EGRIFTA- tesamorelin
Theratechnologies Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use EGRIFTA (1 mg/vial) safely and effectively. See full prescribing information for EGRIFTA (1 mg/vial).
EGRIFTA® (tesamorelin for injection), for subcutaneous use Initial U.S. Approval: 2010 INDICATIONS AND USAGEEGRIFTA is a growth hormone-releasing factor (GHRF) analog indicated for the reduction of excess abdominal fat in HIV-infected adult patients with lipodystrophy. (1) Limitations of use: DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost commonly reported adverse reactions (>5%): Arthralgia, injection site erythema, injection site pruritus, pain in extremity, peripheral edema, and myalgia. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact THERA patient supportTM toll free at 1-833-23THERA (1-833-238-4372) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 7/2019 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
EGRIFTA is indicated for the reduction of excess abdominal fat in HIV-infected adult patients with lipodystrophy.
Limitations of Use:
- Long-term cardiovascular safety of EGRIFTA has not been established. Consider risk/benefit of continuation of treatment in patients who have not had a reduction in visceral adipose tissue.
- EGRIFTA is not indicated for weight loss management as it has a weight neutral effect.
- There are no data to support improved compliance with anti-retroviral therapies in HIV-positive patients taking EGRIFTA.
2 DOSAGE AND ADMINISTRATION
2.1 Dosage and Administration
- The dosage and administration recommendations in this prescribing information only apply to EGRIFTA (tesamorelin for injection) 1 mg per vial formulation. For dosage and administration recommendations for tesamorelin for injection 2 mg per vial formulation, see the EGRIFTA SV prescribing information. These two formulations and strengths have differences in the dosage, the number of vials required to prepare a dose, reconstitution instructions, and storage requirements.
- The dose of EGRIFTA is 2 mg (2 mL of the reconstituted solution [see Dosage and Administration (2.2)]), injected subcutaneously once daily.
- Inject EGRIFTA into the abdomen. Rotate injection sites to different areas of the abdomen [see Warnings and Precautions (5.5)]. Do not inject into scar tissue, bruises or the navel.
2.2 Reconstitution Procedure
- Instruct patients to read the Instructions for Use enclosed in the EGRIFTA Medication Box.
- Two vials of 1 mg of EGRIFTA must be reconstituted to prepare the recommended dose. Use only the diluent provided, Sterile Water for Injection, USP, to reconstitute EGRIFTA.
- Reconstitute the first 1 mg vial of EGRIFTA with 2.2 mL of diluent. Mix by rolling the vial gently in your hands for 30 seconds. Do not shake. Reconstitute the second 1 mg vial of lyophilized powder with the entire solution from the first vial. Mix by rolling the vial gently in your hands for 30 seconds. Do not shake.
- Inspect the reconstituted vial visually for particulate matter and discoloration. Use only if the solution is clear, colorless and without particulate matter.
- Administer 2 mL of EGRIFTA immediately following reconstitution and throw away any unused solution and diluent. If not used immediately, discard the reconstituted solution. Do not freeze or refrigerate the reconstituted solution.
3 DOSAGE FORMS AND STRENGTHS
EGRIFTA for injection is supplied in a single-dose 1 mg vial as a white to off-white lyophilized powder and a diluent of 10 mL Sterile Water for Injection, USP.
4 CONTRAINDICATIONS
EGRIFTA is contraindicated in patients with:
- Disruption of the hypothalamic-pituitary axis due to hypophysectomy, hypopituitarism, pituitary tumor/surgery, head irradiation or head trauma.
- Active malignancy. Any preexisting malignancy should be inactive and its treatment complete prior to instituting therapy [see Warnings and Precautions (5.1)].
- Known hypersensitivity to tesamorelin or the excipients in EGRIFTA [see Warnings and Precautions (5.5)].
- Pregnant women because modifying visceral adipose tissue offers no benefit in a pregnant woman and could result in fetal harm [see Use in Specific Populations (8.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Increased Risk of Neoplasms
New Malignancy
Carefully consider the decision to start treatment with EGRIFTA based on the increased background risk of malignancies in HIV-positive patients.
Active Malignancy
EGRIFTA induces the release of endogenous growth hormone (GH), a known growth factor (GF). Do not treat patients with active malignancy with EGRIFTA [see Contraindications (4)].
History of Malignancy
For patients with a history of non-malignant neoplasms, initiate EGRIFTA therapy after careful evaluation of the potential benefit of treatment. For patients with a history of treated and stable malignancies, initiate EGRIFTA therapy only after careful evaluation of the potential benefit of treatment relative to the risk of re-activation of the underlying malignancy. Discontinue EGRIFTA if there is any evidence of recurrent malignancy.
5.2 Elevated IGF-1 Levels
EGRIFTA stimulates GH production and increases serum IGF-1 a growth factor. The effects of prolonged elevations in IGF-1 levels are unknown. Monitor IGF-1 levels during EGRIFTA therapy. Consider discontinuing EGRIFTA in patients with persistent elevations of IGF-1 levels (e.g., >3 SDS), particularly if the efficacy response is not robust.
Among patients who received EGRIFTA for 26 weeks, 47% had IGF-1 levels greater than 2 standard deviation scores (SDS), and 36% had SDS >3, with this effect seen as early as 13 weeks of treatment. Among those patients who remained on EGRIFTA for a total of 52 weeks, at the end of treatment, 34% had IGF-1 SDS >2 and 23% had IGF-1 SDS >3.
5.3 Fluid Retention
Fluid retention may occur during EGRIFTA therapy and is thought to be related to the induction of GH secretion. This manifests as increased tissue turgor and musculoskeletal discomfort resulting in adverse reactions (e.g. edema, arthralgia, and carpal tunnel syndrome) which are either transient or resolve with discontinuation of treatment.
5.4 Glucose Intolerance or Diabetes Mellitus
EGRIFTA treatment can result in glucose intolerance. During clinical trials, the percentages of patients with elevated HbA1c (≥ 6.5%) from baseline to Week 26 were 5% and 1% in the EGRIFTA and placebo groups, respectively. An increased risk of developing diabetes with EGRIFTA (HbA1c level ≥ 6.5%) relative to placebo was observed [intent-to-treat hazard odds ratio of 3.3 (CI 1.4, 9.6)].
Evaluate glucose status prior to initiating EGRIFTA. Monitor all patients treated with EGRIFTA periodically to diagnose those who develop impaired glucose tolerance or diabetes. If patients treated with EGRIFTA develop glucose intolerance or diabetes, consider discontinuing EGRIFTA in patients who do not show a clear efficacy response.
EGRIFTA increases IGF-1, monitor patients with diabetes who are receiving treatment with EGRIFTA at regular intervals for potential development or worsening of retinopathy.
5.5 Hypersensitivity Reactions
Hypersensitivity reactions occurred in 4% of patients treated with EGRIFTA in clinical trials. Reactions included pruritus, erythema, flushing, urticaria, and rash. In cases of suspected hypersensitivity reactions, advise patients to seek prompt medical attention and immediately discontinue treatment with EGRIFTA.
5.6 Injection Site Reactions
EGRIFTA may cause injection site reactions, including injection site erythema, pruritus, pain, irritation, and bruising. The incidence of injection site reactions was 25% in EGRIFTA-treated patients and 14% in placebo-treated patients during the first 26 weeks of treatment in clinical trials. Rotate injection sites to different areas of the abdomen to decrease injection site reactions [see Dosage and Administration (2.1)].
5.7 Increased Mortality in Patients with Acute Critical Illness
Increased mortality in patients with acute critical illness due to complications following open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure has been reported after treatment with pharmacologic amounts of growth hormone. EGRIFTA is a growth hormone-releasing hormone (GHRH) and since EGRIFTA stimulates growth hormone production, consider discontinuing EGRIFTA in critically ill patients.
6 ADVERSE REACTIONS
The following important adverse reactions are also described elsewhere in the labeling:
- Increased risk of neoplasms [see Warnings and Precautions (5.1)]
- Elevated IGF-1 levels [see Warnings and Precautions (5.2)]
- Fluid retention [see Warnings and Precautions (5.3)]
- Glucose intolerance or diabetes mellitus [see Warnings and Precautions (5.4)]
- Hypersensitivity reactions [see Warnings and Precautions (5.5)]
- Injection site reactions [see Warnings and Precautions (5.6)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Seven hundred and forty (740) HIV-infected patients with lipodystrophy and excess abdominal fat were treated with 2 mg dose of EGRIFTA (1 mg/vial formulation) in clinical trials; of these, 543 received EGRIFTA during the initial 26-week placebo-controlled phase.
