Label: LIVMARLI- maralixibat chloride solution

- NDC Code(s): 79378-110-01
- Packager: Mirum Pharmaceuticals Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated April 1, 2024
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LIVMARLI safely and effectively. See full prescribing information for LIVMARLI.
LIVMARLI® (maralixibat) oral solution
Initial U.S. Approval: 2021RECENT MAJOR CHANGES
INDICATIONS AND USAGE
LIVMARLI is an ileal bile acid transporter (IBAT) inhibitor indicated for:
- the treatment of cholestatic pruritus in patients 3 months of age and older with Alagille syndrome (ALGS). (1.1)
- the treatment of cholestatic pruritus in patients 5 years of age and older with progressive familial intrahepatic cholestasis (PFIC). (1.2)
-
Limitations of Use:
LIVMARLI is not recommended in a subgroup of PFIC type 2 patients with specific ABCB11 variants resulting in non-functional or complete absence of bile salt export pump (BSEP) protein. (14.2)
-
Limitations of Use:
DOSAGE AND ADMINISTRATION
- ALGS: The recommended dosage is 380 mcg/kg once daily, taken 30 minutes before a meal in the morning. (2.1)
- Starting dose is 190 mcg/kg orally once daily, and should be increased to 380 mcg/kg once daily after one week, as tolerated and not to exceed a maximum daily dose of 28.5 mg. (2.1)
- PFIC: The recommended dosage is 570 mcg/kg twice daily. (2.2)
- Starting dose is 285 mcg/kg orally once daily in the morning and should be increased to 285 mcg/kg twice daily, 428 mcg/kg twice daily, and then to 570 mcg/kg twice daily, as tolerated and not to exceed a maximum daily dose of 38 mg. (2.2)
DOSAGE FORMS AND STRENGTHS
Oral solution: 9.5 mg of maralixibat per mL. (3)
CONTRAINDICATIONS
Patients with prior or active hepatic decompensation events (e.g., variceal hemorrhage, ascites, hepatic encephalopathy). (4)
WARNINGS AND PRECAUTIONS
- Hepatotoxicity: Obtain baseline liver tests and monitor patients frequently for the first 6 to 8 months after starting therapy, and as clinically indicated thereafter during treatment. If liver test abnormalities or signs of clinical hepatitis occur, consider dose reduction or treatment interruption. For persistent or recurrent liver test abnormalities relative to baseline, discontinue LIVMARLI. Monitor patients with compensated cirrhosis frequently. Permanently discontinue LIVMARLI if hepatic decompensation event occurs. (5.1)
- Gastrointestinal Adverse Reactions: Consider reducing the dosage or interrupting LIVMARLI treatment if a patient experiences persistent diarrhea or abdominal pain, or has diarrhea with bloody stool, vomiting, dehydration requiring treatment, or fever. Consider stopping LIVMARLI treatment if diarrhea or abdominal pain persists and no alternate etiology is identified. (5.2)
- Fat-Soluble Vitamin (FSV) Deficiency: Obtain baseline levels and monitor during treatment. Supplement if deficiency is observed. If FSV deficiency persists or worsens despite FSV supplementation, consider discontinuing LIVMARLI treatment. (5.3)
- Fracture: Consider interrupting LIVMARLI treatment and supplement with FSV. LIVMARLI can be restarted once FSV deficiency is corrected and maintained at corrected levels.
- Bleeding: Interrupt treatment with LIVMARLI. Treatment can be restarted if the FSV deficiency is corrected and bleeding has resolved.
ADVERSE REACTIONS
Most common adverse reactions (≥5%) are:
- ALGS: diarrhea, abdominal pain, vomiting, fat-soluble vitamin deficiency, liver test abnormalities, and bone fractures. (6.1)
- PFIC: diarrhea, fat soluble vitamin deficiency, abdominal pain, liver test abnormalities, hematochezia, and bone fractures. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Mirum Pharmaceuticals at 1-855-MRM-4YOU or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Treatment of Cholestatic Pruritus in Patients with Alagille Syndrome
1.2 Treatment of Cholestatic Pruritus in Patients with Progressive Familial Intrahepatic Cholestasis
2 DOSAGE AND ADMINISTRATION
2.1 Alagille Syndrome
2.2 Progressive Familial Intrahepatic Cholestasis
2.3 Missed Dose
2.4 Important Administration Instructions
2.5 Dosage Modification for Management of Adverse Events
2.6 Administration Modification for Drug Interaction
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
5.2 Gastrointestinal Adverse Reactions
5.3 Fat Soluble Vitamin (FSV) Deficiency
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on LIVMARLI
7.2 Effects of LIVMARLI on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5. Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Alagille Syndrome
14.2 Progressive Familial Intrahepatic Cholestasis
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Treatment of Cholestatic Pruritus in Patients with Alagille Syndrome
LIVMARLI® is indicated for the treatment of cholestatic pruritus in patients 3 months of age and older with Alagille syndrome (ALGS).
1.2 Treatment of Cholestatic Pruritus in Patients with Progressive Familial Intrahepatic Cholestasis
LIVMARLI is indicated for the treatment of cholestatic pruritus in patients 5 years of age and older with progressive familial intrahepatic cholestasis (PFIC).
Limitations of Use:
LIVMARLI is not recommended in a subgroup of PFIC type 2 patients with specific ABCB11 variants resulting in non-functional or complete absence of bile salt export pump (BSEP) protein [see Clinical Studies (14.2)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Alagille Syndrome
The recommended dosage is 380 mcg/kg once daily, taken 30 minutes before a meal in the morning. Start dosing at 190 mcg/kg administered orally once daily; after one week, increase to 380 mcg/kg once daily, as tolerated. The maximum daily dose should not exceed 28.5 mg (3 mL) per day. Refer to the dosing by weight guidelines presented in Table 1.
