: |
May reduce the analgesic effect of BELBUCA and/or precipitate withdrawal symptoms. |
|
Intervention: |
Avoid concomitant use. |
|
Examples: |
butorphanol, nalbuphine, pentazocine |
|
Muscle Relaxants |
|
|
Clinical Impact: |
Buprenorphine may enhance the neuromuscular blocking action of skeletal muscle relaxants and produce an increased degree of respiratory depression. |
|
Intervention: |
Monitor patients receiving muscle relaxants and BELBUCA for signs of respiratory depression that may be greater than otherwise expected and decrease the dosage of BELBUCA and/or the muscle relaxant as necessary. |
|
Diuretics |
|
|
Clinical Impact: |
Opioids can reduce the efficacy of diuretics by inducing the release of antidiuretic hormone. |
|
Intervention: |
Monitor patients for signs of diminished diuresis and/or effects on blood pressure and increase the dosage of the diuretic as needed. |
|
Anticholinergic Drugs |
|
|
Clinical Impact: |
The concomitant use of opioid analgesics, including buprenorphine, and anticholinergic drugs may increase the risk of urinary retention and/or severe constipation, which may lead to paralytic ileus. |
|
Intervention: |
Monitor patients for signs of urinary retention or reduced gastric motility when BELBUCA is used concomitantly with anticholinergic drugs. |
|
Antiretrovirals: Nucleoside reverse transcriptase inhibitors (NRTIs) |
|
|
Clinical Impact: |
Nucleoside reverse transcriptase inhibitors (NRTIs) do not appear to induce or inhibit the P450 enzyme pathway, thus no interactions with buprenorphine are expected. |
|
Intervention: |
None |
|
Antiretrovirals: Non-nucleoside reverse transcriptase inhibitors (NNRTIs) |
|
|
Clinical Impact: |
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) are metabolized principally by CYP3A4. Efavirenz, nevirapine, and etravirine are known CYP3A inducers, whereas delaviridine is a CYP3A inhibitor. Significant pharmacokinetic interactions between NNRTIs (e.g., efavirenz and delavirdine) and buprenorphine have been shown in clinical studies, but these pharmacokinetic interactions did not result in any significant pharmacodynamic effects. |
|
Intervention: |
Patients who are on chronic BELBUCA treatment should have their dose monitored if NNRTIs are added to their treatment regimen. |
|
Examples: |
efavirenz, nevirapine, etravirine, delavirdine |
|
Antiretrovirals: Protease inhibitors (PIs) |
|
|
Clinical Impact: |
Studies have shown some antiretroviral protease inhibitors (PIs) with CYP3A4 inhibitory activity (nelfinavir, lopinavir/ritonavir, ritonavir) have little effect on buprenorphine pharmacokinetic and no significant pharmacodynamic effects. Other PIs with CYP3A4 inhibitory activity (atazanavir and atazanavir/ritonavir) resulted in elevated levels of buprenorphine and norbuprenorphine, and patients in one study reported increased sedation. Symptoms of opioid excess have been found in post-marketing reports of patients receiving buprenorphine and atazanavir with and without ritonavir concomitantly. |
|
Intervention: |
Monitor patients taking BELBUCA and atazanavir with and without ritonavir, and dose reduction of BELBUCA may be warranted. |
|
Examples: |
atazanavir, ritonavir |
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate and well-controlled studies of BELBUCA or buprenorphine in pregnant women. Limited published data on use of buprenorphine, the active ingredient in BELBUCA, in pregnancy, have not shown an increased risk of major malformations. In animal reproduction studies, embryofetal death was observed in both rats and rabbits administered buprenorphine during the period of organogenesis via the oral route of administration at doses approximately 53 to 11 times the maximum recommended human dose (MRHD), respectively. In pre- and post-natal development studies in rats, dystocia was observed after treatment with buprenorphine via the IM route of administration at a dose approximately 27 times the MRHD, and increased neonatal death was observed after treatment via the oral, IM, and SC routes of administration at doses approximately 4, 3, and 0.5 times the MRHD, respectively. No teratogenic effects were observed in rats treated with buprenorphine via the oral, IM, and IV routes of administration during organogenesis at doses approximately 853, 27, and 4 times the MRHD, respectively, or in rabbits treated with buprenorphine via the oral, SC, and IV routes of administration at doses approximately 267, 53, and 9 times the MRHD, respectively. However, in a few studies, some events such as acephalus, omphalocele, and skeletal abnormalities were observed but these findings were not clearly treatment-related [see Data].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Prolonged use of opioid analgesics during pregnancy for medical or nonmedical purposes can result in physical dependence in the neonate and neonatal opioid withdrawal syndrome shortly after birth. Observe newborns for symptoms of neonatal opioid withdrawal syndrome, including poor feeding, diarrhea, irritability, tremor, rigidity, and seizures, and manage accordingly [see Warnings and Precautions ( 5.3)] .
Labor or Delivery
Opioids cross the placenta and may produce respiratory depression and psycho-physiologic effects in neonates. An opioid antagonist such as naloxone must be available for reversal of opioid-induced respiratory depression in the neonate. BELBUCA is not recommended for use in women immediately prior to labor, when shorter-acting analgesics or other analgesic techniques are more appropriate. Opioid analgesics, including BELBUCA, can prolong labor through actions which temporarily reduce the strength, duration, and frequency of uterine contractions. However, this effect is not consistent and may be offset by an increased rate of cervical dilation, which tends to shorten labor.
Data
Animal Data
Buprenorphine administration during organogenesis was not teratogenic in rats or rabbits after intramuscular (IM) or subcutaneous (SC) doses up to 5 mg/kg/day (estimated exposure was approximately 27 and 53 times, respectively, the maximum recommended human dose (MRHD) for buccal BELBUCA of 1.8 mg on a mg/m 2 basis), after IV doses up to 0.8 mg/kg/day (estimated exposure was approximately 4 and 9 times, respectively, the MRHD), or after oral doses up to 160 mg/kg/day in rats (estimated exposure was approximately 853 times the MRHD) and 25 mg/kg/day in rabbits (estimated exposure was approximately 267 times the MRHD). Significant increases in skeletal abnormalities (e.g., extra thoracic vertebra or thoraco-lumbar ribs) were noted in rats after SC administration of 1 mg/kg/day and up (estimated exposure was approximately 5 times the MRHD), but were not observed at oral doses up to 160 mg/kg/day (estimated exposure was approximately 853 times the MRHD. Increases in skeletal abnormalities in rabbits after IM administration of 5 mg/kg/day (estimated exposure was approximately 53 times the MRHD) or oral administration of 1 mg/kg/day or greater estimated exposure was approximately 11 times the MRHD) were not statistically significant.
In rabbits, buprenorphine produced statistically significant pre-implantation losses at oral doses of 1 mg/kg/day or greater (estimated exposure was approximately 11 times the MRHD) and post-implantation losses that were statistically significant at IV doses of 0.2 mg/kg/day or greater (estimated exposure was approximately 2 times the MRHD).
Dystocia was noted in pregnant rats treated intramuscularly with buprenorphine 5 mg/kg/day (approximately 27 times the MRHD). Fertility, peri- and post-natal development studies with buprenorphine in rats indicated increases in neonatal mortality after oral doses of 0.8 mg/kg/day and up (approximately 4 times the MRHD), after IM doses of 0.5 mg/kg/day and up (approximately 3 times the MRHD), and after SC doses of 0.1 mg/kg/day and up (approximately 0.5 times the MRHD). Delays in the occurrence of righting reflex and startle response were noted in rat pups at an oral dose of 80 mg/kg/day (approximately 427 times the MRHD).
8.2 Lactation
Risk Summary
Based on two studies in 13 lactating women being treated for opioid dependence and their breastfed infants, buprenorphine and its metabolite norbuprenorphine are present in low levels in human milk and infant urine, and available data have not shown adverse reactions in breastfed infants [see Data]. There are no data on the effects of BELBUCA on milk production. Because of the potential for serious adverse reactions, including excess sedation and respiratory depression in a breastfed infant, advise patients that breastfeeding is not recommended during treatment with BELBUCA
Clinical Considerations
Infants exposed to BELBUCA through breast milk should be monitored for excess sedation and respiratory depression. Withdrawal symptoms can occur in breastfed infants when maternal administration of buprenorphine is stopped or when breast-feeding is stopped.