The most commonly reported adverse reactions were hypersensitivity reactions (e.g., rash, urticaria), edema-related reactions (e.g., arthralgia, extremity pain, peripheral edema, and carpal tunnel syndrome), hyperglycemia, and injection site reactions (injection site erythema, pruritus, pain, urticaria, irritation, swelling, and hemorrhage).
Adverse reactions that occurred more frequently with EGRIFTA relative to placebo and had an incidence ≥1% during the first 26 weeks across all studies are presented in Table 1.
|
*Injection site reaction includes: Injection site erythema, Injection site pruritus, Injection site rash, Injection site urticaria, Injection site pain, Injection site swelling, Injection site irritation, Injection site hemorrhage. |
||
| Preferred Term |
Placebo (N=263) | EGRIFTA (N=543) |
| Injection site reaction* Arthralgia Pain in extremity Myalgia Edema peripheral Paresthesia Hypoesthesia Rash Dyspepsia Musculoskeletal pain Pain Pruritus Vomiting Musculoskeletal stiffness Blood creatine phosphokinase increased Carpal tunnel syndrome Joint swelling Muscle strain Night sweats Palpitations | 6 11 5 2 2 2 2 2 1 1 1 1 0 0 0 0 0 0 0 0 | 17 13 6 6 6 5 4 4 2 2 2 2 3 2 1 1 1 1 1 1 |
In the EGRIFTA clinical trials, mean baseline HbA1c was 5.3% among patients in both the EGRIFTA and placebo groups. Patients receiving EGRIFTA had an increased risk of developing diabetes (HbA1c level ≥ 6.5%) compared with placebo (5% vs. 1%), with a hazard ratio of 3.3 (CI 1.4, 9.6).
6.2 Immunogenicity
As with all therapeutic proteins and peptides, there is a potential for development of anti-EGRIFTA antibodies. The observed incidence of antibody positivity in an assay is highly dependent on several factors, including assay sensitivity and specificity, methodology, sample handling, timing of sample collection, concomitant medication and underlying disease. For these reasons, comparison of the incidence of antibodies to EGRIFTA with the incidence of antibodies to other products may be misleading.
In the clinical trials with EGRIFTA 1 mg/vial formulation, anti-tesamorelin IgG antibodies were detected in 50% of patients treated with EGRIFTA for 26 weeks and 47% of patients who received EGRIFTA for 52 weeks. In the subset of patients with hypersensitivity reactions, anti-tesamorelin IgG antibodies were detected in 85%. Cross-reactivity to endogenous growth hormone-releasing hormone (GHRH) was observed in approximately 60% of patients who developed anti-tesamorelin antibodies. Patients with and without anti-tesamorelin IgG antibodies had similar mean reductions in visceral adipose tissue (VAT) and IGF-1 response. In a group of patients who had antibodies to tesamorelin after 26 weeks of treatment (56%) and were re-assessed 6 months later, after stopping EGRIFTA treatment, 18% were still antibody positive.
Neutralizing antibodies to tesamorelin and human GHRH (hGHRH) were detected in vitro at Week 52 in 10% and 5% of EGRIFTA-treated patients, respectively. Changes in VAT and IGF-1 levels in patients with or without in vitro neutralizing antibodies were comparable.
7 DRUG INTERACTIONS
7.1 Cytochrome P450-Metabolized Drugs
Co-administration of EGRIFTA with simvastatin, a CYP3A substrate, showed that EGRIFTA had no significant impact on the pharmacokinetics profiles of simvastatin in healthy subjects [see Clinical Pharmacology (12.3)].
EGRIFTA stimulates GH production. Published data indicate that GH may modulate cytochrome P450 (CYP450) mediated antipyrine clearance. These data suggest that GH may alter the clearance of compounds known to be metabolized by CYP450 liver enzymes (e.g., corticosteroids, sex steroids, anticonvulsants, and cyclosporine). Monitor patients for potential interactions when administering EGRIFTA in combination with other drugs known to be metabolized by CYP450 liver enzymes.
7.2 Glucocorticoids
GH inhibits 11β-hydroxysteroid dehydrogenase type 1 (11βHSD-1), a microsomal enzyme required for conversion of cortisone to its active metabolite, cortisol, in hepatic and adipose tissue. EGRIFTA stimulates GH production; therefore, patients receiving glucocorticoid replacement for previously diagnosed hypoadrenalism may require an increase in maintenance or stress doses following initiation of EGRIFTA. Patients treated with cortisone acetate and prednisone may be affected more than others because conversion of these drugs to their biologically active metabolites is dependent on the activity of 11βHSD-1.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
EGRIFTA is contraindicated in pregnant women because modifying visceral adipose tissue offers no benefit in pregnant women and could result in fetal harm [see Clinical Considerations and Contraindications (4)]. Administration of tesamorelin acetate to rats during organogenesis resulted in
hydrocephaly in offspring at a dose of approximately two and four times the clinical dose, based on measured drug exposure (AUC). If EGRIFTA is used during pregnancy, or if the patient becomes pregnant while taking it, discontinue EGRIFTA.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
During pregnancy, visceral adipose tissue increases due to normal metabolic and hormonal changes. Modifying pregnancy-associated physiologic changes in visceral adipose tissue with EGRIFTA offers no known benefit and could result in fetal harm.
Data
Animal Data
Tesamorelin acetate administration to rats during organogenesis and lactation resulted in hydrocephaly in offspring at a dose of approximately two and four times the clinical dose, respectively, based on measured drug exposure (AUC). Actual animal dose was 1.2 mg/kg. During organogenesis, lower doses approximately 0.1 to 1 times the clinical dose caused delayed skull ossification in rats. Actual animal doses were 0.1 to 0.6 mg/kg. No adverse developmental effects occurred in rabbits using doses up to approximately 500 times the clinical dose.
8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommend that HIV-infected mothers in the United States not breastfeed their infants to avoid risking postnatal transmission of HIV-1 infection. There are no data on the presence of tesamorelin in human milk, the effects on the breastfed child, or the effects on milk production. Because of both the potential for (1) HIV-1 infection transmission (in HIV-negative infants), (2) developing viral resistance (in HIV-positive patients), and (3) any possible adverse effects of tesamorelin, mothers should not breastfeed if they receive EGRIFTA.
8.4 Pediatric Use
The safety and effectiveness of EGRIFTA in pediatric patients have not been established.
In pediatric patients with open epiphyses, treatment with EGRIFTA may result in linear growth acceleration and excessive growth. EGRIFTA is not indicated for use in pediatric patients with open or closed epiphyses.
11 DESCRIPTION
EGRIFTA contains tesamorelin (as the acetate salt), a human growth hormone-releasing factor (hGHRF) analog. The peptide precursor of tesamorelin acetate is produced synthetically and is comprised of the 44 amino acid sequence of human GHRF. Tesamorelin acetate is made by attaching a hexenoyl moiety, a C6 chain with a double bond at position 3, to the tyrosine residue at the N-terminal part of the molecule. The molecular formula of tesamorelin acetate is C221H366N72O67S • x C2H4O2 (x ≈ 7) and its molecular weight (free base) is 5135.9 Daltons. The structural formula of tesamorelin acetate is:

EGRIFTA is a sterile, white to off-white, preservative-free lyophilized powder for subcutaneous injection. After reconstitution with the supplied diluent (Sterile Water for Injection, USP), the solution is clear and colorless. Each single-dose vial of EGRIFTA contains 1 mg of tesamorelin (equivalent to 1.1 mg of tesamorelin acetate) and the following inactive ingredient: 50 mg mannitol, USP.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
In vitro, tesamorelin binds and stimulates human GHRF receptors with similar potency as the endogenous GRF [see Clinical Pharmacology (12.2)].
Growth hormone-releasing factor (GHRF), also known as growth hormone-releasing hormone (GHRH), is a hypothalamic peptide that acts on the pituitary somatotroph cells to stimulate the synthesis and pulsatile release of endogenous growth hormone (GH), which is both anabolic and lipolytic. GH exerts its effects by interacting with specific receptors on a variety of target cells, including chondrocytes, osteoblasts, myocytes, hepatocytes, and adipocytes, resulting in a host of pharmacodynamic effects. Some, but not all these effects, are primarily mediated by IGF-1 produced in the liver and in peripheral tissues.