Table 1: Volume per Dose (mL) by Patient Weight Patient Weight
(kg)Days 1-7
(190 mcg/kg once daily)Beginning Day 8
(380 mcg/kg once daily)Volume per Dose
(mL)Volume per Dose
(mL)5 to 6 0.1 0.2 7 to 9 0.15 0.3 10 to 12 0.2 0.45 13 to 15 0.3 0.6 16 to 19 0.35 0.7 20 to 24 0.45 0.9 25 to 29 0.5 1 30 to 34 0.6 1.25 35 to 39 0.7 1.5 40 to 49 0.9 1.75 50 to 59 1 2.25 60 to 69 1.25 2.5 70 or higher 1.5 3 2.2 Progressive Familial Intrahepatic Cholestasis
The recommended dosage is 570 mcg/kg twice daily 30 minutes before a meal. The starting dose is 285 mcg/kg orally once daily in the morning, and should be increased to 285 mcg/kg twice daily, 428 mcg/kg twice daily, and then to 570 mcg/kg twice daily, as tolerated. The maximum daily dose should not exceed 38 mg (4 mL) per day. Refer to the dosing by weight guidelines presented in Table 2.
Table 2: Volume per Dose (mL) by Patient Weight Patient Weight
(kg)285 mcg/kg 428 mcg/kg 570 mcg/kg Volume per Dose (mL) Volume per Dose (mL) Volume per Dose (mL) 10 to 12 0.35 0.5 0.6 13 to 15 0.4 0.6 0.8 16 to 19 0.5 0.8 1 20 to 24 0.6 1 1.25 25 to 29 0.8 1.25 1.5 30 to 34 0.9 1.5 2 35 to 39 1.25 1.5 2 40 to 49 1.25 2 2 50 to 59 1.5 2 2 60 or higher 2 2 2 2.3 Missed Dose
For once daily dosing: If a dose is missed, it should be taken as soon as possible within 12 hours of the time it is usually taken, and the original dosing schedule should be resumed. If a dose is missed by more than 12 hours, the dose can be omitted and the original dosing schedule resumed.
For twice daily dosing: If a dose is missed, it should be taken as soon as possible within 6 hours of the time it is usually taken, and the original dosing schedule should be resumed. If a dose is missed by more than 6 hours, the dose can be omitted and the original dosing schedule resumed.
2.4 Important Administration Instructions
Administer LIVMARLI 30 minutes before a meal [see Clinical Pharmacology (12.3)].
A calibrated measuring device (0.5 mL, 1 mL or 3 mL oral dosing dispenser) will be provided by the pharmacy to measure and deliver the prescribed dose accurately.
After opening the LIVMARLI bottle, store below 30°C (86°F) and discard any remaining LIVMARLI after 100 days.
2.5 Dosage Modification for Management of Adverse Events
Establish the baseline pattern of variability of liver tests prior to starting LIVMARLI, so that potential signs of liver injury can be identified. Monitor liver tests (e.g., ALT [alanine aminotransferase], AST [aspartate aminotransferase], TB [total bilirubin]), DB [direct bilirubin], and International Normalized Ratio [INR]) during treatment with LIVMARLI. Reduce the dosage or interrupt LIVMARLI if new onset liver test abnormalities occur. Once the liver test abnormalities either return back to baseline values or stabilize at a new baseline value, consider restarting LIVMARLI at the last tolerated dose, and increase the dose as tolerated. Consider discontinuing LIVMARLI permanently if liver test abnormalities recur or symptoms consistent with clinical hepatitis are observed [see Warnings and Precautions (5.1)].
LIVMARLI has not been studied in patients with hepatic decompensation. Discontinue LIVMARLI permanently if a patient experiences a hepatic decompensation event (e.g., variceal hemorrhage, ascites, hepatic encephalopathy).
2.6 Administration Modification for Drug Interaction
Bile Acid Binding Resins
Administer LIVMARLI at least 4 hours before or 4 hours after administering the bile acid binding resins [see Drug Interactions (7.1)] when used concomitantly.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
LIVMARLI is contraindicated in patients with prior or active hepatic decompensation events (e.g., variceal hemorrhage, ascites, hepatic encephalopathy) [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
LIVMARLI treatment is associated with a potential for drug-induced liver injury.
In the PFIC trial, treatment-emergent hepatic decompensation events and elevations of liver tests or worsening of liver tests occurred. Two patients experienced drug-induced liver injury (DILI) attributable to LIVMARLI. Two additional patients experienced DILI in the open-label extension portion of the trial. Of these four patients, one patient required liver transplant and another patient died.
In the ALGS trial, treatment-emergent elevations of liver tests or worsening of liver tests occurred. Most abnormalities included elevations in ALT, AST, and/or TB/DB. One patient whose TB was elevated at baseline discontinued LIVMARLI after 28 weeks due to increased TB above baseline. Four patients had ALT increases that led to dose modification (n=1), dose interruption (n=2), or permanent discontinuation (n=2) of LIVMARLI during the long-term, open-label extension period [see Adverse Reactions (6.1)].
Obtain baseline liver tests because some ALGS and PFIC patients have abnormal liver tests at baseline. Monitor patients frequently for the first 6 to 8 months after starting therapy and as clinically indicated thereafter during treatment with LIVMARLI. Monitor for elevations in liver tests, for the development of liver-related adverse reactions, and for physical signs of hepatic decompensation. If liver test abnormalities or signs of clinical hepatitis occur in the absence of other causes, consider dose reduction or treatment interruption.
Permanently discontinue LIVMARLI if a patient experiences the following:
- persistent or recurrent liver test abnormalities, or
- upon rechallenge, signs and symptoms consistent with clinical hepatitis, or
- a hepatic decompensation event.