Data
Based on limited data from a study of six lactating women being treated for opioid dependence who were taking a median oral dose of buprenorphine of 0.29 mg/kg/day 5-8 days after delivery, breast milk contained a median infant dose of 0.42 mcg/kg/day of buprenorphine and 0.33 mcg/kg/day of norbuprenorphine, which are equal to 0.2% and 0.12% of the maternal weight-adjusted dose. The median concentrations of buprenorphine and norbuprenorphine in infant urine were 1.0 nmol/L and 2.3 nmol/L, respectively.
Based on limited data from a study of seven lactating women being treated for opioid dependence who were taking a median oral dose of buprenorphine of 7 mg/day an average of 1.12 months after delivery, the mean milk concentrations of buprenorphine and norbuprenorphine were 3.65 mcg/L and 1.94 mcg/L, respectively. Based on the limited data from this study, and assuming milk consumption of 150 mL/kg/day, an exclusively breastfed infant would receive an estimated mean of 0.55 mcg/kg/day of buprenorphine and 0.29 mcg/kg/day of norbuprenorphine, which are 0.38% and 0.18% of the maternal weight-adjusted dose.
No adverse reactions were observed in the infants in these two studies.
8.4 Pediatric Use
The safety and efficacy of BELBUCA have not been established in pediatric patients.
8.5 Geriatric Use
Of the total number of patients that were treated with BELBUCA in controlled and open-label chronic pain trials (2,127), 340 patients were 65 years and older. Of those, 49 patients were aged 75 years and older. The incidences of selected BELBUCA-related adverse effects were higher in older subjects.
No notable differences in pharmacokinetics were observed from population pharmacokinetic analysis in subjects aged 65 compared to younger subjects. Other reported clinical experience with buprenorphine has not identified differences in responses between the elderly and younger patients. Although specific dose adjustments on the basis of advanced age are not required for pharmacokinetic reasons, use caution in the elderly population to ensure safe use [see Clinical Pharmacology ( 12.3)] .
8.6 Hepatic Impairment
BELBUCA has not been evaluated in patients with severe hepatic impairment.
The effects of hepatic impairment on the pharmacokinetics of buprenorphine were evaluated in a pharmacokinetic study. Buprenorphine is extensively metabolized in the liver and buprenorphine plasma levels were found to be higher and the half-life was found to be longer in subjects with moderate and severe hepatic impairment, but not in subjects with mild hepatic impairment.
Given that increased buprenorphine plasma levels are associated with a greater risk of toxicity and overdose, a dosage reduction in patients with severe hepatic impairment (i.e., Child-Pugh C) is recommended [ see Dosage and Administration ( 2.5) ]. Monitor patients with severe hepatic impairment for signs and symptoms of overdose. A dosage reduction in patients with moderate hepatic impairment (Child-Pugh B) is not needed, however, monitor these patients for signs and symptoms of toxicity or overdose. A dosage reduction in patients with mild hepatic impairment (Child-Pugh A) is not needed [see Dosage and Administration ( 2.5), Warnings and Precautions ( 5.11) and Clinical Pharmacology ( 12.3)] .
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
BELBUCA contains buprenorphine hydrochloride, a Schedule III controlled substance with an abuse potential similar to other Schedule III opioids. BELBUCA can be abused and is subject to misuse, abuse, addiction, and criminal diversion [see Warnings and Precautions ( 5.1)] .
9.2 Abuse
All patients treated with opioids, including BELBUCA, require careful monitoring for signs of abuse and addiction, because use of opioid analgesic products carry the risk of addiction, even under appropriate medical use.
Prescription drug abuse is the intentional, non-therapeutic use of a prescription drug, even once, for its rewarding psychological or physiological effects.
Drug addiction is a cluster of behavioral, cognitive, and physiological phenomena that develop after repeated substance use and includes a strong desire to take the drug, difficulties in controlling its use, persisting in its use despite harmful consequences, a higher priority given to drug use than to other activities and obligations, increased tolerance, and sometimes a physical withdrawal.
“Drug-seeking” behavior is very common in persons with substance use disorders . Drug-seeking tactics include emergency calls or visits near the end of office hours, refusal to undergo appropriate examination, testing, or referral, repeated “loss” of prescriptions, tampering with prescriptions and reluctance to provide prior medical records or contact information for other treating health care providers(s). “Doctor shopping” (visiting multiple prescribers) to obtain additional prescriptions is common among drug abusers and people suffering from untreated addiction. Preoccupation with achieving adequate pain relief can be appropriate behavior in a patient with poor pain control.
Abuse and addiction are separate and distinct from physical dependence and tolerance. Health care providers should be aware that addiction may not be accompanied by concurrent tolerance and symptoms of physical dependence in all persons with substance use disorders. In addition, abuse of opioids can occur in the absence of true addiction.
BELBUCA, like other opioids, can be diverted for non-medical use into illicit channels of distribution. Careful record-keeping of prescribing information including quantity, frequency, and renewal requests, as required by state and federal law, is strongly advised.
Proper assessment of the patient, proper prescribing practices, periodic re-evaluation of therapy, and proper dispensing and storage are appropriate measures that help to reduce abuse of opioid drugs.
Risks Specific to Abuse of BELBUCA
BELBUCA is intended for buccal use only. Abuse of BELBUCA poses a risk of overdose and death. This risk is increased with concurrent abuse of BELBUCA with alcohol and other substances, including other opioids and benzodiazepines [see Warnings and Precautions ( 5.4), Drug Interactions ( 7)] . Intentional compromise of the buccal film might result in the uncontrolled delivery of buprenorphine and pose a significant risk to the abuser that could result in overdose and death [see Warnings and Precautions ( 5.1)]. Abuse may occur by applying the buccal film in the absence of legitimate purpose, or by swallowing, snorting, or injecting buprenorphine extracted from the buccal film.
9.3 Dependence
Both tolerance and physical dependence can develop during chronic opioid therapy. Tolerance is the need for increasing doses of opioids to maintain a defined effect such as analgesia (in the absence of disease progression or other external factors). Tolerance may occur to both the desired and undesired effects of drugs and may develop at different rates for different effects.
Physical dependence results in withdrawal symptoms after abrupt discontinuation or a significant dose reduction of a drug. Withdrawal also may be precipitated through the administration of drugs with opioid antagonist activity, e.g., naloxone, nalmefene, or mixed agonist/antagonist analgesics (pentazocine, butorphanol, nalbuphine). Physical dependence may not occur to a clinically significant degree until after several days to weeks of continued opioid usage.
BELBUCA should not be abruptly discontinued [see Dosage and Administration ( 2.4)]. If BELBUCA is abruptly discontinued in a physically-dependent patient, a withdrawal syndrome may occur. Some or all of the following can characterize this syndrome: restlessness, lacrimation, rhinorrhea, yawning, perspiration, chills, myalgia, and mydriasis. Other signs and symptoms also may develop including: irritability, anxiety, backache, joint pain, weakness, abdominal cramps, insomnia, nausea, anorexia, vomiting, or diarrhea or increased blood pressure, respiratory rate, or heart rate.
Infants born to mothers physically dependent on opioids will also be physically dependent and may exhibit respiratory difficulties and withdrawal symptoms [see Warnings and Precautions ( 5.3) and Use in Specific Populations ( 8.1)].
10 OVERDOSAGE
Clinical Presentation
Acute overdosage with BELBUCA is manifested by respiratory depression, somnolence progressing to stupor or coma, skeletal muscle flaccidity, cold and clammy skin, constricted pupils, bradycardia, hypotension, partial or complete airway obstruction, atypical snoring, and death. Marked mydriasis rather than miosis may be seen due to severe hypoxia in overdose situations.
Treatment of Overdose
In case of overdose, priorities are the re-establishment of a patent and protected airway and institution of assisted or controlled ventilation, if needed. Employ other supportive measures (including oxygen, vasopressors) in the management of circulatory shock and pulmonary edema, as indicated. Cardiac arrest or arrhythmias will require advanced life support techniques.
Naloxone may not be effective in reversing any respiratory depression produced by buprenorphine. High doses of naloxone, 10-35 mg/70 kg, may be of limited value in the management of buprenorphine overdose. The onset of naloxone effect may be delayed by 30 minutes or more. Doxapram hydrochloride (a respiratory stimulant) has also been used.