12.2 Pharmacodynamics
Tesamorelin stimulates growth hormone secretion, and subsequently increases IGF-1 and IGFBP-3 levels. No clinically significant changes in the levels of other pituitary hormones, including thyroid-stimulating hormone (TSH), luteinizing hormone (LH), adrenocorticotropic hormone (ACTH) and prolactin, were observed in patients receiving EGRIFTA in clinical trials.
12.3 Pharmacokinetics
Absorption
The absolute bioavailability of EGRIFTA after subcutaneous administration of a 2 mg dose of EGRIFTA (1 mg/vial formulation) was determined to be less than 4% in healthy adult subjects.
Single and multiple dose pharmacokinetics of EGRIFTA have been characterized in healthy subjects and HIV-infected patients without lipodystrophy using a 2 mg dose of EGRIFTA (1 mg/vial formulation). Tesamorelin mean extent of absorption (AUC) was 34% higher in HIV-infected patients than healthy subjects. Tesamorelin peak plasma concentration (Cmax) was similar in HIV-infected patients and healthy subjects. The median peak plasma tesamorelin concentration (Tmax) was 0.15 h in both populations.
The mean values [coefficient of variation (CV)] of the extent of absorption (AUC) for tesamorelin were 634.6 (72.4) and 852.8 (91.9) pg.h/mL in healthy subjects and HIV-infected patients, respectively, after a single subcutaneous administration of a 2 mg dose of EGRIFTA (1 mg/vial formulation). The mean (CV) peak tesamorelin concentration (Cmax) values were 2874.6 (43.9) pg/mL in healthy subjects and 2822.3 (48.9) pg/mL in HIV-infected patients. The median peak plasma tesamorelin concentration (Tmax)was 0.15 h in both populations.
Distribution
The mean volume of distribution (±SD) of tesamorelin following a single subcutaneous administration of the 2 mg dose of EGRIFTA (1 mg/vial formulation) was 9.4±3.1 L/kg in healthy subjects and 10.5±6.1 L/kg in HIV-infected patients.
Metabolism
No formal metabolism studies have been performed in humans.
Elimination
Mean elimination half-life (T1/2) of tesamorelin was 26 and 38 minutes in healthy subjects and HIV-infected patients, respectively, after subcutaneous administration of a 2 mg dose of EGRIFTA (1 mg/vial formulation) for 14 consecutive days.
Specific Populations
Pharmacokinetics of tesamorelin in patients with renal or hepatic impairment, in pediatric patients, or in elderly patients has not been established.
Drug Interactions
Simvastatin
The effect of multiple dose administration of EGRIFTA on the pharmacokinetics of simvastatin and simvastatin acid was evaluated in healthy subjects. Co-administration with simvastatin (a CYP3A substrate) resulted in 8% decrease in extent of absorption (AUCinf) and 5% increase in rate of absorption (Cmax) of simvastatin. For simvastatin acid there was a 15% decrease in AUCinf and 1% decrease in Cmax [see Drug Interactions (7.1)].
Ritonavir
The effect of multiple dose administration of EGRIFTA on the pharmacokinetics of ritonavir was evaluated in healthy subjects. Co-administration with ritonavir resulted in 9% decrease in AUCinf and 11% decrease in Cmax of ritonavir [see Drug Interactions (7.1)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Life-time carcinogenicity studies in rodents have not been conducted with tesamorelin acetate. No potential mutagenicity of tesamorelin acetate was revealed in a battery of tests including induction of gene mutations in bacteria (the Ames test), gene mutations in mammalian cells grown in vitro (hamster CHO K1 cells), and chromosomal damage in intact animals (bone marrow cells in mice). There was no effect on fertility in male or female rats following administration of tesamorelin acetate at doses up to 0.6 mg/kg (approximately equal to clinical exposure) for 28 days in males or 14 days in females.
14 CLINICAL STUDIES
Two multicenter, randomized, double-blind, placebo-controlled studies were conducted in HIV-infected patients with lipodystrophy and excess abdominal fat (abdominal lipohypertrophy). Study 1 and Study 2 consisted of a 26-week Main Phase and a 26-week Extension Phase, respectively. Main inclusion criteria were age 18 to 65 years, a waist circumference ≥95 cm (37.4 inches) and a waist-to-hip ratio ≥0.94 for men and ≥94 cm (37.0 inches) and ≥0.88 for women, respectively, and fasting blood glucose (FBG) <150 mg/dL (8.33 mmol/L). Main exclusion criteria included BMI ≤ 20 kg/m2, type 1 diabetes mellitus, type 2 diabetes mellitus, previous treatment with insulin or with oral hypoglycemic or insulin-sensitizing agents, history of malignancy, and hypopituitarism. Patients were on a stable anti-retroviral regimen for at least 8 weeks prior to randomization. Patients meeting the inclusion/exclusion criteria were randomized in a 2:1 ratio to receive a 2 mg dose of EGRIFTA (1 mg/vial formulation) or placebo subcutaneously daily for 26 weeks. The primary efficacy assessment for each of these studies was the percent change from baseline to Week 26 in visceral adipose tissue (VAT), as assessed by computed tomography (CT) scan at L4-L5 vertebral level. Secondary endpoints included changes from baseline in patient-reported outcomes related to body image, triglycerides, ratio of total cholesterol to HDL cholesterol, IGF-1 levels, and safety parameters. Other endpoints included changes from baseline in waist circumference, abdominal subcutaneous tissue (SAT), trunk fat, and lean body mass. In both studies, EGRIFTA-treated patients completing the 26-week treatment period were re-randomized to blinded therapy with either daily placebo or a 2 mg dose of EGRIFTA (1 mg/vial formulation) for an additional 26-week treatment period (Extension Phase) in order to assess maintenance of VAT reduction and to gather long-term safety data. For inclusion in the Extension Phase studies, subjects must have completed the Main Phase with FBG ≤ 150 mg/dL.
Main Phase (Baseline to Week 26):
Study 1 (NCT 00123253): This study randomized 412 HIV-infected patients with lipodystrophy and excess abdominal fat to receive either a 2 mg dose of EGRIFTA (1 mg/vial formulation) (N=273) or placebo (N=137). At baseline for the two groups combined, mean age was 48 years; 86% were male; 75% were white, 14% were Black/African American, and 8% were Hispanic; mean weight was 90 kg; mean BMI was 29 kg/m2; mean waist circumference was 104 cm; mean hip circumference was 100 cm; mean VAT was 176 cm2; mean CD4 cell count was 606 cells/mm3; 69% had undetectable viral load (<50 copies/mL); and 33.7% randomized to EGRIFTA and 36.6% randomized to placebo had impaired glucose tolerance, while 5.6% randomized to EGRIFTA and 6.7% randomized to placebo had diet-controlled diabetes mellitus. The twenty-six week completion rate in Study 1 was 80%.
Study 2 (NCT 00435136): This study randomized 404 HIV-infected patients with lipodystrophy and excess abdominal fat to receive either a 2 mg dose of EGRIFTA (1 mg/vial formulation) (N=270) or placebo (N=126). At baseline for the two groups combined, mean age was 48 years; 84% were male; 77% were white, 12% were Black/African American, and 9% were Hispanic; mean weight was 88 kg; mean BMI was 29 kg/m2; mean waist circumference was 105 cm; mean hip circumference was 100 cm; mean VAT was 189 cm2; mean CD4 cell count was 592 cells/mm3; 83% had undetectable viral load (<50 copies/mL); and 44% randomized to EGRIFTA and 40% randomized to placebo had impaired glucose tolerance, while 9.3% randomized to EGRIFTA and 10% randomized to placebo had diet-controlled type 2 diabetes mellitus. The twenty-six week completion rate in Study 2 was 74%.