The safety and effectiveness of LIVMARLI have not been established in patients with decompensated cirrhosis. Monitor patients with compensated cirrhosis frequently and discontinue LIVMARLI if hepatic decompensation occurs. LIVMARLI is contraindicated in patients with prior or active hepatic decompensation events [see Contraindications (4)].
5.2 Gastrointestinal Adverse Reactions
Diarrhea and abdominal pain were reported as the most common adverse reactions in patients treated with LIVMARLI [see Adverse Reactions (6.1)].
Consider reducing the dosage or interrupting LIVMARLI treatment if a patient experiences persistent diarrhea or abdominal pain, or has diarrhea with bloody stool, vomiting, dehydration requiring treatment, or fever. Consider stopping LIVMARLI treatment if diarrhea or abdominal pain persists and no alternate etiology is identified. Monitor for dehydration due to diarrhea and treat promptly. LIVMARLI was not evaluated in PFIC patients with chronic diarrhea requiring intravenous fluids.
When diarrhea or abdominal pain resolves, restart LIVMARLI at the last tolerated dose and increase the dose as tolerated. Consider stopping LIVMARLI treatment if they recur upon re-challenge with LIVMARLI.
5.3 Fat Soluble Vitamin (FSV) Deficiency
LIVMARLI may adversely affect absorption of fat-soluble vitamins (FSV). FSV include vitamins A, D, E, and K (measured using INR levels). ALGS and PFIC patients can have FSV deficiency at baseline and are frequently supplemented with FSV.
In ALGS patients in Trial 1, treatment-emergent FSV deficiency was reported in 3 (10%) patients during 48 weeks of treatment.
In PFIC patients in Trial 2, treatment-emergent FSV deficiency (assessed biochemically) was reported in 13 (28%) of LIVMARLI-treated patients versus 16 (35%) of placebo-treated patients during 26 weeks of treatment.
Bone Fracture
Treatment-emergent bone fracture events have been observed more frequently with LIVMARLI-treated patients compared to placebo-treated patients [see Adverse Reactions (6.1)]. If fracture occurs, consider interrupting LIVMARLI treatment and supplement with FSV. LIVMARLI can be restarted once FSV deficiency is corrected and maintained at corrected levels.
Bleeding
If bleeding occurs, interrupt treatment with LIVMARLI. LIVMARLI can be restarted if FSV deficiency is corrected and bleeding has resolved.
Obtain serum FSV levels prior to initiation of LIVMARLI and monitor the levels periodically during treatment, along with any clinical manifestations of FSV deficiency. Supplement with FSV if FSV deficiency is diagnosed. Consider discontinuing LIVMARLI if FSV deficiency persists or worsens despite adequate FSV supplementation.
If complications of FSV deficiency occur, such as fracture or bleeding, consider interrupting LIVMARLI treatment and supplement with FSV. LIVMARLI can be restarted once FSV deficiency is corrected and maintained at corrected levels.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in labeling:
- Hepatotoxicity [see Warnings and Precautions (5.1)]
- Gastrointestinal Adverse Reactions [see Warnings and Precautions (5.2)]
- Fat Soluble Vitamin (FSV) Deficiency [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
ALGS:
In the Alagille syndrome clinical development program, which includes five clinical studies comprising 86 patients, patients received doses of LIVMARLI up to 760 mcg/kg per day with a median duration of exposure of 32.3 months (range: 0.03 - 60.9 months). In Trial 1, the 4-week placebo control period occurred after 18 weeks of LIVMARLI treatment. In two supportive studies that included long-term open-label extensions, only 13 weeks of placebo-controlled treatment occurred which evaluated doses lower than 380 mcg/kg/day. The majority of LIVMARLI exposure in the development program occurred without a placebo control in open-label trial extensions.
The most common adverse reactions (≥5%) for ALGS patients treated with LIVMARLI are presented in Table 3 below. Treatment interruptions or dose reductions occurred in 5 (6%) patients due to diarrhea, abdominal pain, or vomiting.
Table 3: Adverse Reactions Occurring in ≥ 5% of Patients Treated with LIVMARLI in the ALGS Clinical Development Program LIVMARLI (n=86) Adverse Reaction Any Grade
n (%)Number of events per 100 person-years* - *
- Exposure adjusted incidence rate for each adverse reaction type was calculated using the first occurrence of this adverse reaction per patient
- †
- Terms were defined as:
Fat-Soluble Vitamin deficiency includes: A, D, E, or K deficiency, or INR increase
Abdominal Pain includes: abdominal discomfort, abdominal distension, abdominal pain, abdominal pain lower, abdominal pain upper
Transaminases increased includes: ALT abnormal, ALT increased, AST abnormal, AST increased
Bone Fracture includes: tibia fracture, rib fracture, hand fracture, humerus fracture, pathological fracture, forearm fracture, clavicle fracture
Diarrhea 48 (55.8%) 41.6 Abdominal pain† 46 (53.5%) 38.6 Vomiting 35 (40.7%) 19.8 Nausea 7 (8.1%) 2.9 Fat-Soluble Vitamin deficiency† 22 (25.6%) 11.1 Transaminases increased (ALT, AST)† 16 (18.6%) 6.9 Bone Fractures† 8 (9.3%) 3.3 Liver Test Abnormalities
Increase in Transaminases
In a pooled analysis of patients with ALGS (N=86) administered LIVMARLI, increases in hepatic transaminases (ALT) were observed. Seven (8.1%) patients discontinued LIVMARLI due to ALT increases. Three (3.5%) patients had a decrease in dose or interruption of LIVMARLI in response to ALT increases. In the majority of cases, the elevations resolved or improved after discontinuation or dose modification of LIVMARLI. In some cases, the elevations resolved or improved without change in LIVMARLI dosing. Increases to more than three times baseline in ALT occurred in 26% of patients treated with LIVMARLI and increases to more than five times baseline occurred in 3%. AST increases to more than three times baseline occurred in 16% of patients treated with LIVMARLI, and an increase to more than five times baseline occurred in one patient. Elevations in transaminases were asymptomatic and not associated with bilirubin elevations or other laboratory abnormalities.