Because the duration of reversal would be expected to be less than the duration of action of buprenorphine from BELBUCA, carefully monitor the patient until spontaneous respiration is reliably re-established. Even in the face of improvement, continued medical monitoring is required for at least 24 hours because of the possibility of extended effects of buprenorphine.
In an individual physically dependent on opioids, administration of an opioid receptor antagonist may precipitate an acute withdrawal. The severity of the withdrawal produced will depend on the degree of physical dependence and the dose of the antagonist administered. If a decision is made to treat serious respiratory depression in the physically dependent patient with an opioid antagonist, administration of the antagonist should be begun with care and by titration with smaller than usual doses of the antagonist.
11 DESCRIPTION
BELBUCA is a buccal film providing transmucosal delivery of buprenorphine hydrochloride, a partial opioid agonist. BELBUCA is a rectangular bi-layer, peppermint-flavored, buccal film with rounded corners, consisting of a white to off-white backing layer with strength identifier printed in black ink and a light yellow to yellow active mucoadhesive layer containing buprenorphine hydrochloride. The yellow side of the buccal film is applied to the inside of the cheek where it adheres to the moist buccal mucosa to deliver the drug as the film dissolves.
The chemical name of buprenorphine hydrochloride is 6,14-ethenomorphinan-7-methanol, 17-(cyclopropylmethyl)- α-(1,1-dimethylethyl)-4, 5-epoxy-18,19-dihydro-3-hydroxy-6-methoxy-α-methyl-, hydrochloride, [5α, 7α, (S)]. The structural formula is:
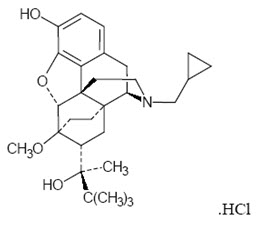
The molecular weight of buprenorphine hydrochloride is 504.10; the empirical formula is C 29H 41NO 4∙HCl. Buprenorphine hydrochloride occurs as a white or off white crystalline powder. It is sparingly soluble in water, freely soluble in methanol, soluble in alcohol, and practically insoluble in cyclohexane. The pKa is 8.5 for the amine function and 10.0 for the phenol function.
BELBUCA is available as 75 mcg, 150 mcg, 300 mcg, 450 mcg, 600 mcg, 750 mcg, and 900 mcg buprenorphine per film. The strength of each film is dependent on the buprenorphine concentration in the formulation and the surface area of the film. Unique identifiers and film size for each strength are listed in Table 6.
|
Buprenorphine Strength (mcg) |
BELBUCA Identifier |
Film Size (cm 2) |
|
75 |
E0 |
1.215 |
|
150 |
E1 |
2.431 |
|
300 |
E3 |
0.934 |
|
450 |
E4 |
1.400 |
|
600 |
E6 |
1.867 |
|
750 |
E7 |
2.334 |
|
900 |
E9 |
2.801 |
The active ingredient in BELBUCA is buprenorphine hydrochloride. Each buccal film also contains carboxymethylcellulose sodium USP, citric acid anhydrous USP, hydroxyethylcellulose NF, hydroxypropylcellulose NF, methylparaben NF, monobasic sodium phosphate anhydrous USP, peppermint oil NF, polycarbophil USP, propylene glycol USP, propylparaben NF, sodium benzoate NF, sodium hydroxide NF, saccharin sodium NF, titanium dioxide USP, vitamin E acetate USP, yellow iron oxide, purified water USP, and TekPrint TM SW-9008 black ink (shellac NF, black iron oxide NF).
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Buprenorphine is a partial agonist at the mu-opioid receptor and an antagonist at the kappa-opioid receptor.
12.2 Pharmacodynamics
Effects on the Central Nervous System
The principal action of therapeutic value of buprenorphine is analgesia and is thought to be due to buprenorphine binding with high affinity to opioid receptors on neurons in the brain and spinal cord. Buprenorphine may also cause sedation or somnolence via an action in the brain.
Buprenorphine produces mu opioid receptor-mediated respiratory depression by direct action on brainstem respiratory centers by reducing sensitivity of the brainstem to increases in carbon dioxide tension and to electrical stimulation.
Buprenorphine causes miosis, even in total darkness, and little tolerance develops to this effect. Pinpoint pupils are a sign of opioid overdose but are not pathognomonic (e.g., pontine lesions of hemorrhagic or ischemic origins may produce similar findings). Marked mydriasis rather than miosis may be seen with worsening hypoxia in the setting of buprenorphine overdose.
Unlike other opioids, buprenorphine appears to exhibit a dose-ceiling effect.
Effects on the Gastrointestinal Tract and Other Smooth Muscle
Gastric, biliary, and pancreatic secretions are decreased by buprenorphine. Buprenorphine causes a reduction in motility associated with an increase in tone in the stomach and duodenum. Digestion of food in the small intestine is delayed and propulsive contractions are decreased. Propulsive peristaltic waves in the colon are decreased while tone is increased to the point of spasm. The end result is constipation. Buprenorphine may cause an increase in biliary tract pressure as a result of spasm of the sphincter of Oddi.
Effects on the Cardiovascular System
Buprenorphine may cause a reduction in blood pressure.
Effects on Cardiac Electrophysiology
QTc prolongation with BELBUCA has been observed. Of the 1590 patients that were treated with BELBUCA in controlled and open-label chronic pain trials at doses up to 900 mcg every 12 hours, 2% demonstrated a prolongation of QTcF to a post-baseline value between 450 - 480 msec during therapy.
Effects on the Endocrine System
Opioids inhibit the secretion of ACTH, cortisol, and luteinizing hormone (LH) in humans. They also stimulate prolactin, growth hormone (GH) secretion, and pancreatic secretion of insulin and glucagon.
Effects on the Immune System
Opioids have been shown to have a variety of effects on components of the immune system in in vitro and animal models. The clinical significance of these findings is unknown. Overall, the effects of opioids appear to be modestly immunosuppressive.
12.3 Pharmacokinetics
Absorption
Systemic plasma levels of buprenorphine increased in a linear manner (C max and AUC) over the single dose range of 75 to 1200 mcg as shown in Table 7. The absolute bioavailability of BELBUCA ranged from 46 to 65%.
|
Regimen | Dosage (mcg) |
C max (ng/mL) |
AUC 0-t (h∙ng/mL) |
AUC0-∞ (h∙ng/mL) |
T max* (hr) |
|
Single Dose |
75 |
0.17±0.30 |
0.46±0.22 |
0.63±0.24 |
3.00 (1.50-4.00) |
|
300 |
0.47±0.47 |
2.00±0.68 |
2.3±0.68 |
2.50 (0.50-4.00) |
|
|
1200 |
1.43±0.45 |
9.6±2.9 |
10.5±3.32 |
3.00 (1.00-4.00) |
|
| * T max values reported as median and range | |||||
Following the multiple dose administration (60 to 240 mcg every 12 hours) of BELBUCA, apparent steady-state buprenorphine plasma concentrations were achieved prior to the 6 th dose. Buprenorphine steady-state C max and AUC increased proportional to dose.
Systemic exposure to buprenorphine from BELBUCA film was reduced by 23-27% by the ingestion of liquids (cold, hot and room temperature water) during film administration; additionally coadministration with low pH liquid, such as decaffeinated cola, decreased buprenorphine exposure from BELBUCA by approximately 37%. The consumption of liquids should be avoided until the buccal film has completely dissolved [see Dosage and Administration ( 2.7)] .
Distribution
Buprenorphine is approximately 96% protein bound, primarily to alpha and beta globulin.
Elimination
Metabolism
Buprenorphine undergoes both N-dealkylation to norbuprenorphine and glucuronidation. The N-dealkylation pathway is mediated primarily by CYP3A4. Norbuprenorphine, the major metabolite, can further undergo glucuronidation. Norbuprenorphine has been found to bind opioid receptors in vitro; however, it has not been studied clinically for opioid-like activity
Excretion
A mass balance study of buprenorphine showed complete recovery of radiolabel in urine (30%) and feces (69%) collected up to 11 days after dosing. Almost all of the dose was accounted for in terms of buprenorphine, norbuprenorphine, and two unidentified buprenorphine metabolites. In urine, most of buprenorphine and norbuprenorphine was conjugated (buprenorphine, 1% free and 9.4% conjugated; norbuprenorphine, 2.7% free and 11% conjugated). In feces, almost all of the buprenorphine and norbuprenorphine was free (buprenorphine, 33% free and 5% conjugated; norbuprenorphine, 21% free and 2% conjugated).