Results for the Main Phases of Studies 1 and 2 are presented in Tables 2 and 3.
|
Baseline data are expressed as mean (SD); Change refers to least-squares mean (LSM); CI: confidence interval. |
|||||
|
1 Results derived from the statistical model: Ln(VAT Week 26/VAT Baseline) = Ln(VAT Baseline) + treatment group |
|||||
| MAIN PHASE (Baseline-Week 26) | |||||
| Study 1 | Study 2 | ||||
|
2 mg EGRIFTA (1 mg/vial) | Placebo (N=137) |
2 mg EGRIFTA (1 mg/vial) | Placebo (N=126) |
||
| Baseline (cm2) | 178 (77) | 171 (77) | 186 (87) | 195 (95) | |
| Change (cm2) | -27 | 4 | -21 | -0 | |
| Mean treatment difference (95% CI) | -31 (-39,-24) | -21 (-29,-12) | |||
| Mean change (%)1 | -18 | 2 | -14 | -2 | |
| Mean treatment difference (95% CI)1 | -20 (-24, -15) | -12 (-16, -7) | |||
|
Baseline data are expressed as mean (SD); Change refers to least-squares mean (LSM); CI: confidence interval. |
|||||
| MAIN PHASE (Baseline-Week 26) | |||||
| Study 1 | Study 2 | ||||
| 2 mg EGRIFTA (1 mg/vial) | Placebo (N=137) | 2 mg EGRIFTA (1 mg/vial) | Placebo (N=126) |
||
| IGF-1 (ng/mL) | Baseline | 161 (59) | 168 (75) | 146 (66) | 149 (59) |
| Change | 107 | -15 | 108 | 3 | |
| Mean treatment difference (95% CI) | 122 (101, 141) | 105 (85, 126) | |||
| IGFBP-3 (mg/L) | Baseline | 3 (1) | 3 (1) | 3 (1) | 3 (1) |
| Change | 0.4 | -0.2 | 0.8 | -0.0 | |
| Mean treatment difference (95% CI) | 0.6 (0.5, 0.8) | 0.8 (0.5, 1.0) | |||
| Weight (kg) | Baseline | 90 (14) | 90 (14) | 89 (14) | 87 (16) |
| Change | -0.4 | 0.0 | 0.5 | 0.3 | |
| Mean treatment difference (95% CI) | -0.4 (-1.3, 0.5) | 0.2 (-0.7, 1.3) | |||
| Waist circumference (cm) | Baseline | 104 (10) | 105 (9) | 105 (9) | 105 (9) |
| Change | -3 (5) | -1 (4) | -2 (5) | -1 (5) | |
| Mean treatment difference (95% CI) | -2 (-2.8, -0.9) | -1 (-2.5, -0.3) | |||
At Week 26, treatment with a 2 mg dose of EGRIFTA (1 mg/vial formulation) resulted in a reduction from baseline in mean trunk fat of 1.0 kg in Study 1 and 0.8 kg in Study 2, respectively (compared with an increase of 0.4 kg in Study 1 and of 0.2 kg in Study 2, respectively, in patients receiving placebo). Treatment with EGRIFTA resulted in an increase from baseline in mean lean body mass of 1.3 kg in Study 1 and of 1.2 kg in Study 2, respectively (compared with a decrease of 0.2 kg in Study 1 and of 0.03 kg in Study 2, respectively, in patients receiving placebo).
Patient Reported Outcomes
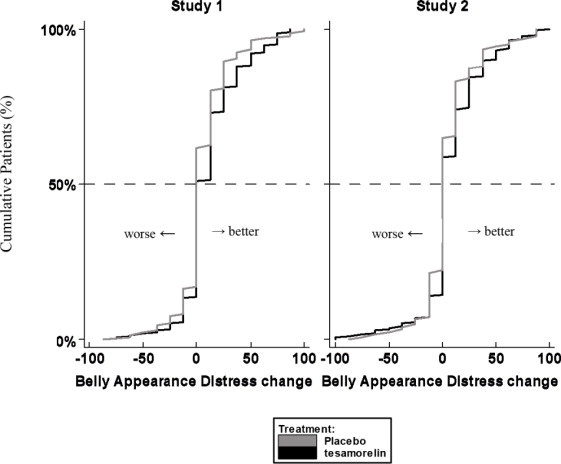
Patients rated the degree of distress associated with their belly appearance on a 9-point rating scale that was then transformed to a score from 0 (extremely upsetting and distressing) to 100 (extremely encouraging). A score of 50 indicated neutral (no feeling either way). A positive change from baseline score indicated improvement, i.e., less distress.
The cumulative distribution of response (change from baseline to 26 weeks) is shown in Figure 1 for both treatment groups. A curve shifted to the right on this scale indicates a greater percentage of patients reporting improvement.
Figure 1. Cumulative Distribution of Response for Belly Appearance Distress
Extension Phase (Weeks 26-52):
In the double-blind Extension Phase, patients on a 2 mg dose of EGRIFTA (1 mg/vial formulation) completing the 26-week Main Phase were re-randomized to receive a 2 mg dose of EGRIFTA or placebo.
Study 1 (NCT 00123253)
This study re-randomized 207 HIV-infected patients with lipodystrophy who completed a 2 mg dose of EGRIFTA (1 mg/vial formulation) in the Main Phase to receive either EGRIFTA(N=154) or placebo (N=50) for an additional 26-week duration (3:1 randomization ratio). At baseline (Week 26) for the two groups combined, mean age was 48 years; 88% were male; 78% were white, 12% were Black/African American, and 8% were Hispanic; mean weight was 90 kg; mean BMI was 29 kg/m2; mean waist circumference was 102 cm; mean hip circumference was 100 cm; mean VAT was 145 cm2; mean CD4 cell count was 639 cells/mm3;68% had undetectable viral load (<50 copies/mL); and for those EGRIFTA-treated patients completing the 26-week treatment period that were re-randomized to EGRIFTA (T-T group) or re-randomized to placebo, 37% and 32%, respectively, had impaired glucose tolerance, while 2% re-randomized to EGRIFTA and 6% re-randomized to placebo had diet-controlled type 2 diabetes mellitus. The completion rate for patients randomized into the extension phase of Study 1 was 83%.
Study 2 (NCT 00435136)
This study re-randomized 177 HIV-infected patients with lipodystrophy who completed EGRIFTA treatment in the Main Phase to receive either a 2 mg dose of EGRIFTA (1 mg/vial formulation) (N=92) or placebo (N=85) for an additional 26-week duration (1:1 randomization ratio). At baseline (Week 26) for the two groups combined, mean age was 48 years; 90% were male; 84% were white, 9% were Black/African American, and 7% were Hispanic; mean weight was 89 kg; mean BMI was 28 kg/m2; mean waist circumference was 105 cm; mean hip circumference was 100 cm; mean VAT was 172 cm2; mean CD4 cell count was 579 cells/mm3; 82% had undetectable viral load (<50 copies/mL); and for those EGRIFTA-treated patients completing the 26-week treatment period that were re-randomized to EGRIFTA (T-T group) or re-randomized to placebo, 49% and 51%, respectively, had impaired glucose tolerance, while 4% re-randomized to EGRIFTA and 13% re-randomized to placebo had diet-controlled diabetes mellitus. The completion rate for patients randomized into the extension phase of Study 2 was 81%.
Results for the Extension Phases of Studies 1 and 2 are presented in Tables 4 and 5.
|
Week 26 baseline data are expressed as mean (SD). Change refers to least-squares mean (LSM); CI: confidence interval. |
||||
|
1T-T = tesamorelin for Weeks 0-26 and tesamorelin for Weeks 26-52 |
||||
|
2T-P = tesamorelin for Weeks 0-26 and placebo for Weeks 26-52 |
||||
|
3Results derived from the statistical model: Ln(VAT Week 52/Week 26) = Ln(Week 26 VAT) + treatment group |
||||
| EXTENSION PHASE (Week 26-52) | ||||
| Study 1 | Study 2 | |||
| T-T1
(Week 26-52) (N=154) | T-P2
(Week 26-52) (N=50) | T-T1
(Week 26-52) (N=92) | T-P2
(Week 26-52) (N=85) |
|
| Week 26 (cm2) | 145 (72) | 144 (72) | 166 (89) | 177 (88) |
| Change (cm2) | 3 | 25 | -11 | 24 |
| Mean treatment difference (95% CI) | -22 (-34, -10) | -35 (-48, -22) | ||
| Mean change (%)3 | 0 | 22 | -5 | 16 |
| Mean treatment difference (95% CI)3 | -17 (-24, -10) | -18 (-24, -11) | ||
Figure 2 shows the percent change in VAT from baseline (Week 0) over time until 52 weeks in completer patients.