PFIC:
In Trial 2, which enrolled 93 patients, 47 patients received doses of LIVMARLI up to 570 mcg/kg BID, with a median duration of exposure of 6 months (range: 0.3-6.7 months).
The most common adverse reactions (≥5%) for PFIC patients treated with LIVMARLI at a rate greater than placebo are presented in Table 4 below. Diarrhea was the most frequent adverse reaction; the majority of episodes were mild and transient with a median duration of 5.5 days. Nineteen (40.4%) LIVMARLI-treated subjects had diarrhea greater than or equal to 7 days. Placebo-treated subjects had a median duration of 3 days of diarrhea and two subjects (4.3%) experienced diarrhea with a duration greater than or equal to 7 days. There were no severe events reported. The majority of abdominal pain events were mild based on AE grading, and associated with concurrent diarrhea.
One LIVMARLI-treated patient with an event of mild diarrhea discontinued treatment. Treatment interruptions or dose reductions occurred in 3 (6.4%) LIVMARLI-treated patients due to diarrhea or abdominal pain. No placebo-treated subjects discontinued treatment or had dose reductions or interruptions due to diarrhea.
Table 4: Adverse Reactions (any grade) Occurring in ≥5% and at a Rate Greater Than Placebo in Patients Treated with LIVMARLI in the PFIC Trial Adverse Reaction
(Any Grade)Placebo
n=46LIVMARLI
n=47- *
-
Terms were defined as:
Abdominal Pain includes: abdominal pain, abdominal pain upper, abdominal distension
Transaminases increased includes: Hypertransaminasaemia, ALT abnormal, ALT increased, AST abnormal, AST increased, Transaminases increased, Hepatic enzyme increased.
Bone Fracture includes: upper limb fracture, lower limb fracture, radius fracture, ulna fracture, femur fracture, foot fracture
Diarrhea 9 (19.6%) 27 (57.4%) Abdominal pain* 7 (15.2%) 13 (27.7%) Transaminases increased (ALT or AST) * 3 (6.5%) 8 (17%) Hematochezia or rectal hemorrhage 1 (2.2%) 4 (8.5%) Bone Fractures* 0 3 (6.4%) Hepatotoxicity
LIVMARLI treatment is associated with a potential for drug-induced liver injury. In PFIC patients in Trial 2, treatment-emergent elevations of liver tests or worsening of liver tests, relative to baseline values, and hepatic decompensation events were observed. Two patients experienced drug-induced liver injury (DILI) attributable to LIVMARLI; one patient received 570 mcg/kg twice daily and the second patient required dose interruption and reduction. Two additional patients experienced DILI in the open-label extension portion of the trial. Of these four patients, one patient required liver transplant and another patient died.
Biliary Complications
In the placebo-controlled portion of Trial 2, two LIVMARLI-treated patients developed cholangitis or cholecystitis within 3-weeks of drug discontinuation (after 84 days and 130 days after initiating LIVMARLI treatment, respectively). Four LIVMARLI-treated patients developed cholecystitis or cholangitis in the open-label extension portion of Trial 2; the average time to onset was 281 days.
Bone Fracture
In PFIC patients in Trial 2, treatment-emergent bone fracture events were observed. Three receiving LIVMARLI experienced bone fractures relative to none in placebo-treated patients. The median time to onset of fractures was 73 days. Two LIVMARLI-treated patients developed bone fractures in the open-label portion of Trial 2, with average time to onset of fracture was 204 days.
Bleeding
In PFIC patients in Trial 2, treatment-emergent events of hematochezia (4 [8.5%] versus 1 [2.2%]), decrease in hemoglobin greater than or equal to 2 grams/dL from baseline (8 [17%] versus 1 [2.2%]), were reported more frequently in LIVMARLI-treated patients relative to placebo-treated patients.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of LIVMARLI. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal disorders: hematemesis, liver transplant, post-endoscopy hemorrhage, post-liver biopsy hemorrhage
General disorders and administration site conditions: drug ineffective
Injury, poisoning and procedural complications: off label use
Investigations: gamma-glutamyltransferase increased
Nervous system disorders: intracranial hemorrhage
-
7 DRUG INTERACTIONS
7.2 Effects of LIVMARLI on Other Drugs
OATP2B1 substrates
Maralixibat is an OATP2B1 inhibitor based on in vitro studies. A decrease in the oral absorption of OATP2B1 substrates (e.g., statins) due to OATP2B1 inhibition in the GI tract cannot be ruled out. Consider monitoring the drug effects of OATP2B1 substrates (e.g. statins) as needed [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Maternal use at the recommended clinical dose of LIVMARLI is not expected to result in measurable fetal exposure because systemic absorption following oral administration is low [see Clinical Pharmacology (12.3)]. Maralixibat may inhibit the absorption of fat-soluble vitamins [see Warnings and Precautions (5.3) and Clinical Considerations]. In animal reproduction studies, no developmental effects were observed (see Data).
The estimated background risk of major birth defects for ALGS is higher than the general population because ALGS is an autosomal dominant condition. The background risk of miscarriage for ALGS is unknown. The background risk of birth defects and miscarriage for PFIC is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Maralixibat may inhibit the absorption of fat-soluble vitamins (FSV). Monitor for FSV deficiency and supplement as needed. Increased supplementation of FSVs may be needed during pregnancy [see Warnings and Precautions (5.3)].
