Label: PROBENECID tablet, film coated

-
Contains inactivated NDC Code(s)
NDC Code(s): 71626-999-08 - Packager: Medstone Pharma LLC
- This is a repackaged label.
- Source NDC Code(s): 0527-1367
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated September 4, 2020
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
Probenecid is a uricosuric and renal tubular transport blocking agent.
The chemical name for probenecid is 4-[(dipropylamino) sulfonyl] benzoic acid. It has the following structural formula:
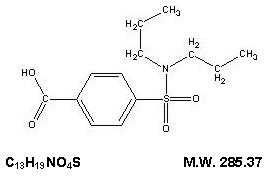
Probenecid, USP is a white or nearly white, fine, crystalline powder. Probenecid is soluble in dilute alkali, in alcohol, in chloroform, and in acetone; it is practically insoluble in water and in dilute acids.
Each tablet for oral administration contains 500 mg of probenecid and the following inactive ingredients: microcrystalline cellulose, sodium lauryl sulfate, sodium starch glycolate, starch (corn), povidone, colloidal silicon dioxide, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, D&C Yellow #10 Aluminum Lake, FD&C Yellow #6 Aluminum Lake, and FD&C Blue #2 Aluminum Lake.
-
CLINICAL PHARMACOLOGY
Probenecid is a uricosuric and renal tubular blocking agent. It inhibits the tubular reabsorption of urate, thus increasing the urinary excretion of uric acid and decreasing serum urate levels. Effective uricosuria reduces the miscible urate pool, retards urate deposition, and promotes resorption of urate deposits.
Probenecid inhibits the tubular secretion of penicillin and usually increases penicillin plasma levels by any route the antibiotic is given. A 2-fold to 4-fold elevation has been demonstrated for various penicillins.
Probenecid also has been reported to inhibit the renal transport of many other compounds including aminohippuric acid (PAH), aminosalicylic acid (PAS), indomethacin, sodium iodomethamate and related iodinated organic acids, 17-ketosteroids, pantothenic acid, phenolsulfonphthalein (PSP), sulfonamides, and sulfonylureas. See also Drug Interactions.
Probenecid decreases both hepatic and renal excretion of sulfobromophtalein (BSP). The tubular reabsorption of phosphorus is inhibited in hypoparathyroid but not in euparathyroid individuals.
Probenecid does not influence plasma concentrations of salicylates, nor the excretion of streptomycin, chloramphenicol, chlortetracycline, oxytetracycline, or neomycin.
-
INDICATIONS AND USAGE
Probenecid tablets are indicated for the treatment of the hyperuricemia associated with gout and gouty arthritis.
As an adjuvant to therapy with penicillin or with ampicillin, methicillin, oxacillin, cloxacillin, or nafcillin, for elevation and prolongation of plasma levels by whatever route the antibiotic is given.
- CONTRAINDICATIONS
-
WARNINGS
Exacerbation of gout following therapy with probenecid may occur; in such cases colchicine or other appropriate therapy is advisable.
Probenecid increases plasma concentrations of methotrexate in both animals and humans. In animal studies, increased methotrexate toxicity has been reported. If probenecid is given with methotrexate, the dosage of methotrexate should be reduced and serum levels may need to be monitored.
In patients on probenecid the use of salicylates in either small or large doses is contraindicated because it antagonizes the uricosuric action of probenecid. The biphasic action of salicylates in the renal tubules accounts for the so-called "paradoxical effect" of uricosuric agents. In patients on probenecid who require a mild analgesic agent the use of acetaminophen rather than small doses of salicylates would be preferred.
Rarely, severe allergic reactions and anaphylaxis have been reported with the use of probenecid. Most of these have been reported to occur within several hours after readministration following prior usage of the drug.
The appearance of hypersensitivity reactions requires cessation of therapy with probenecid.
-
PRECAUTIONS
General
Hematuria, renal colic, costovertebral pain, and formation of uric acid stones associated with the use of probenecid in gouty patients may be prevented by alkalization of the urine and a liberal fluid intake (see DOSAGE AND ADMINISTRATION). In these cases when alkali is administered, the acid-base balance of the patient should be watched.