Figure 2. Percent Change from Baseline in VAT over Time
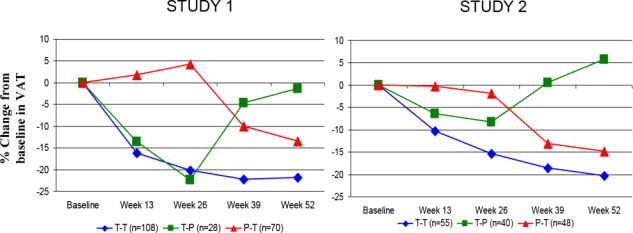
Data in Figure 2 are expressed as mean values. T-T (tesamorelin to tesamorelin) refers to the group of patients who received tesamorelin for Weeks 0-26 and were re-randomized to tesamorelin for Weeks 26-52. T-P (tesamorelin to placebo) refers to the group of patients who received tesamorelin for Weeks 0-26 and were re-randomized to placebo for Weeks 26-52. P-T (placebo to tesamorelin) refers to the group of patients who received placebo for Weeks 0-26 and were switched to tesamorelin (treated open label) for Weeks 26-52.
|
Week 26 baseline data are expressed as mean (SD); Change refers to least-squares mean (LSM); CI: confidence interval. |
|||||
|
1T-T = tesamorelin for Week 0-26 and tesamorelin for Week 26-52 |
|||||
|
2T-P = tesamorelin for Week 0-26 and placebo for Week 26-52 |
|||||
| EXTENSION PHASE (Weeks 26-52) | |||||
| Study 1 | Study 2 | ||||
| T-T1
(Week 26-52) (N=154) | T-P2
(Week 26-52) (N=50) | T-T1
(Week 26-52) (N=92) | T-P2
(Week 26-52) (N=85) |
||
| IGF-1 (ng/mL) | Week 26 | 291 (124) | 281 (105) | 280 (134) | 269 (110) |
| Change | -59 | -137 | -25 | -135 | |
| Mean treatment difference (95% CI) | 78 (50, 106) | 110 (87, 134) | |||
| IGFBP-3 (mg/L) | Week 26 | 3 (1) | 3 (1) | 3 (1) | 3 (1) |
| Change | -0.2 | -0.5 | -0.3 | -0.9 | |
| Mean treatment difference (95% CI) | 0.3 (-0.0, 0.6) | 0.6 (0.3, 0.9) | |||
| Weight (kg) | Week 26 | 89 (14) | 92 (17) | 89 (13) | 90 (14) |
| Change | 0.2 | 0.6 | -0.5 | 0.1 | |
| Mean treatment difference (95% CI) | -0.4 (-2, 1) | -0.6 (-2, 1) | |||
| Waist circumference (cm) | Week 26 | 101 (10) | 102 (12) | 101 (9) | 103 (11) |
| Change | -0.2 | 2.4 | -1.1 | 0.2 | |
| Mean treatment difference (95% CI) | -2.6 (-4, -1) | -1.3 (-2, 0) | |||
Patients treated with a 2 mg dose of EGRIFTA (1 mg/vial formulation) for 52 weeks (T-T group) showed no change between Weeks 26 and 52 in mean trunk fat (increase of 0.1 kg in Study 1 and decrease of 0.5 kg in Study 2, respectively, compared with an increase of 1.4 kg in patients in the T-P group in Study 1 and an increase of 1.09 kg in Study 2, respectively) nor was there a change from Week 26 baseline in mean lean body mass (decrease of 0.1 kg in Study 1 and increase of 0.1 kg in Study 2, respectively, compared with a decrease of 1.8 kg in patients in the T-P group in Study 1 and a decrease of 1.7 kg in Study 2, respectively).
16 HOW SUPPLIED/STORAGE AND HANDLING
EGRIFTA for injection is supplied as a white to off-white lyophilized powder in a 1 mg single-dose vial with a diluent of 10 mL vial of Sterile Water for Injection, USP.
EGRIFTA (NDC 62064-011-60) is available in a package comprised of two boxes containing 60 (sixty) 1 mg single-dose vials of EGRIFTA in the Medication Box and a 30 single-dose 10 mL bottles of Sterile Water for Injection, USP diluent with a 30 day supply of disposable syringes, and needles in the Injection Box.
Protect EGRIFTA from light and keep in the original box until time of use. Store EGRIFTA 1 mg/vial at refrigerated temperature, between 2°C to 8°C (36°F to 46°F). Store the Sterile Water for Injection, USP, syringes and needles at controlled room temperature of 20°C to 25°C (68°F to 77°F).
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Increased Risk of Malignancy
Inform patients about the increased background risk of malignancies in HIV-positive patients and for patients with a history of neoplasms, inform them about the risk of malignancy reoccurrence [see Warnings and Precautions (5.1)].
Elevated IGF-1 Levels
Inform patients that treatment with EGRIFTA increases IGF-1 levels and that they will need periodic monitoring of their IGF-1 levels [see Warnings and Precautions (5.2)].
Fluid Retention
Inform patients that treatment with EGRIFTA may cause fluid retention, resulting in adverse reactions including edema, arthralgia, and carpal tunnel syndrome [see Warnings and Precautions (5.3)].
Glucose Intolerance or Diabetes Mellitus
Inform patients that treatment with EGRIFTA may result in glucose intolerance or diabetes mellitus. Advise patients that they will need to be monitored to see if impaired glucose tolerance or diabetes mellitus develops, and that if they have pre-existing diabetes mellitus, they may need adjustments to their anti-diabetic medications [see Warnings and Precautions (5.4)].
Hypersensitivity Reactions
Inform patients that hypersensitivity reactions (e.g., rash, urticaria) may occur during treatment with EGRIFTA. Advise patients to seek prompt medical attention and to immediately discontinue treatment with EGRIFTA if a reaction occurs [see Warnings and Precautions (5.5)].
Injection Site Reactions
Inform patients that injection site reactions may occur with EGRIFTA, including injection site erythema, pruritus, pain, irritation, and bruising. Advise patients to rotate the site of injection to reduce the risk of injection site reactions [see Warnings and Precautions (5.6)].
Pregnancy
Advise women to discontinue EGRIFTA if pregnancy occurs, as the drug offers no known benefit to pregnant women and could result in fetal harm [see Contraindications (4) and Use in Specific Populations (8.1)].
Lactation
Because of both the potential for HIV-1 infection transmission and serious adverse reactions in nursing infants, mothers receiving EGRIFTA should be instructed not to breastfeed [see Use in Specific Populations (8.2)].
Administration
Counsel patients that they should never share an EGRIFTA syringe with another person, even if the needle is changed. Sharing of syringes or needles between patients may pose a risk of transmission of infection.
|
PATIENT INFORMATION EGRIFTA® (eh-GRIF-tuh) (tesamorelin for injection) for subcutaneous use 1 mg vial |
|||||
|
Read the Patient Information that comes with EGRIFTA before you start to take EGRIFTA and each time you get a refill. There may be new information. This leaflet does not take the place of talking to your healthcare provider about your medical condition or your treatment. |
|||||
|
What is EGRIFTA?
The long-term safety of EGRIFTA on the heart and blood vessels (cardiovascular) is not known. EGRIFTA is not for weight loss management. It is not known whether taking EGRIFTA helps improve how well you take (compliance with) antiretroviral medicines. It is not known if EGRIFTA is safe and effective in children. EGRIFTA is not recommended to be used in children with open or closed bone growth plates (epiphyses). |
|||||
|
Who should not use EGRIFTA? Do not use EGRIFTA if you:
|
|||||
|
What should I tell my healthcare provider before using EGRIFTA? Before using EGRIFTA, tell your healthcare provider about all of your medical conditions, including if you:
|
|||||
|
How should I use EGRIFTA?
|
|||||
|
What are the possible side effects of EGRIFTA? EGRIFTA may cause serious side effects, including:
|
|||||
|
○ a rash over your body ○ shortness of breath or trouble breathing |
○ hives ○ fast heartbeat ○ itching |
○ swelling of your face or throat ○ feeling of faintness or fainting ○ reddening or flushing of the skin |
|||
|
|||||
|
○ redness ○ irritation ○ swelling |
○ itching ○ bruising or bleeding |
○ pain ○ rash |
|||
|
|||||
|
The most common side effects of EGRIFTA include: |
|||||
|
• pain in legs and arms |
|
||||
|
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800- FDA-1088. You may also report side effects to THERA patient supportTM toll-free at 1-833-23THERA (1-833-238-4372). |
|||||
|
How should I store EGRIFTA 1 mg vials, Sterile Water for Injection, syringes and needles?
Keep EGRIFTA and all medicines out of the reach of children. |
|||||
|
General information about the safe and effective use of EGRIFTA. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use EGRIFTA for a condition for which it was not prescribed. Do not give EGRIFTA to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about EGRIFTA that is written for health professionals. |
|||||
|
What are the ingredients in EGRIFTA? Active ingredient: tesamorelin acetate Inactive ingredients: mannitol Distributed by: Theratechnologies Inc., Montréal, Québec, Canada H3A 1T8 For more information about EGRIFTA, go to www.EGRIFTA.com or call toll-free at 1-833-23THERA (1-833-238-4372). |
|||||
This Patient Information has been approved by the U.S. Food and Drug Administration Revised: 07/2019
Instructions for Use
EGRIFTA® (eh-GRIF-tuh)
(tesamorelin for injection)
for subcutaneous use
1 mg vial
This Instructions for Use contains step-by-step information on how to use the 1 mg vial of EGRIFTA. Two vials of EGRIFTA 1 mg must be mixed with a total of 2.2 mL of the Sterile Water for Injection provided in the Injection Box given to you by the pharmacy. You need two EGRIFTA 1 mg vials from the Medication Box to prepare your dose. The recommended dose is 2 mg (2 mL).