Based on multiple dose studies performed with BELBUCA, the mean plasma elimination half-life of buprenorphine was 27.6±11.2 hours.
Drug Interactions
CYP3A4 Inhibitors and Inducers: Buprenorphine undergoes N-dealkylation pathway mediated primarily by CYP3A4, so its metabolism can be inhibited by CYP3A4 inhibitors. The interaction of buprenorphine with all CYP3A4 inducers has not been studied. [see Drug Interactions ( 7)] .
Buprenorphine has been found to be a CYP2D6 and CYP3A4 inhibitor and its major metabolite,norbuprenorphine, has been found to be a moderate CYP2D6 inhibitor in in vitro studies employing human liver microsomes. However, the relatively low plasma concentrations of buprenorphine and norbuprenorphine resulting from therapeutic doses are not expected to raise significant drug-drug interaction concerns.
Specific Populations
Hepatic Impairment
BELBUCA has not been evaluated in patients with severe hepatic impairment. The pharmacokinetics of buprenorphine following an IV infusion of 0.3 mg of buprenorphine were compared in 8 patients with mild hepatic impairment (Child-Pugh A), 4 patients with moderate impairment (Child-Pugh B), and 12 subjects with normal hepatic function. Buprenorphine and norbuprenorphine plasma levels did not increase in mild or moderately impaired patient cohorts.
In another pharmacokinetic study, the disposition of buprenorphine was determined after administering a 2.0/0.5 mg buprenorphine/naloxone sublingual tablet in subjects with varied degrees of hepatic impairment as indicated by Child-Pugh criteria. The disposition of buprenorphine in patients with hepatic impairment was compared to disposition in subjects with normal hepatic function. In subjects with mild hepatic impairment, the changes in mean C max, AUC 0-last, and half-life values of buprenorphine were not clinically significant. No dose adjustment is needed in patients with mild hepatic impairment.
For subjects with moderate and severe hepatic impairment, mean C max , AUC 0-last, and half-life values of buprenorphine were increased. [see Dosage and Administration ( 2.5), Warnings and Precautions ( 5.11), and Use in Specific Populations ( 8.6)] .
|
Hepatic Impairment |
PK Parameters |
Increase in buprenorphine compared to healthy subjects |
|
Moderate |
C max |
8% |
|
AUC 0-last |
64% |
|
|
Half-life |
35% |
|
|
Severe |
C max |
72% |
|
AUC 0-last |
181% |
|
|
Half-life |
57% |
Oral Mucositis
In an open-label pharmacokinetic study in 6 cancer patients with Grade 3 mucositis, buprenorphine was absorbed more rapidly from BELBUCA resulting in a higher C max (~79%) and AUC (~56%) compared to age- and gender-matched healthy control patients [see Dosage and Administration ( 2.6), Warnings and Precautions ( 5.15) ].
Geriatric Patients
No notable differences in pharmacokinetics were observed from population PK analysis in subjects aged 65 compared to younger subjects. Other reported clinical experience with buprenorphine has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Pediatric Patients
BELBUCA has not been studied in children and is not recommended for pediatric use.
Sex
No notable sex differences in pharmacokinetics were observed from population PK analysis.
Renal Impairment
No studies in patients with renal impairment have been performed with BELBUCA. In an independent study, the effect of impaired renal function on buprenorphine pharmacokinetics after IV bolus and after continuous IV infusion administration was evaluated; and no notable differences in plasma buprenorphine concentrations were identified in patients with normal renal function compared to impaired renal function or renal failure.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies of buprenorphine were conducted in Sprague-Dawley rats and CD-1 mice. Buprenorphine was administered in the diet to rats at doses of 0.6, 5.5, and 56 mg/kg/day for 27 months (estimated exposure was approximately 3, 29, and 299 times the maximum recommended human dose (MRHD) of buccal Belbuca of 1.8 mg on a mg/m 2 basis, respectively). Statistically significant dose-related increases in testicular interstitial (Leydig’s) cell tumors occurred. In an 86-week study in CD-1 mice, buprenorphine was not carcinogenic at dietary doses up to 100 mg/kg/day (estimated exposure was approximately 267 times the MRHD).
Mutagenesis
Buprenorphine was studied in a series of tests utilizing gene, chromosome, and DNA interactions in both prokaryotic and eukaryotic systems. Results were negative in yeast ( S. cerevisiae) for recombinant, gene convertant, or forward mutations; negative in Bacillus subtilis “rec” assay, negative for clastogenicity in CHO cells, Chinese hamster bone marrow and spermatogonia cells, and negative in the mouse lymphoma L5178Y assay.
Results were equivocal in the Ames test: negative in studies in two laboratories, but positive for frame shift mutation at a high dose (5 mg/plate) in a third study. Results were positive in the Green-Tweets ( E. coli) survival test, positive in a DNA synthesis inhibition (DSI) test with testicular tissue from mice, for both in vivo and in vitro incorporation of [ 3H]thymidine, and positive in unscheduled DNA synthesis (UDS) test using testicular cells from mice.
Impairment of Fertility
Reproduction studies of buprenorphine in rats demonstrated no evidence of impaired fertility at daily oral doses up to 80 mg/kg/day (estimated exposure approximately 427 times the MRHD) or up to 5 mg/kg/day IM or SC (estimated exposure was approximately 27 times the MRHD).
14 CLINICAL STUDIES
The efficacy of BELBUCA has been evaluated in three 12-week double-blind, placebo-controlled clinical trials in opioid-naïve and opioid-experienced patients with moderate-to-severe chronic low back pain using pain scores as the primary efficacy variable. Two of these studies, described below, demonstrated efficacy in patients with low back pain. One study in low back pain did not show a statistically significant pain reduction for BELBUCA compared to placebo.
12-Week Study in Opioid-Naïve Patients with Chronic Low Back Pain
A total of 749 patients with chronic low back pain entered an open-label, dose-titration period for up to eight weeks. Potential subjects were excluded from participation for QTcF interval of 450 ms or more, hypokalemia, clinically unstable cardiac disease, a history of Long QT Syndrome or an immediate family member with this condition, or taking Class IA or Class III antiarrhythmic medications. Patients initiated therapy with a single 75 mcg dose of BELBUCA on Day 1 and continued taking BELBUCA 75 mcg either once daily or every 12 hours for 4-8 days as tolerated. The dose was then increased to 150 mcg every 12 hours, and patients could continue to dose escalate in 150 mcg dose increments every 4-8 days for up to 6 weeks if the adverse effects were tolerable and the analgesic effects were not adequate. Patients who achieved adequate analgesia and tolerable adverse effects on BELBUCA for at least 2 weeks were then randomized to continue their titrated dose of BELBUCA or matching placebo. Sixty-one percent (61%) of the patients who entered the open-label dose titration period were able to titrate to a tolerable and effective dose and were randomized into a 12-week, double-blind treatment period. Fifteen percent of patients discontinued due to an adverse event and 4% discontinued due to lack of a therapeutic effect. The remaining 20% of patients discontinued due to various non-drug related administrative reasons.
During the first 2 weeks of double-blind treatment, patients were allowed up to 2 tablets per day of hydrocodone/acetaminophen 5/325 mg as supplemental analgesia to minimize opioid withdrawal symptoms in patients randomized to placebo. Thereafter, the supplemental analgesia was limited to 1 to 2 tablets of acetaminophen 500 mg per day. Seventy-six percent of the patients treated with BELBUCA completed the 12-week treatment compared to 73% of the patients treated with placebo. Of the 209 patients randomized to BELBUCA, 4% discontinued due to lack of efficacy and 8% due to adverse events. Of the 211 patients randomized to placebo, 11% discontinued due to lack of efficacy and 4% due to adverse events.
Of the patients who were randomized, the mean pain (SD) scores on a 0 to 10 numeric rating scale (NRS) were 7.1 (1.06) and 7.2 (1.05) prior to open-label titration and 2.8 (1.01) and 2.8 (1.12) at the beginning of the double-blind period for BELBUCA and placebo, respectively. The change from double-blind baseline to week 12 in mean pain (SD) NRS score was statistically significant favoring patients treated with BELBUCA, compared with patients treated with placebo.