Data
Animal Data
No effects on embryo-fetal development were observed in pregnant rats treated orally with up to 1000 mg/kg/day (approximately 3300 to 12000 times the maximum recommended dose based on AUC [area under the plasma concentration-time curve]) or in pregnant rabbits treated orally with up to 250 mg/kg/day (approximately 1200 to 4700 times the maximum recommended dose based on AUC) during the period of organogenesis. No effects on postnatal development were observed in a pre- and postnatal development study, in which female rats were treated orally with up to 750 mg/kg/day during organogenesis through lactation. Maternal systemic exposure to maralixibat at the maximum dose tested was approximately 2500 to 9400 times the maximum recommended dose based on AUC.
8.2 Lactation
Risk Summary
LIVMARLI has low absorption following oral administration, and breastfeeding is not expected to result in exposure of the infant to LIVMARLI at the recommended dose [see Clinical Pharmacology (12.3)]. There are no data on the presence of LIVMARLI in human milk, the effects on the breastfed infant, or the effects on milk production. Patients with ALGS or PFIC can have FSV deficiency as part of their disease. Maralixibat may reduce absorption of fat-soluble vitamins [see Warnings and Precautions (5.3)]. Monitor FSV levels and supplement FSV intake, if FSV deficiency is observed during lactation. The developmental and health benefits of breastfeeding should be considered along with the mother's need for LIVMARLI and any potential adverse effects on the breastfed child from LIVMARLI or from the underlying maternal condition.
8.4 Pediatric Use
ALGS
The safety and effectiveness of LIVMARLI for the treatment of cholestatic pruritus in Alagille syndrome have been established in pediatric patients aged 3 months of age and older. Use of LIVMARLI in this population is supported by evidence from a study of patients 1 to 15 years of age (N=31) that included 18 weeks of open-label treatment followed by a 4 week placebo-controlled randomized withdrawal period and a subsequent 26-week open-label treatment period. Additional safety information was obtained from four studies in patients up to 21 years of age (N=55) [see Adverse Reactions (6) and Clinical Studies (14.1)]. Use of LIVMARLI in patients 3 to <12 months of age is supported by an open-label, multicenter study of LIVMARLI which showed a similar safety, tolerability and pharmacokinetic profile to patients with ALGS ≥12 months of age.
The safety and effectiveness of LIVMARLI have not been established in patients with ALGS less than 3 months of age.
PFIC
The safety and effectiveness of LIVMARLI for the treatment of cholestatic pruritus in PFIC have been established in pediatric patients aged 5 years of age and older. Use of LIVMARLI in this population is supported by evidence from Trial 2 in patients 1 to <18 years of age that included 26 weeks of placebo-controlled safety and efficacy data [see Adverse Reactions (6) and Clinical Studies (14.2)].
The safety and effectiveness of LIVMARLI have not been established in patients with PFIC younger than 12 months of age.
8.5 Geriatric Use
The safety and effectiveness of LIVMARLI for the treatment of pruritus in ALGS or PFIC in adult patients, 65 years of age and older, have not been established.
8.6 Hepatic impairment
Clinical studies of LIVMARLI included ALGS or PFIC patients with impaired hepatic function at baseline. The efficacy and safety in ALGS or PFIC patients with clinically significant portal hypertension and in patients with decompensated cirrhosis have not been established. LIVMARLI is contraindicated in patients with prior or active hepatic decompensation events [see Dosage and Administration (2.5), Contraindications (4), Warnings and Precautions (5.1), and Clinical Studies (14)].
-
10 OVERDOSAGE
Single doses of maralixibat up to 500 mg, approximately 18-fold higher than the recommended dose, have been administered in healthy adults and were tolerated without a meaningful increase in adverse effects when compared to lower doses. If an overdose occurs, discontinue LIVMARLI, monitor the patient for any signs and symptoms and institute general supportive measures if needed.
LIVMARLI contains propylene glycol as an excipient. In cases of suspected overdose, monitor for signs of propylene glycol toxicity, including hemolysis, hyperosmolarity with anion gap metabolic acidosis, acute kidney injury, CNS toxicity, and multi-organ failure. Discontinue LIVMARLI if propylene glycol toxicity is suspected.
-
11 DESCRIPTION
LIVMARLI (maralixibat) oral solution is an ileal bile acid transporter (IBAT) inhibitor. Maralixibat is present as a chloride salt with the chemical name 1-[[4-[[4-[(4R,5R)-3,3-dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro-4-hydroxy-1,1-dioxido-1-benzothiepin-5-yl]phenoxy]methyl]phenyl]methyl]-4-aza-1-azoniabicyclo[2.2.2]octane chloride. The molecular formula of maralixibat chloride is C40H56ClN3O4S with a molecular weight of 710.42. It has the following chemical structure:
LIVMARLI is supplied in a multiple-dose bottle containing 9.5 mg of maralixibat per mL (equivalent to 10 mg of maralixibat chloride per mL). The oral solution contains the following inactive ingredients: edetate disodium, grape flavor, propylene glycol, purified water, and sucralose. The pH of the oral solution is 3.8 – 4.8.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Maralixibat is a reversible inhibitor of the ileal bile acid transporter (IBAT). It decreases the reabsorption of bile acids (primarily the salt forms) from the terminal ileum.
Pruritus is a common symptom in patients with ALGS or PFIC and the pathophysiology of pruritus in patients with ALGS or PFIC is not completely understood. Although the complete mechanism by which maralixibat improves pruritus in ALGS or PFIC patients is unknown, it may involve inhibition of the IBAT, which results in decreased reuptake of bile salts, as observed by a decrease in serum bile acids [see Clinical Pharmacology (12.2)].