Be sure that you read, understand, and follow this Instructions for Use before using EGRIFTA. Your healthcare provider should show you how to mix and inject EGRIFTA before you inject it for the first time. Ask your healthcare provider if you have any questions.
Keep this Instructions for Use in case you need to look at it again later.
Important information for use of EGRIFTA
- Do not use a syringe or needle more than 1 time.
- Do not share your syringe or needles with other people, even if the needle has been changed. You may give other people a serious infection, or get a serious infection from them.
-
If you are missing any supplies from the boxes given to you by the pharmacy or if any of the supplies look damaged, call your pharmacist or contact THERA patient supportTM toll-free at 1-833-23THERA (1-833-238-4372) right away.
Preparing for your EGRIFTA injection
Step 1: Find a well-lit, clean, and flat surface, such as a table.
Step 2: Gather your supplies:
- Medication Box that contains 60 EGRIFTA 1 mg single-dose vials
- Injection Box that contains the following:
- 30 single-dose 10 mL bottles of Sterile Water for Injection, used for mixing
- 30 sterile 3 mL syringes with sterile needle attached
- 30 1½” 18-gauge sterile needles, used for mixing
- 30 ½” 27-gauge sterile needles, used for injection
- Other Supplies Needed
- Two alcohol pads
- One sterile gauze
- One adhesive bandage
- Sharps disposal container or a puncture resistant container for throwing away used needles and syringes after you are done with them. (See “How should I dispose of the used syringes, needles, bottles and vials?”)
For each injection using EGRIFTA 1 mg vial you will need: (See Figure A)
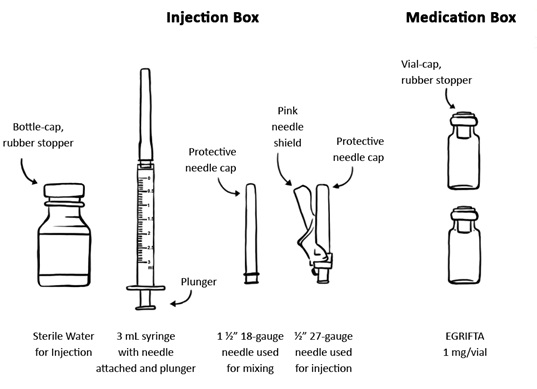
Figure A
Step 3: Take out the following from the Injection Box:
- One Sterile Water for Injection bottle
- One 3 mL syringe with needle attached
- One 1½" 18-gauge needle used for mixing
- One ½" 27-gauge needle used for injection
Step 4: Take 2 vials of EGRIFTA 1 mg from the Medication Box. Put the box with the remaining vials back in the refrigerator right away.
Step 5: Prepare to use your supplies:
- Wash your hands with soap and water. Dry your hands with a clean towel.
- Take off the plastic caps from both of the EGRIFTA 1 mg vials and the Sterile Water for Injection bottle.
- Clean the rubber stoppers on the top of both of the EGRIFTA 1 mg vials and Sterile Water for Injection bottle with an alcohol pad.
How to mix EGRIFTA 1 mg vial
Step 1: Pick up the 3 mL syringe with the needle attached. Carefully remove the protective needle cap by pulling it straight off. Do not twist the needle cap.
Insert the needle through the rubber stopper of the Sterile Water for Injection bottle. (See Figure B)
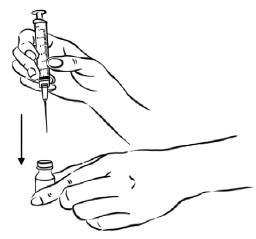
Figure B
Step 2: With the needle still in the Sterile Water for Injection bottle, turn the bottle and syringe upside down.
Pull back the plunger until the Sterile Water reaches the 2.2 mL mark on the syringe. (See Figure C)
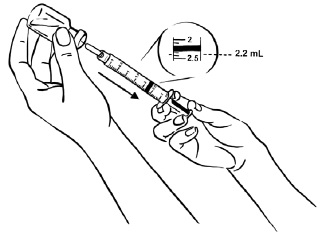
Figure C
- Throw away the bottle containing the unused Sterile Water for Injection. (See “How should I dispose of the used syringes, needles, bottles and vials?”)
Step 3: Remove the syringe with needle attached from the Sterile Water bottle.
Insert the needle into the rubber stopper of one of the EGRIFTA 1 mg vials.
With the needle at a slight angle, slowly push the plunger of the syringe all the way down so that the Sterile Water goes down the inside wall of the EGRIFTA 1 mg vial instead of directly onto the powder to avoid foaming. (See Figure D)
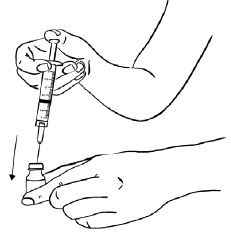
Figure D
Step 4: While keeping the syringe with needle attached in the EGRIFTA 1 mg vial and the vial upright, roll the vial gently in your hands for 30 seconds, until the Sterile Water and EGRIFTA powder are mixed well. Do not shake the vial. (See Figure E)
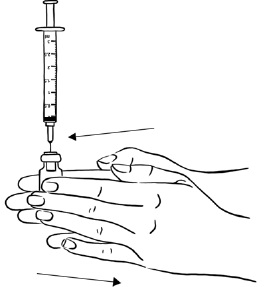
Figure E
Step 5: With the needle still in the EGRIFTA 1 mg vial, turn the vial and syringe upside down. Pull the syringe back until you see just the tip of the needle going through the rubber stopper.
Pull back on the plunger of the syringe until all of the liquid inside the vial is in the syringe. The medicine should be at around the 2.2 mL mark on the syringe. (See Figure F)
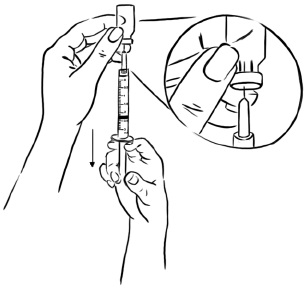
Figure F
Step 6: Remove the needle from the vial. (See Figure G)
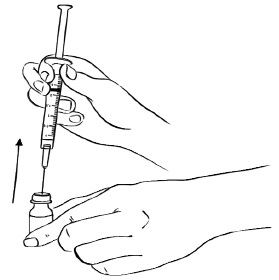
Figure G
Step 7: Place the protective needle cap on its side on a clean flat surface. Without touching the needle, hold the syringe and slide the needle carefully into the needle cap. (See Figure H)
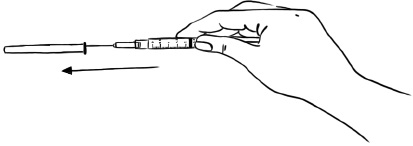
Figure H
Step 8: Push the protective needle cap all the way in or until it snaps shut (See Figure I). Do not touch the needle cap until it covers the needle completely.
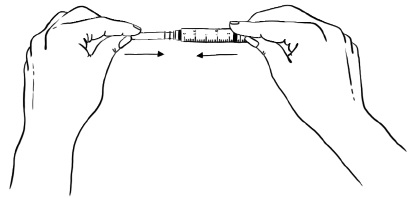
Figure I
Step 9: With the protective needle cap securely on the needle, remove the needle by holding the syringe firmly and twisting the needle cap counterclockwise (to the left). (See Figure J)
Throw away the needle. (See "How should I dispose of the used syringes, needles, bottles and vials?")
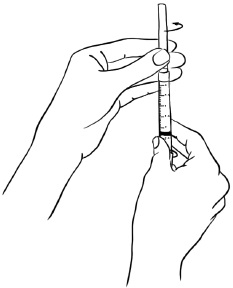
Figure J
Step 10: Place the 1½" 18-gauge needle to be used for mixing, with its protective needle cap in place, onto the syringe. Hold the syringe firmly and twist the needle cap clockwise (to the right) until it closes securely. (See Figure K)
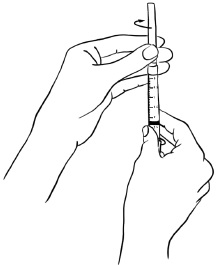
Figure K
Step 11: Carefully remove the protective needle cap by pulling it straight off. Do not twist the needle cap.