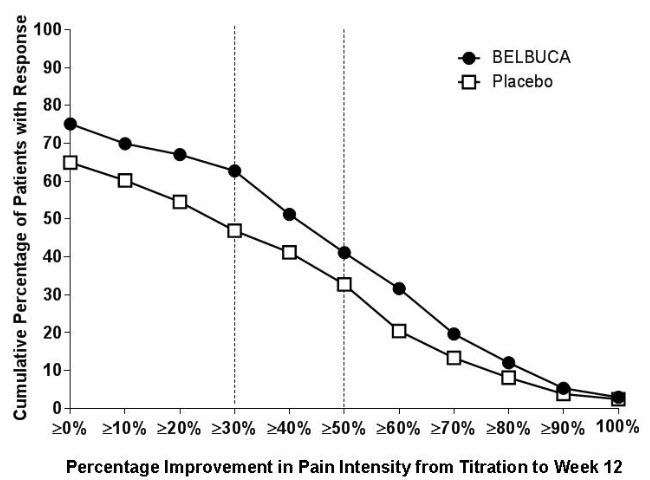
A higher proportion of BELBUCA patients (62%) had at least a 30% reduction in pain score from prior to open-label titration to study endpoint when compared to patients who received placebo buccal film (47%). A higher proportion of BELBUCA patients (41%) also had at least a 50% reduction in pain score from prior to open-label titration to study endpoint compared to patients who received placebo (33%).
The proportion of patients with various degrees of improvement, from prior to open-label titration (Titration-Baseline) to study endpoint, is shown in Figure 1 below.
12-Week Study in Opioid-Experienced Patients with Chronic Low Back Pain
Eight hundred and ten (810) patients on chronic opioid therapy (total daily dose 30-160 mg in oral morphine sulfate equivalents (MSE) for at least 4 weeks) entered an open-label, dose-titration period with BELBUCA for up to 8 weeks, following taper of their prior opioids to 30 mg oral MSE daily. Potential subjects were excluded from participation for QTcF interval of 450 ms or more, hypokalemia, clinically unstable cardiac disease, a history of Long QT Syndrome or an immediate family member with this condition, or taking Class IA or Class III antiarrhythmic medications. Patients were initiated with BELBUCA 150 mcg every 12 hours if they were on 30 to 89 mg oral MSE daily and 300 mcg every 12 hours if they were on 90 to160 mg oral MSE daily prior to taper. If a patient tolerated the adverse events and the analgesic effects were not adequate, the dose was increased in increments of 150 mcg every 12 hours after 4 to 8 days for up to 6 weeks. Patients were permitted to take hydrocodone/acetaminophen 5/325 mg as analgesic rescue as needed up to a maximum of 4 doses per day during the open-label dose titration period. After a dose was reached with adequate analgesia and tolerable adverse effects for a period of 2 weeks, patients were randomized to continue their titrated dose of BELBUCA or matching placebo. Sixty-three percent (63%) of the patients who entered the open-label titration period were able to titrate to a tolerable and effective dose and were randomized into a 12-week double-blind treatment phase. Ten percent (10%) of patients discontinued due to an adverse event, 8% discontinued due to lack of a therapeutic effect, and 0.1% discontinued due to opioid withdrawal during the open-label titration period. The remaining 20% of patients discontinued due to various non drug related administrative reasons.
During the double-blind period, patients were permitted to take up to 2 doses of 5/325 mg or 10/650 mg of hydrocodone/acetaminophen per day for the first 2 weeks to minimize opioid withdrawal symptoms in patients randomized to placebo. After the first 2 weeks, patients were permitted to take 1 dose of 5/325 mg or 10/650 mg per day. Eighty-three percent of patients treated with BELBUCA and 57% of patients treated with placebo buccal film completed the 12-week treatment period. Of the 243 patients randomized to BELBUCA, 8% discontinued due to lack of efficacy and 2% due to adverse events. Of the 248 patients randomized to placebo buccal film, 25% discontinued due to lack of efficacy and 5% due to adverse events.
Of the patients who were randomized into the double-blind period, the mean pain (SD) NRS scores were 6.8 (1.28) and 6.6 (1.32) prior to open-label titration and 2.9 (0.985) and 2.8 (1.05) at the beginning of the double-blind period for BELBUCA and placebo, respectively. The change from baseline to week 12 in mean pain (SD) NRS score was statistically significant in favor of patients treated with BELBUCA compared with patients treated with placebo.
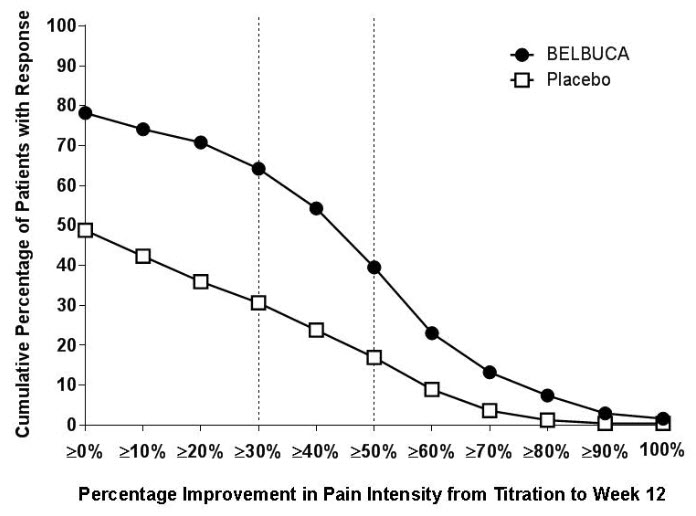
A higher proportion of BELBUCA patients (64%) had at least a 30% reduction in pain score from prior to open-label titration to study endpoint when compared to patients who received placebo buccal film (31%). A higher proportion of BELBUCA patients (39%) also had at least a 50% reduction in pain score from prior to open-label titration to study endpoint compared to patients who received placebo (17%).
The proportion of patients with various degrees of improvement from prior to open-label titration (Titration-Baseline) to study endpoint is shown in Figure 2 below.
16 HOW SUPPLIED/STORAGE AND HANDLING
BELBUCA (buprenorphine) buccal films are supplied in cartons containing 60 individual child-resistant foil packages as follows
|
Strength |
NDC Number |
Foil Color |
|
The 75 mcg buccal film is printed with E0 |
63481-161-60 |
Red |
|
The 150 mcg buccal film is printed with E1 |
63481-207-60 |
Green |
|
The 300 mcg buccal film is printed with E3 |
63481-348-60 |
Gray |
|
The 450 mcg buccal film is printed with E4 |
63481-519-60 |
Purple |
|
The 600 mcg buccal film is printed with E6 |
63481-685-60 |
Blue |
|
The 750 mcg buccal film is printed with E7 |
63481-820-60 |
Lt. Blue |
|
The 900 mcg buccal film is printed with E9 |
63481-952-60 |
Orange |
Store at 25°C (77°F), excursions permitted to 15° to 30°C (59°F to 86ºF). [See USP Controlled Room Temperature.]
Advise patients to store buprenorphine-containing medications safely and out of sight and reach of children. Destroy any unused medication appropriately [ see Patient Counseling Information ( 17)].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling ( Medication Guide)
Addiction, Abuse, and Misuse
Inform patients that the use of BELBUCA, even when taken as recommended, can result in addiction, abuse, and misuse, which could lead to overdose and death [see Warnings and Precautions ( 5.1)]. Instruct patients not to share BELBUCA with others and to take steps to protect BELBUCA from theft or misuse.
Life-Threatening Respiratory Depression
Inform patients of the risk of life-threatening respiratory depression, including information that the risk is greatest when starting BELBUCA or when the dose is increased and that it can occur even at recommended doses [see Warnings and Precautions ( 5.2)] . Advise patients how to recognize respiratory depression and to seek medical attention if breathing difficulties develop.
Accidental Exposure
Inform patients that accidental exposure, especially in children, may result in respiratory depression or death [see Warnings and Precautions ( 5.2)]. Instruct patients to take steps to store BELBUCA securely and to dispose of unused BELBUCA by opening unused packages and flushing the film down the toilet.