12.2 Pharmacodynamics
ALGS
In Trial 1, pediatric patients with ALGS were administered open-label treatment with LIVMARLI 380 mcg/kg once daily for 13 weeks after an initial 5-week dose-escalation period [see Clinical Studies (14.1)]. At baseline, serum bile acids were highly variable among patients ranging from 20 to 749 µmol/L and mean (SD) serum bile acid level was 283 (210.6) µmol/L. Serum bile acid levels decreased from baseline in the majority of patients as early as at Week 12 and the reduction in serum bile acids was generally maintained for the treatment period.
PFIC
In Trial 2, pediatric patients with PFIC were administered LIVMARLI 570 mcg/kg or placebo twice daily for up to 22 weeks after an initial 4–6-week dose escalation period [see Clinical Studies (14.2)]. At baseline, serum bile acids concentrations were highly variable among patients ranging from 4 to 504 µmol/L and mean (SD) serum bile acid level was 253 (136) µmol/L. Serum bile acid concentrations decreased from baseline in the majority of patients as early as at Week 2; while the concentrations fluctuated, the reduction in serum bile acids was generally maintained for the treatment period.
12.3 Pharmacokinetics
Because of the low systemic absorption of maralixibat, pharmacokinetic parameters cannot be reliably calculated at the recommended dose. Concentrations of maralixibat in the pediatric ALGS and PFIC patients were below the limit of quantification (0.25 ng/mL) in the majority of plasma samples.
Following single oral administration of maralixibat in healthy adults at doses ranging from 1 mg to 500 mg, plasma concentrations of maralixibat were below the limit of quantification (0.25 ng/mL) at doses less than 20 mg and PK parameters could not be reliably estimated.
Following a single dose administration of 30 mg under fasted condition, median Tmax was 0.75 and mean (SD) Cmax and AUClast were 1.65 (1.10) ng/ml and 3.43 (2.13) ng∙h/mL, respectively.
Absorption
Maralixibat is minimally absorbed and plasma concentrations are often below the limit of quantification (0.25 ng/mL) after single or multiple doses at recommended doses. Following a single oral administration of maralixibat 30, 45, and 100 mg liquid formulation under fasted condition, AUClast and Cmax increased in a dose-dependent manner with increase of 4.6-and 2.4-fold, respectively, following a 3.3-fold dose increase from 30 to 100 mg.
No accumulation of maralixibat was observed following repeated oral administration of maralixibat in healthy adults at doses up to 100 mg once-daily.
Effect of Food
Concomitant administration of a high-fat meal with a single oral dose of maralixibat decreased both the rate and extent of absorption. AUC and Cmax of maralixibat values in the fed state were 64.8% to 85.8% lower relative to oral administration of 30 mg in fasted conditions. The effect of food on the changes of systemic exposures to maralixibat is not clinically significant [see Dosage and Administration (2.1, 2.3)].
Elimination
Following a single oral dose of 30 mg maralixibat in healthy adults, the mean half-life (t1/2) was 1.6 hours.
Drug Interaction Studies
Effect of Other Drugs on Maralixibat
Maralixibat is not a substrate of the drug transporters MDR1 (P-gp), BCRP, OATP1B1, OATP1B3, or OATP2B2; therefore, concomitant drug products are not predicted to affect the disposition of maralixibat.
Effect of Maralixibat on Other Drugs
In vitro, maralixibat did not induce CYP isoforms 1A2, 2B6, or 3A4, nor inhibit CYP isoforms 1A2, 2B6, 2C8, 2C9, 2C19 or 2D6 at clinically relevant concentrations. Maralixibat inhibits CYP3A4 in vitro, however clinically relevant effects on the pharmacokinetics of CYP3A4 substrates are unlikely. In vitro, maralixibat did not inhibit the transporters MDR1 (P-gp), BCRP, OAT1, OAT3, OATP1B1, OATP1B3, PEPT1, OCT1, OCT2, OCT3, OCTN1, OCTN2, MRP2, MATE1, or MATE2-K at clinically relevant concentrations.
Maralixibat inhibits the drug transporter OATP2B1 in vitro, which can potentially result in reduced absorption of drugs that rely on OATP2B1-mediated uptake in the GI tract. In clinical studies coadministration of 4.75 mg maralixibat (once daily in the morning) with daily doses of either simvastatin, or lovastatin in the evening, did not have a clinically relevant effect on the pharmacokinetics of these statins and their metabolites. Coadministration of 4.75 mg maralixibat did not affect pharmacokinetics of atorvastatin. However, the effect of maralixibat on the pharmacokinetics of OATP2B1 substrates at higher doses has not been evaluated in a clinical study.
12.5. Pharmacogenomics
PFIC is a heterogenous disease caused by homozygous or compound heterozygous variants, with different PFIC subtypes occurring in the general population. PFIC1 is caused by variants in the aminophospholipid flippase (ATP8B1) gene, which encodes the Familial Intrahepatic Cholestasis 1 (FIC1) protein, while PFIC2 results from variants in the ABCB11 gene, which encodes the Bile Salt Export Pump (BSEP) protein. PFIC2 is further categorized into BSEP subgroups based on specific variants. The BSEP-1 subgroup includes patients with at least one p.D482G (c.1445A>G) or p.E297G (c.890A>G) variant, BSEP-2 includes patients with at least one missense variant other than p.D482G or p.E297G (non BSEP-1), and BSEP-3 includes patients with variants that are predicted to encode a non-functional protein. PFIC3 is caused by variants in the ABCB4 gene, which encodes multidrug resistance protein 3 (MDR3). PFIC4 is caused by variants in the tight junction protein 2 gene (TJP2), which encodes TJP2. PFIC6 is caused by variants in myosin 5B (MYO5B), which encodes MYO5B. Patients can be clinically diagnosed with PFIC without a known pathogenic variant.