Insert the needle into the rubber stopper of the second EGRIFTA 1 mg vial. With the needle at a slight angle, slowly push the plunger of the syringe all the way down so that the liquid goes down the inside wall of the EGRIFTA vial instead of directly onto the powder to avoid foaming. (see Figure L).
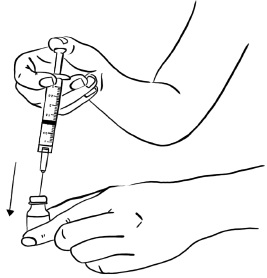
Figure L
Step 12: While keeping the syringe with the needle attached in the EGRIFTA 1 mg vial and the vial upright, roll the vial gently in your hands for 30 seconds, until the Sterile Water and EGRIFTA powder are mixed well. Do not shake the vial. (See Figure M)
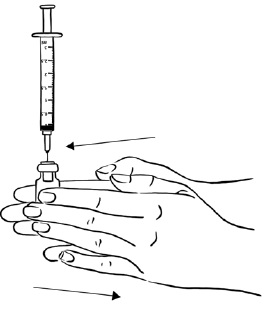
Figure M
Note:
- After mixing, the solution should look clear and colorless, with no particles in it.
- Do not use EGRIFTA if it looks cloudy, discolored, or if you see particles in it.
- Call your healthcare provider, pharmacist, or contact THERA patient supportTM toll-free at 1-833-23THERA (1-833-238-4372) right away if the solution is cloudy, discolored, or contains particles.
Step 13: With the needle still in the EGRIFTA 1 mg vial, turn the vial and syringe upside down. Pull the syringe back until you see just the tip of the needle going through the rubber stopper.
Pull back on the plunger of the syringe until all of the liquid inside the vial is in the syringe. The medicine should be at around the 2.2 mL mark on the syringe. (See Figure N)
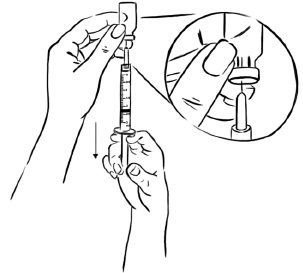
Figure N
Step 14: Remove the needle from the vial. (See Figure O)
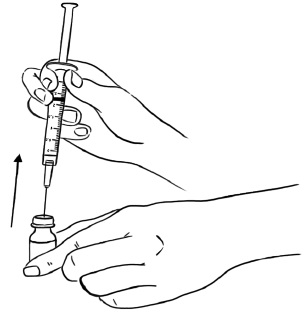
Figure O
Step 15: Place the protective needle cap on its side on a clean flat surface. Without touching the needle, hold the syringe and slide the needle carefully into the needle cap. (See Figure P)
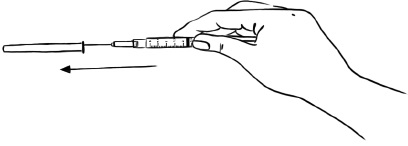
Figure P
Step 16: Push the protective needle cap all the way in or until it snaps shut (See Figure Q). Do not touch the needle cap until it covers the needle completely.
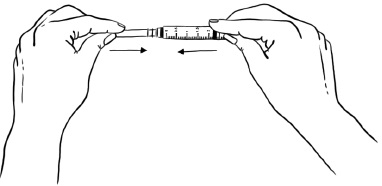
Figure Q
Step 17: With the protective needle cap securely on the needle, remove the needle by holding the syringe firmly and twisting the needle cap counterclockwise (to the left). (See Figure R)
Throw away the needle. (See "How should I dispose of the used syringes, needles, bottles and vials?")
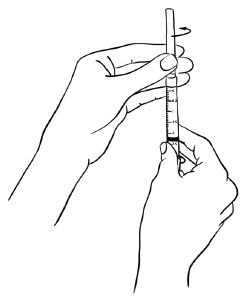
Figure R
Step 18: Place the ½" 27-gauge needle to be used for your injection, with its protective needle cap and pink needle shield in place, onto the syringe. Hold the syringe firmly and twist the needle cap and pink needle shield clockwise (to the right) until it closes securely. (See Figure S)
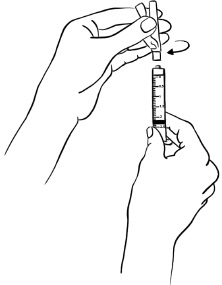
Figure S
Where do I inject EGRIFTA 1 mg vial?
Inject EGRIFTA into the stomach-area (abdomen). (See Figure T)
- Choose an injection site that is at least 2 inches (5 cm) away from your belly button. (See the shaded area in Figure T)
- Do not inject into areas with scar tissue or bruises.
- Do not inject into your belly button.
- Avoid areas with any hard bumps from previous injections.
- Rotate the site for each injection. This may help prevent bruising or irritation. You may want to write down the date and location of each daily injection to help you remember.
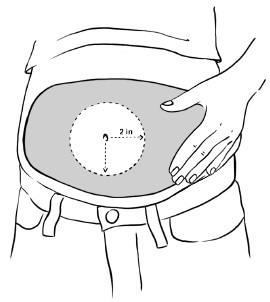
Figure T
How to inject your 2 mg dose of EGRIFTA
Do not remove the needle cap until you are ready to inject.
Step 1: Hold the syringe with the needle facing up. Pull the pink needle shield down away from the protective needle cap.
Carefully remove the needle cap by pulling it straight off the needle. Do not twist the needle cap. (See Figure U)
Do not touch the needle.
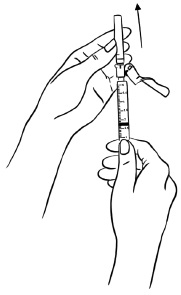
Figure U
Step 2: Tap the syringe gently with your finger to force any air bubbles to rise to the top. Slowly push up on the plunger to remove any air and bubbles and until the medicine in the syringe is at the 2 mL mark of the syringe. (See Figure V)
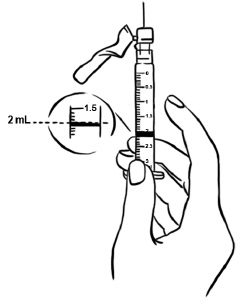
Figure V
Step 3: Clean the injection site you have selected with an alcohol pad and let it dry. Hold the syringe in one hand. With your other hand, gently pinch a fold of skin at your cleaned injection site. Hold the skin between your thumb and fingers. (See Figure W)
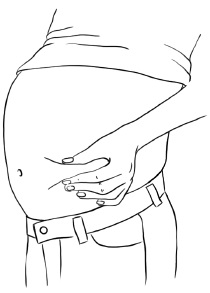
Figure W
Step 4: Hold the syringe at a 90 degree angle to the skin.
With a quick, dart-like motion, insert the needle into the skin. Most of the needle should go beneath the skin. (See Figure X)
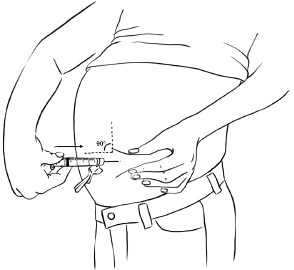
Figure X
Step 5: Remove your hand from the pinched area of skin after the needle is inserted. Be careful not to remove the needle from the skin.
Step 6: Slowly push the plunger of the syringe all the way down until all of the medicine in the syringe has been injected. (See Figure Y)
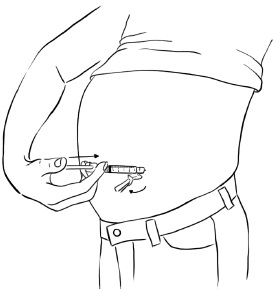
Figure Y
Pull the needle out of your skin. Be careful to pull the needle out at the same angle as it was inserted.
Step 7: While holding the syringe in one hand, gently press the pink needle shield against a hard, flat surface until you hear a “click” and the injection needle is covered by the pink needle shield. (See Figure Z)
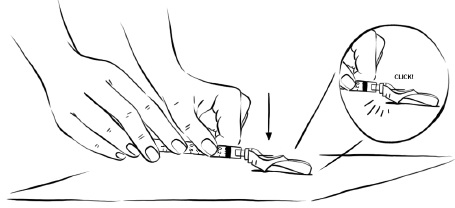
Figure Z
- Apply pressure to the injection site with gauze for 30 seconds. If there is bleeding, apply an adhesive bandage to the site.