Interaction with Alcohol and other CNS Depressants
Inform patients that potentially serious additive effects may occur if BELBUCA is used with alcohol or other CNS depressants and not to use such drugs unless supervised by a health care provider [see Warnings and Precautions ( 5.4)] .
Interaction with Benzodiazepines
Warn patients that it is extremely dangerous to self-administer benzodiazepines while taking BELBUCA, and warn patients to use benzodiazepines concurrently with BELBUCA only as directed by their physician [see Drug Interactions ( 7)] .
Hypotension
Inform patients that BELBUCA may cause orthostatic hypotension and syncope. Instruct patients how to recognize symptoms of low blood pressure and how to reduce the risk of serious consequences should hypotension occur (e.g., sit or lie down, carefully rise from a sitting or lying position) [see Warnings and Precautions ( 5.12)].
Constipation
Advise patients of the potential for severe constipation, including management instructions andwhen to seek medical attention [see Warnings and Precautions ( 5.13)] .
Driving or Operating Heavy Machinery
Inform patients that BELBUCA may impair the ability to perform potentially hazardous activities such as driving a car or operating heavy machinery. Advise patients not to perform such tasks until they know how they will react to the medication [see Warnings and Precautions ( 5.16)] .
Anaphylaxis
Inform patients that anaphylaxis has been reported with ingredients contained in BELBUCA. Advise patients how to recognize such a reaction and when to seek medical attention [ see Warnings and Precautions (5.12)].
Pregnancy
Neonatal Opioid Withdrawal Syndrome
Inform female patients of reproductive potential that prolonged use of BELBUCA during pregnancy can result in neonatal opioid withdrawal syndrome, which may be life-threatening if not recognized and treated [see Warnings and Precautions ( 5.3), Use in Specific Populations ( 8.1)] .
Embryofetal Toxicity
Advise female patients that BELBUCA can cause fetal harm and to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations ( 8.1)].
Lactation
Advise patients that breastfeeding is not recommended during treatment with BELBUCA [see Use in Specific Populations ( 8.2)] .
Important Administration Instructions
Instruct patients how to properly use BELBUCA, including the following:
- To carefully follow instructions for the application of BELBUCA and to avoid eating or drinking until it dissolves.
- To apply BELBUCA once daily, or every twelve (12) hours at the same time or times each day.
- To avoid applying BELBUCA to areas of the mouth with any open sores or lesions.
- To not use BELBUCA if the pouch seal is broken or the buccal film is cut, damaged, or changed in any way.
Disposal
Instruct patients to dispose of BELBUCA as soon as it is no longer needed. To dispose of unused BELBUCA film, inform the patient to:
- Remove all BELBUCA films from their foil packages.
- Drop the BELBUCA films into toilet and flush.
- Discard foil packaging in trash.
Instruct patients not to flush BELBUCA down the toilet in thefoil packages or cartons [s ee Dosage and Administration ( 2.8)] .
Healthcare professionals can telephone Endo Pharmaceuticals (1-800-462-3636) for information on this product.
Distributed by:
Endo Pharmaceuticals Inc.
Malvern, PA 19355
BELBUCA is a trademark of Endo International plc or one of its affiliates.
©2015 Endo Pharmaceuticals Inc. All rights reserved.
116340
Medication Guide
BELBUCA TM (bel-BUE-kuh)
(buprenorphine) buccal film, CIII
BELBUCA is:
- A strong prescription pain medicine that contains an opioid (narcotic) that is used to manage pain severe enough to require daily around-the-clock, long-term treatment with an opioid, when other pain treatments such as non-opioid pain medicines or immediate-release opioid medicines do not treat your pain well enough or you cannot tolerate them.
- A long-acting opioid pain medicine that can put you at risk for overdose and death. Even if you take your dose correctly as prescribed, you are at risk for opioid addiction, abuse, and misuse that can lead to death.
- Not for use to treat pain that is not around-the-clock.
Important information about BELBUCA:
- Get emergency help right away if you take too much BELBUCA (overdose). When you first start taking BELBUCA, when your dose is changed, or if you take too much (overdose), serious or life-threatening breathing problems that can lead to death may occur.
- Never give anyone else your BELBUCA. They could die from taking it. Store BELBUCA away from children and in a safe place to prevent stealing or abuse. Selling or giving away BELBUCA is against the law.
Do not use BELBUCA if you have:
- severe asthma, trouble breathing, or other lung problems.
- a bowel blockage or have narrowing of the stomach or intestines.
Before applying BELBUCA, tell your healthcare provider if you have a history of:
- head injury, seizures
- heart rhythm problems (long QT syndrome)
- liver, kidney, thyroid problems
- pancreas or gallbladder problems
- problems urinating
- abuse of street or prescription drugs, alcohol addiction, or mental health problems.
Tell your healthcare provider if you are:
- pregnant or planning to become pregnant. Prolonged use of BELBUCA during pregnancy can cause withdrawal symptoms in your newborn baby that could be life-threatening if not recognized and treated.
- breastfeeding. Not recommended during treatment with BELBUCA. It may harm your baby.
- taking prescription or over-the-counter medicines, vitamins, or herbal supplements. Taking BELBUCA with certain other medicines can cause serious side effects and could lead to death.
While using BELBUCA:
- Do not change your dose. Apply BELBUCA exactly as prescribed by your healthcare provider.
- See the detailed Instructions for Use for information about how to apply BELBUCA.
- Do not apply BELBUCA if the package seal is broken or the film is cut, damaged, or changed in any way.
- After the film has adhered to your cheek, avoid eating or drinking until the film has completely dissolved, usually within 30 minutes.
- Avoid touching or moving the buccal film with your tongue or fingers.
- Call your healthcare provider if the dose you are using does not control your pain.
- Do not stop using BELBUCA without talking to your healthcare provider.
- After you stop using BELBUCA, remove any unused film from the foil pack and flush down the toilet. Throw the empty foil packaging in the trash.
While using BELBUCA DO NOT:
- Drive or operate heavy machinery, until you know how BELBUCA affects you. BELBUCA can make you sleepy, dizzy, or lightheaded.
- Drink alcohol or use prescription or over-the-counter medicines containing alcohol. Using products containing alcohol during treatment with BELBUCA may cause you to overdose and die.
The possible side effects of BELBUCA are:
- nausea, constipation, headache, vomiting, dizziness, and sleepiness. Call your healthcare provider if you have any of these symptoms and they are severe.
Get emergency medical help if you have:
- trouble breathing, shortness of breath, fast heartbeat, chest pain, swelling of your face, tongue or throat, extreme drowsiness, light-headedness when changing positions, or you are feeling faint.
These are not all the possible side effects of BELBUCA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. For more information go to dailymed.nlm.nih.gov
Marketed by: Endo Pharmaceuticals Inc., Malvern, PA 19355, www.endo.com or call 1-800-462-3636.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Issue: October 2015
116116
Instructions for Use
BELBUCA (bel-BUE-kuh)
(buprenorphine)
buccal film, CIII
Before you use BELBUCA buccal film, it is important that you read the Medication Guide and these Patient Instructions for Use so that you use BELBUCA the right way. Ask your healthcare provider or pharmacist if you have any questions about the right way to use BELBUCA.
Important:
- BELBUCA buccal film is sealed in a foil package. Do not open the package until ready to use. After opening, use the entire BELBUCA buccal film right away.
- Do not apply BELBUCA buccal film if the package seal is broken or the film is cut, damaged, or changed in any way.
- BELBUCA buccal film is available in different strengths. Make sure you have the strength that has been prescribed for you.
- Avoid placing BELBUCA buccal film to areas of the mouth with any open sores or lesions.
Open the BELBUCA package:
- Hold the foil package as shown below
(see Figure A). Fold along the dotted line at the top of the foil package.

- Keep folded and tear down or cut with scissors at the notch in the direction of the scissors on the dotted line
(See Figure B). Tear all the way to the bottom. Be careful to avoid cutting and damaging the BELBUCA buccal film when using scissors.