Use with caution in patients with a history of peptic ulcer.
Probenecid has been used in patients with some renal impairment, but dosage requirements may be increased. Probenecid may not be effective in chronic renal insufficiency particularly when the glomerular filtration rate is 30 mL/minute or less. Because of its mechanism of action, probenecid is not recommended in conjunction with a penicillin in the presence of known renal impairment.
A reducing substance may appear in the urine of patients receiving probenecid. This disappears with discontinuance of therapy. Suspected glycosuria should be confirmed by using a test specific for glucose.
Drug Interactions
When probenecid is used to elevate plasma concentrations of penicillin or other beta-lactams, or when such drugs are given to patients taking probenecid therapeutically, high plasma concentrations of the other drug may increase the incidence of adverse reactions associated with that drug. In the case of penicillin or other beta-lactams, psychic disturbances have been reported.
The use of salicylates antagonizes the uricosuric action of probenecid (see WARNINGS). The uricosuric action of probenecid is also antagonized by pyrazinamide.
Probenecid produces an insignificant increase in free sulfonamide plasma concentrations, but a significant increase in total sulfonamide plasma levels. Since probenecid decreases the renal excretion of conjugated sulfonamides, plasma concentrations of the latter should be determined from time to time when a sulfonamide and probenecid are coadministered for prolonged periods. Probenecid may prolong or enhance the action of oral sulfonylureas and thereby increase the risk of hypoglycemia.
It has been reported that patients receiving probenecid require significantly less thiopental for induction of anesthesia. In addition, ketamine and thiopental anesthesia were significantly prolonged in rats receiving probenecid.
The concomitant administration of probenecid increases the mean plasma elimination half-life of a number of drugs which can lead to increased plasma concentrations. These include agents such as indomethacin, acetaminophen, naproxen, ketoprofen, meclofenamate, lorazepam, and rifampin. Although the clinical significance of this observation has not been established, a lower dosage of the drug may be required to produce a therapeutic effect, and increases in dosage of the drug in question should be made cautiously and in small increments when probenecid is being coadministered. Although specific instances of toxicity due to this potential interaction have not been observed to date, physicians should be alert to this possibility.
Probenecid given concomitantly with sulindac had only a slight effect on plasma sulfide levels, while plasma levels of sulindac and sulfone were increased. Sulindac was shown to produce a modest reduction in the uricosuric action of probenecid, which probably is not significant under most circumstances.
In animals and in humans, probenecid has been reported to increase plasma concentrations of methotrexate (see WARNINGS).
Falsely high readings for theophylline have been reported in an in vitro study, using the Schack and Waxler technique, when therapeutic concentrations of theophylline and probenecid were added to human plasma.
-
ADVERSE REACTIONS
The following adverse reactions have been observed and within each category are listed in order of decreasing severity.
Central Nervous System: headache, dizziness.
Metabolic: precipitation of acute gouty arthritis.
Gastrointestinal: hepatic necrosis, vomiting, nausea, anorexia, sore gums.
Genitourinary: nephrotic syndrome, uric acid stones with or without hematuria, renal colic, costovertebral pain, urinary frequency.
Hypersensitivity: anaphylaxis, fever, urticaria, pruritus.
Hematologic: aplastic anemia, leukopenia, hemolytic anemia which in some patients could be related to genetic deficiency of glucose -6- phosphate dehydrogenase in red blood cells, anemia.
Integumentary: dermatitis, alopecia, flushing.
-
DOSAGE AND ADMINISTRATION
Gout
Therapy with probenecid should not be started until an acute gouty attack has subsided. However, if an acute attack is precipitated during therapy, probenecid may be continued without changing the dosage, and full therapeutic dosage of colchicine, or other appropriate therapy, should be given to control the acute attack.
The recommended adult dosage is 250 mg (½ probenecid tablet), twice a day for one week, followed by 500 mg (1 tablet) twice a day thereafter.