PFIC2 is the most common subtype accounting for 37-90% of patients with PFIC. The prevalence of BSEP-1, BSEP-2, and BSEP-3 subgroups are approximately 27.3%, 51.5%, and 21.2%, respectively, based on data from a global consortium characterizing the natural history of severe BSEP deficiency.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No drug-related tumors were observed following oral administration of maralixibat chloride to TgRasH2 mice at doses of up to 25 (males) or 75 (females) mg/kg/day for 26 weeks.
-
14 CLINICAL STUDIES
14.1 Alagille Syndrome
The efficacy of LIVMARLI was assessed in Trial 1 (NCT02160782), which consisted of an 18-week open-label treatment period; a 4-week randomized, double-blind, placebo-controlled drug-withdrawal period; a subsequent 26-week open-label treatment period; and a long-term open-label extension period.
Thirty-one pediatric ALGS patients with cholestasis and pruritus were enrolled, with 90.3% of patients receiving at least one medication to treat pruritus at study entry. All patients had JAGGED1 mutation. Patients were administered open-label treatment with LIVMARLI 380 mcg/kg once daily for 13 weeks after an initial 5-week dose-escalation period; two patients discontinued treatment during this first 18 weeks of open-label treatment. The 29 patients who completed the open-label treatment phase were then randomized to continue treatment with LIVMARLI or receive matching placebo during the 4-week drug withdrawal period at Weeks 19-22 (n=16 placebo, n=13 LIVMARLI). All 29 patients completed the randomized, blinded drug withdrawal period; subsequently, patients received open-label LIVMARLI at 380 mcg/kg once daily for an additional 26 weeks.
Randomized patients had a median age of 5 years (range: 1 to 15 years) and 66% were male. The baseline mean (standard deviation [SD]) of liver test parameters were as follows: serum bile acid levels 280 (213) µmol/L, AST 158 (68) U/L, ALT 179 (112) U/L, Gamma Glutamyl Transferase (GGT) 498 (399) U/L, and TB 5.6 (5.4) mg/dL.
Given the patients' young age, a single-item observer-reported outcome was used to measure patients' pruritus symptoms as observed by their caregiver twice daily (once in the morning and once in the evening) on the Itch Reported Outcome Instrument (ItchRO[Obs]). Pruritus symptoms were assessed on a 5-point ordinal response scale, with scores ranging from 0 (none observed or reported) to 4 (very severe). Patients were included in Trial 1 if their average pruritus score was greater than 2.0 (moderate) in the 2 weeks prior to baseline.
The average of the worst daily ItchRO(Obs) pruritus scores was computed for each week. For randomized patients, the mean (SD) at baseline (pre-treatment) was 3.1 (0.5) and the mean (SD) at Week 18 (pre-randomized withdrawal period) was 1.4 (0.9). On average, patients administered LIVMARLI for 22 weeks maintained pruritus reduction whereas those in the placebo group who were withdrawn from LIVMARLI after Week 18 returned to baseline pruritus scores by Week 22. Results from the placebo-controlled period are presented in Table 5. After re-entering the open-label treatment phase, both randomized treatment groups had similar mean pruritus scores by Week 28, the first week placebo patients received the full dosage of LIVMARLI after withdrawal. These observer-rated pruritus results are supported by similar results on patient-rated pruritus in patients 5 years of age and older who were able to self-report their itching severity.
Table 5: Weekly Average of Worst Daily ItchRO(Obs) Pruritus Severity Scores in Trial 1 Maralixibat
(N=13)Placebo
(N=16)Mean Difference Results based on an analysis of covariance model with treatment group and Week 18 average worst daily pruritus score as covariates Week 22, Mean (95% CI) 1.6 (1.1, 2.1) 3.0 (2.6, 3.5) Change from Week 18 to Week 22, Mean (95% CI) 0.2 (-0.3, 0.7) 1.6 (1.2, 2.1) -1.4 (-2.1, -0.8) 14.2 Progressive Familial Intrahepatic Cholestasis
The efficacy of LIVMARLI was assessed in Trial 2 (NCT03905330), a 26-week randomized, placebo-controlled trial.
Efficacy was evaluated in 64 patients with documented molecular diagnosis of PFIC with presence of biallelic known pathogenic variants; this included 31 non-truncated PFIC2 patients (i.e., excluding patients with BSEP3) and 33 patients with PFIC1 (n=13), PFIC3 (n=9), PFIC4 (n=7), or PFIC6 (n=4) [see Clinical Pharmacology (12.5)]. Additional patients with BSEP3 (n=9), prior surgical diversion (n=8), heterozygous subjects (n=2), or no variants associated with cholestasis (n=8) were included for evaluation in a supplemental cohort [see Adverse Reactions (6.1)].
Patients were randomized to receive maralixibat orally 570 mcg/kg (n=33) or placebo (n=31) twice daily. 53% of pediatric patients were females. Most patients were on stable ursodeoxycholic acid (89.1%) or rifampicin (51.6%) therapy at baseline. The baseline mean (standard deviation [SD]) of liver test parameters were as follows: serum bile acid levels 263 (143) μmol/L, AST 113 (82) U/L, ALT 107 (87) U/L, and TB 4.1 (4.1) mg/dL, DB 3.0 (3.1) mg/dL.
Given the patients' young age, a single-item observer-reported outcome was used to measure patients' pruritus symptoms as observed by their caregiver twice daily (once in the morning and once in the evening) on the Itch Reported Outcome Instrument (ItchRO[Obs]). Pruritus symptoms were assessed on a 5-point ordinal response scale, with scores ranging from 0 (none observed or reported) to 4 (very severe). Patients were included in Trial 2 if their average pruritus score was greater than or equal to 1.5 in the 4 weeks prior to baseline.