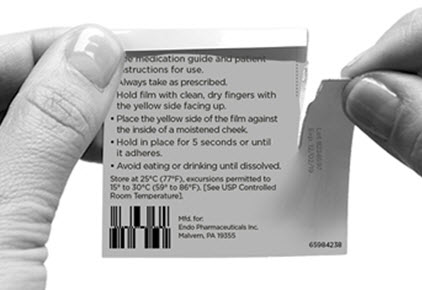
- Remove BELBUCA film from the foil package
(see Figure C).

Use BELBUCA buccal film as follows:
- Use your tongue to wet the inside of your cheek or rinse your mouth with water to moisten the area in your mouth before you place BELBUCA.
- Hold the BELBUCA buccal film with clean, dry fingers with the yellow side facing up
(see Figure D).

- Using a finger, place the yellow side of the BELBUCA buccal film against the inside of your moistened cheek. Press and hold the BELBUCA buccal film in place for 5 seconds and then take your finger away
(see Figure E).
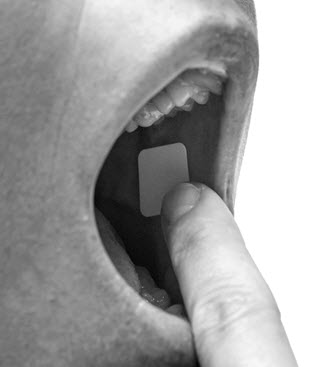
- The BELBUCA buccal film will stick to the inside of your cheek
(see Figure F).

- Leave the BELBUCA buccal film in place until it has completely dissolved, usually within 30 minutes after you apply it.
- Avoid eating food or drinking liquids until BELBUCA buccal film has dissolved.
- Avoid touching or moving BELBUCA buccal film with your tongue or finger after it is in place.
- Do not chew or swallow BELBUCA.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
For more information call Endo Pharmaceuticals at 1-800-462-3636.
Marketed by:
Endo Pharmaceuticals Inc.
Malvern, PA 19355
BELBUCA is a trademark of Endo International plc or one of its affiliates.