- Throw away the syringe. (See "How should I dispose of the used syringes, needles, bottles and vials?")
How should I dispose of the used syringes, needles, bottles and vials?
- Do not recap the needle or remove the needle from the syringe after you inject EGRIFTA.
- Put your used EGRIFTA needles and syringes in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
- If another person receives a needle stick with a used needle, that person should be told to contact a healthcare provider right away.
- Keep the sharps container away from children and pets.
|
If you have any questions, call your healthcare provider. You can call THERA patient supportTM toll-free at 1-833-23THERA (1-833-238-4372) or visit the EGRIFTA website at: www.EGRIFTA.com for more information. |
How should I store EGRIFTA 1 mg vials, Sterile Water for Injection, syringes and needles?
- You will be given 2 boxes from the pharmacy when you get your prescription of EGRIFTA:
- Store the 1 mg EGRIFTA vials in the Medication Box they come in, in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Store the Sterile Water for Injection, syringes and needles that come in the Injection Box at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep EGRIFTA vials out of the light.
- After mixing, use EGRIFTA right away. Throw away any unused EGRIFTA after mixing.
- Do not store, freeze or refrigerate EGRIFTA after it has been mixed with the Sterile Water.
- Throw away any Sterile Water for Injection left in the bottle after use.
- Do not use EGRIFTA after the expiration date (EXP) printed on the carton and vial labels.
Keep EGRIFTA and all medicines out of the reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
THERA
technologies
EGRIFTA® is a registered trademark of Theratechnologies Inc.
Distributed by: Theratechnologies Inc., Montréal, Québec, Canada H3A 1T8
Revised: 07/2019
Principal Display Panel - Medication Box
MEDICATION BOX
(Box 1 of 2)
NDC 62064-011-60
1 mg/vial
Rx only
EGRIFTA
tesamorelin for injection
For subcutaneous injection only
EGRIFTA 1 mg/vial Formulation, Dose,
Mixing Instructions, and Storage conditions are different
than the EGRIFTA SV™ 2 mg/vial Formulation.
Important: Refer to Patient Instructions for Use.
Use 2 vials of EGRIFTA 1 mg to prepare the recommended
dose of 2 mg (2 mL of the reconstituted solution).
READ BEFORE OPENING BOX:
This is the MEDICATION BOX – it contains 60 vials of
EGRIFTA 1 mg/vial and the Patient Instructions for Use.
You will also need the INJECTION BOX for EGRIFTA 1 mg/vial –
it contains syringes and other materials needed for reconstitution
and administration.
Protect
from light.
Keep in
original box.
Store
refrigerated
at 2°C to 8°C
(36°F to 46°F)
60 vials
THERAtechnologies
EGRIFTA® is a registered trademark
of Theratechnologies Inc.
EGRIFTA SV™ is a trademark of
Theratechnologies Inc.
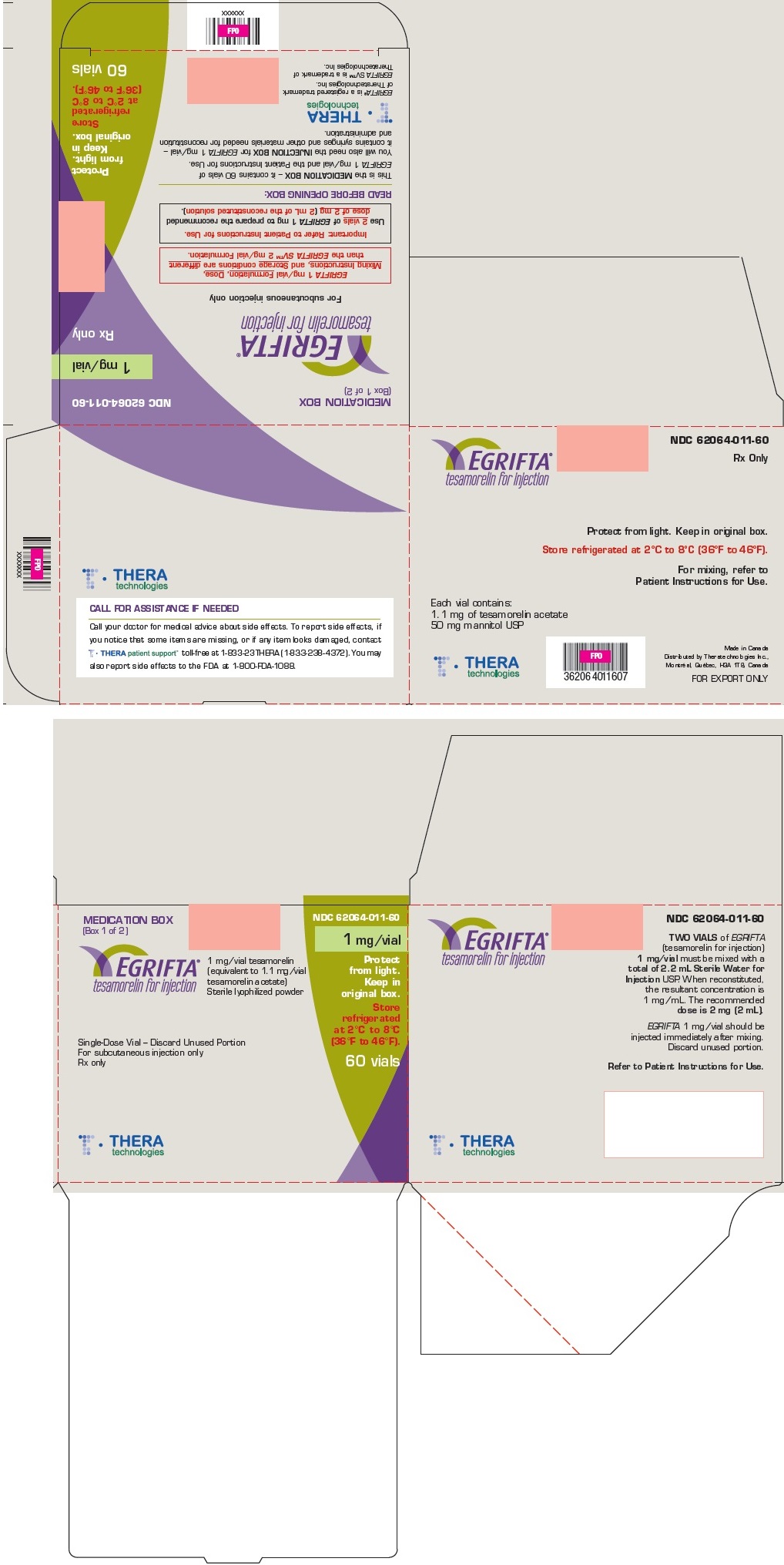
Principal Display Panel - Injection Box
INJECTION BOX (Box 2 of 2)
For EGRIFTA 1 mg/vial (tesamorelin for injection)
For mixing, refer to Patient
Instructions for Use included
in the EGRIFTA® 1 mg/vial
Medication Box.
Rx Only
READ BEFORE OPENING BOX
This is your INJECTION BOX for EGRIFTA (tesamorelin for injection) 1 mg/vial.
This box contains sterile syringes, Sterile Water for Injection USP, and materialsneeded for injecting EGRIFTA®.
Note this INJECTION BOX does NOT contain your EGRIFTA 1 mg/vial medication.
Before removing contents, make sure you also have the EGRIFTA 1 mg/vial
MEDICATION BOX.
Contents of INJECTION BOX:
a) 30 single-dose, 10 mL bottles of Sterile Water for Injection
b) 30 sterile, 3-mL syringes with needle already attached
c) 30 individual 1½" 18-gauge sterile needles used for mixing
d) 30 individual ½" 27-gauge sterile injection needles
Store at
controlled room
temperature
20°C to 25°C
(68°F to 77°F).
Made in USA
Distributed by Theratechnologies Inc., Montréal, Québec, H3A 1T8, Canada
THERAtechnologies
EGRIFTA® is a registered trademark of Theratechnologies Inc.
EGRIFTA SV™ is a trademark of Theratechnologies Inc.

Principal Display Panel - 1 mg Vial Label
Refer to Instructions for Use
EGRIFTA®
tesamorelin for injection
1 mg/vial
Rx only
STERILE
Store at
36°F to 46°F
For subcutaneous injection only
Each vial contains 1.1 mg of tesamorelin acetate
Single-Dose Vial - Discard Unused Portion
Distributed by Theratechnologies Inc.
XXXXXX

| EGRIFTA
tesamorelin kit |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Theratechnologies Inc. (252017520) |