Some degree of renal impairment may be present in patients with gout. A daily dosage of 1000 mg may be adequate. However, if necessary, the daily dosage may be increased by 500 mg increments every 4 weeks within tolerance (and usually not above 2000 mg per day) if symptoms of gouty arthritis are not controlled or the 24 hour uric acid excretion is not above 700 mg. As noted, probenecid may not be effective in chronic renal insufficiency particularly when the glomerular filtration rate is 30 mL/minute or less.
Gastric intolerance may be indicative of overdosage, and may be corrected by decreasing the dosage.
As uric acid tends to crystallize out of an acid urine, a liberal fluid intake is recommended, as well as sufficient sodium bicarbonate (3 to 7.5 g daily), or potassium citrate (7.5 g daily) to maintain an alkaline urine (see PRECAUTIONS).
Alkalization of the urine is recommended until the serum urate level returns to normal limits and tophaceous deposits disappear, i.e., during the period when urinary excretion of uric acid is at a high level. Thereafter, alkalization of the urine and the usual restriction of purine-producing foods may be somewhat relaxed.
Probenecid should be continued at the dosage that will maintain normal serum urate levels. When acute attacks have been absent for 6 months or more and serum urate levels remain within normal limits, the daily dosage may be decreased by 500 mg every 6 months. The maintenance dosage should not be reduced to the point where serum urate levels tend to rise.
Probenecid and Penicillin Therapy (General)
Adults: The recommended dosage is 2000 mg (4 tablets of probenecid) daily in divided doses. This dosage should be reduced in older patients in whom renal impairment may be present.
Children: 2-14 years of age:
Initial dose: 25 mg/kg body weight (or 0.7 g/square meter body surface).
Maintenance Dose: 40 mg/kg body weight (or 1.2 g/square meter body surface) per day, divided into 4 doses.
For children weighing more than 50 kg (110 lb) the adult dosage is recommended.
Probenecid is contraindicated in children under 2 years of age.
The PSP excretion test may be used to determine the effectiveness of probenecid in retarding penicillin excretion and maintaining therapeutic levels. The renal clearance of PSP is reduced to about one-fifth the normal rate when dosage of probenecid is adequate.
Penicillin Therapy (Gonorrhea)1
In uncomplicated gonococcal infections in men and women (urethral, cervical, rectal), 1 g of probenecid should be given orally with 4.8 million units of aqueous procaine penicillin G2 (given IM), or 3 g of amoxicillin2 (given orally), or 3.5 g of ampicillin2 (given orally).
For further guidance, see CDC recommendations for definition of regimens of choice, alternative regimens, treatment of hypersensitive patients, and other aspects of therapy.
-
HOW SUPPLIED
Probenecid Tablets, USP are available containing 500 mg of Probenecid, USP.
The tablets are capsule shaped, film-coated yellow, debossed LCI on one side and 1367 on the other side. They are available as follows:
NDC 0527-1367-01 bottles of 100 tablets
NDC 0527-1367-10 bottles of 1000 tablets
STORE AT 20°-25°C (68°-77°F) [See USP Controlled Room Temperature]
PROTECT FROM LIGHT.
Dispense in a tight, light-resistant container as defined in the USP using a child-resistant closure.
Distributed by:
Lannett Company, Inc.
Philadelphia, PA 19136CIB70317B
Rev 04/16 - PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
PROBENECID
probenecid tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:71626-999(NDC:0527-1367) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PROBENECID (UNII: PO572Z7917) (PROBENECID - UNII:PO572Z7917) PROBENECID 500 mg Inactive Ingredients Ingredient Name Strength CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) SODIUM LAURYL SULFATE (UNII: 368GB5141J) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) STARCH, CORN (UNII: O8232NY3SJ) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) ALUMINUM OXIDE (UNII: LMI26O6933) Product Characteristics Color YELLOW (Yellow) Score no score Shape OVAL Size 18mm Flavor Imprint Code LCI;1367 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:71626-999-08 8 in 1 BOTTLE; Type 0: Not a Combination Product 09/04/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA080966 07/29/1976 Labeler - Medstone Pharma LLC (080783388)