©2015 Endo Pharmaceuticals Inc. All rights reserved.
10/2015
116116


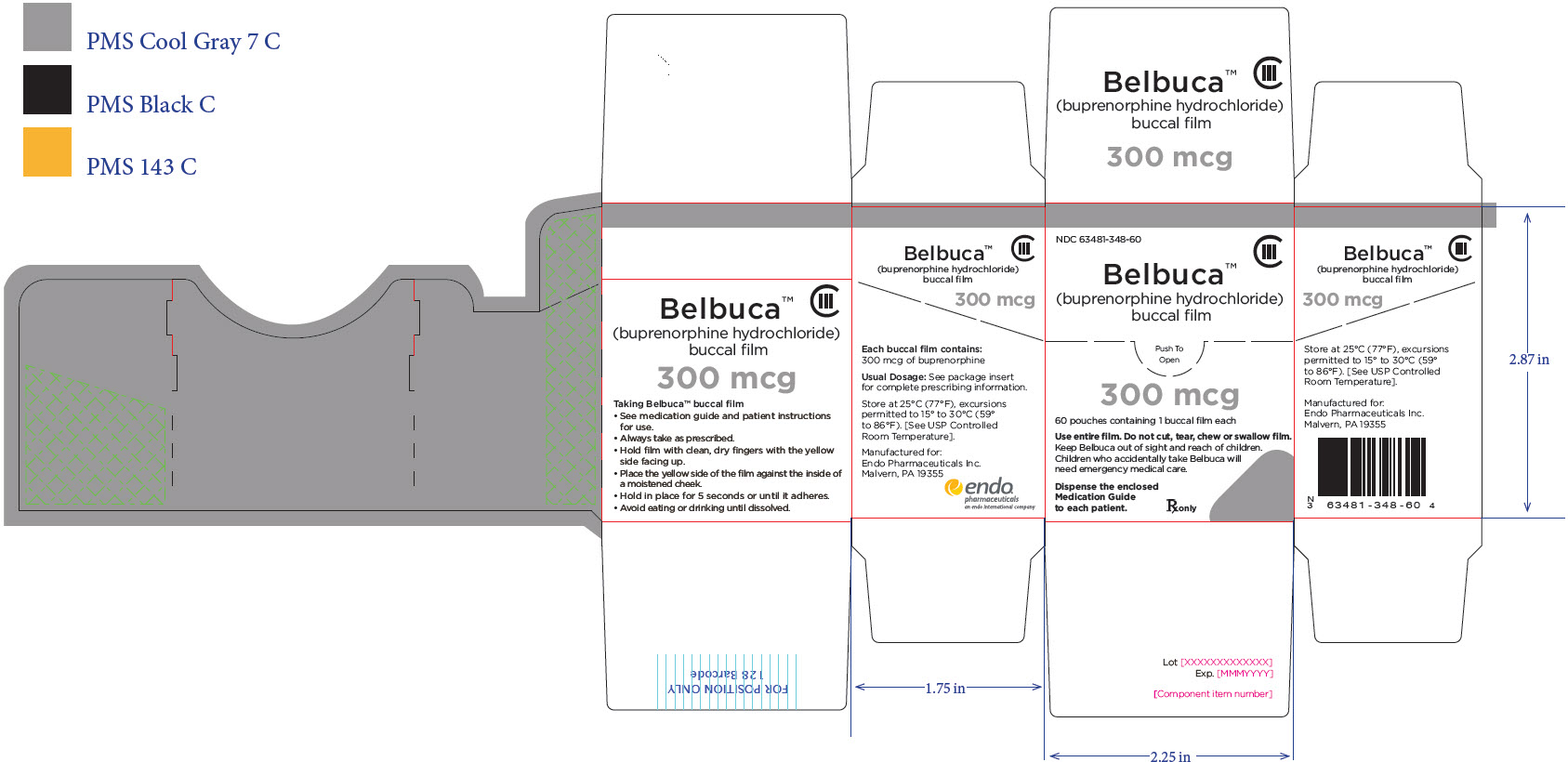

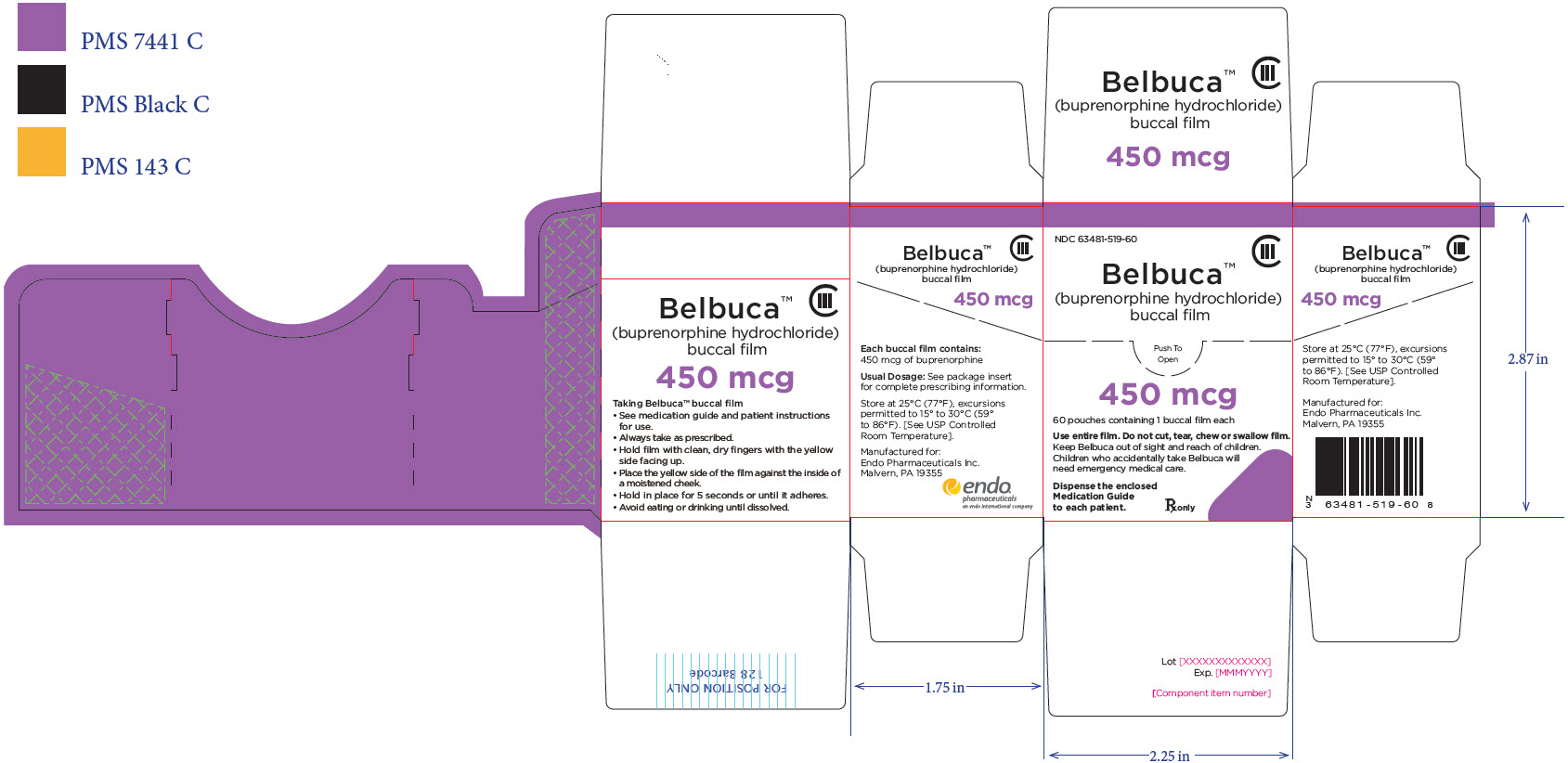



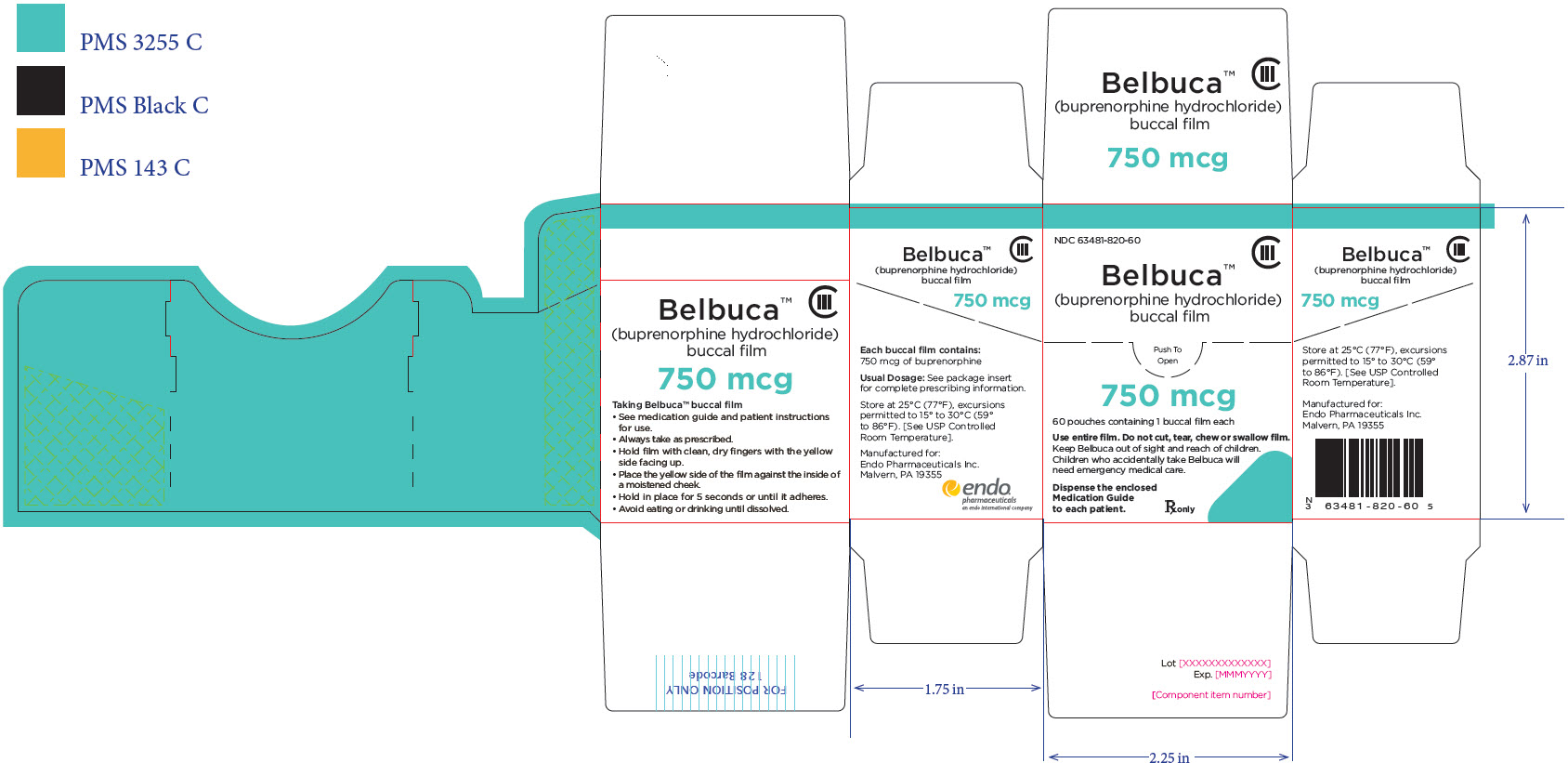
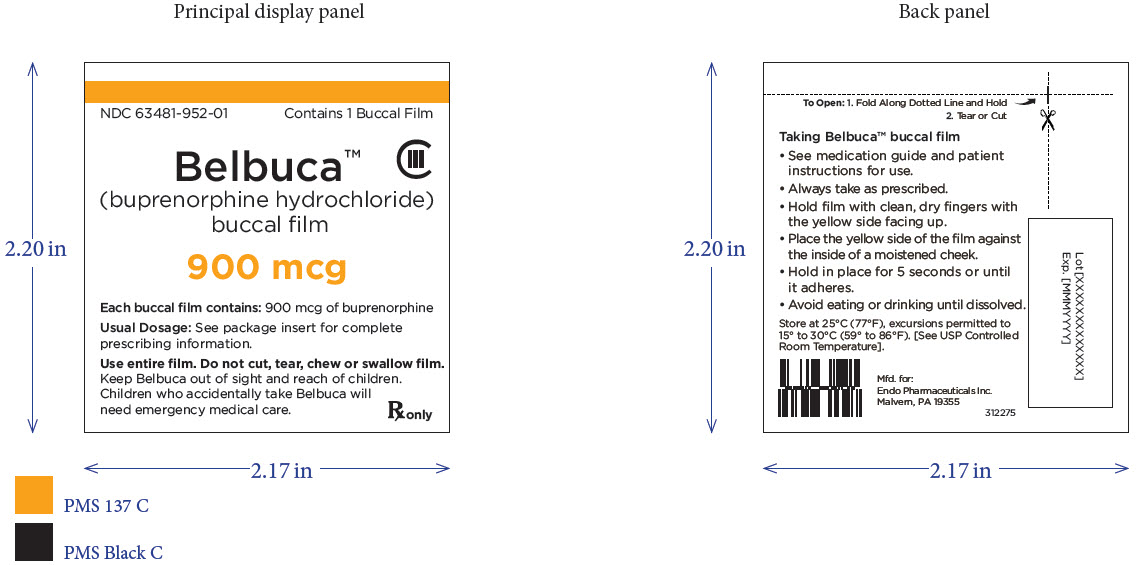

| BELBUCA
buprenorphine hydrochloride film |
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
| BELBUCA
buprenorphine hydrochloride film |
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
| BELBUCA
buprenorphine hydrochloride film |
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
| BELBUCA
buprenorphine hydrochloride film |
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
| BELBUCA
buprenorphine hydrochloride film |
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
| BELBUCA
buprenorphine hydrochloride film |
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
| BELBUCA
buprenorphine hydrochloride film |
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
| Labeler - Endo Pharmaceuticals (178074951) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Tapemark Company | 006154595 | manufacture(63481-161, 63481-952, 63481-207, 63481-348, 63481-519, 63481-685, 63481-820) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| ARx, LLC | 806798018 | manufacture(63481-161, 63481-952, 63481-207, 63481-348, 63481-519, 63481-685, 63481-820) | |