Table 6 presents the results of the comparison between LIVMARLI and placebo on the mean change in the average morning ItchRO(Obs) severity score between baseline and Weeks 15–26. For the outcome summarized over Weeks 15-26, the average morning ItchRO(Obs) severity score for each patient was calculated by: (Step 1) averaging the morning scores within a week; (Step 2) averaging 4 weekly morning scores to yield a single 4-week score; and finally (Step 3) averaging the three 4-week average morning scores (Weeks 15-18, Weeks 19-22, Weeks 23-26). The baseline average itching scores for each patient was calculated by averaging the morning ItchRO(Obs) score obtained in Step 2 across the 4 weeks prior to the first dose of the study. Patients treated with LIVMARLI demonstrated greater improvement in pruritus compared with placebo.
Table 6: Average Morning ItchRO(Obs) Pruritus Severity Scores in Trial 2 Maralixibat
(N=33)Placebo
(N=31)- *
- Results based on least squares means from an analysis of a mixed-effect model for repeated measures (MMRM). Model adjusts for treatment group, visit, treatment group-by-visit interaction, baseline 4-week average morning ItchRO(Obs) severity score, baseline score-by-visit interaction, and PFIC type.
Baseline Mean 2.9 2.7 Change from baseline to Weeks 15-26* Mean (95% CI) -1.8 (-2.2, -1.4) -0.6 (-1.0, -0.2) Mean difference from Placebo (95% CI) -1.2 (-1.7, -0.7) p-value <0.0001 Figure 1 displays the means (95% confidence intervals) of patients' average morning ItchRO(Obs) severity scores in each treatment group for each 4-6 week time period.
Figure 1. Mean of the Average Morning ItchRO(Obs) Pruritus Severity Scores Over Time in PFIC 1, 2, 3, 4, and 6
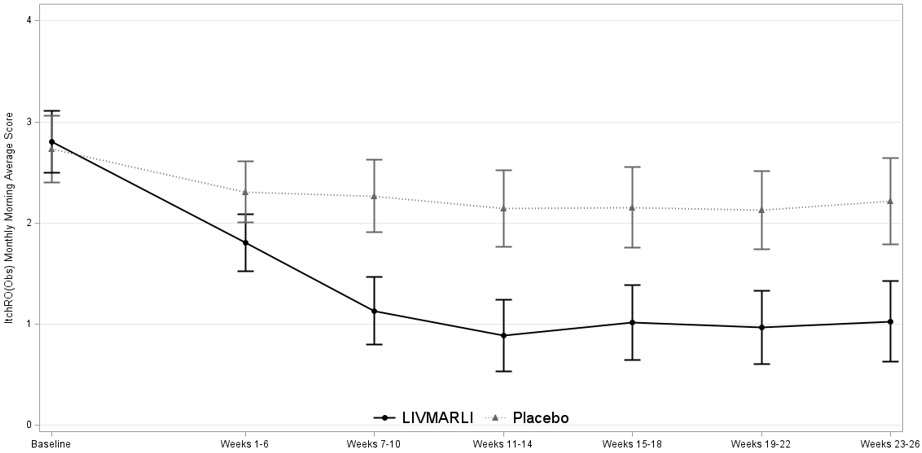
Note: Figure 1 presents least squares mean (95% CI) estimates from a mixed model for repeated measures (MMRM) with observed value as the dependent variable and fixed categorical effects of treatment group, time period, treatment-by-time period interaction, and PFIC type.
Although the number of patients with BSEP3 in Trial 2 were limited, improvement in pruritus was not observed in 5 patients with BSEP3 who received LIVMARLI compared to 4 patients with BSEP3 who received placebo for 26 weeks.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Oral Solution
LIVMARLI is a clear, colorless to yellow oral solution.
Each amber plastic bottle contains LIVMARLI oral solution at a concentration of 9.5 mg per mL.
One 30 mL amber plastic bottle: NDC 79378-110-01
Storage and Handling
Store unopened LIVMARLI between 20°C and 25°C (68°F and 77°F), excursion permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature]. After opening the LIVMARLI bottle, store below 30°C (86°F) [see Dosage and Administration (2.4)].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient or their caregiver(s) to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Administration Instructions
Advise patients or their caregivers(s) to:
- Take LIVMARLI 30 minutes prior to a meal once or twice daily as prescribed using a calibrated measuring device (0.5 mL, 1 mL or 3 mL oral dispenser) provided by the pharmacist to measure and deliver the prescribed dose accurately [see Dosage and Administration (2.1, 2.2 2.4)].
- Take LIVMARLI at least 4 hours before or 4 hours after taking a bile acid binding resin (e.g., cholestyramine, colesevelam, or colestipol) [see Drug Interactions (7.1)].
- Store the opened bottle below 30⁰C (86⁰F). Discard any unused LIVMARLI 100 days after opening the bottle [see How Supplied/Storage and Handling (16)].
Hepatotoxicity
Advise patients or their caregiver(s) that liver tests should be obtained before starting LIVMARLI and periodically during LIVMARLI therapy. Inform patients or their caregiver(s) of the risk of hepatotoxicity that could be fatal and that they will need to undergo monitoring for liver injury. Instruct patients or their caregiver(s) to immediately report any signs or symptoms of severe liver injury to their healthcare provider [see Warnings and Precautions (5.1)].
Gastrointestinal Adverse Reactions
Advise patients or their caregiver(s) to notify their healthcare provider if they experience a new onset or worsening of gastrointestinal symptoms (abdominal pain, vomiting, bloody stool, and diarrhea) [see Warnings and Precautions (5.2)].
Fat Soluble Vitamin (FSV) Deficiency
Advise patients or their caregiver(s) that INR (for vitamin K) and serum levels of vitamins A, D, E will be obtained before starting treatment and periodically during treatment to assess for FSV deficiency [see Warnings and Precautions (5.3)]. Inform patients or their caregiver(s) that they may bleed more easily, may bleed longer, or have a bone fracture. Advise patients or their caregiver(s) to call their healthcare provider for any signs or symptoms of bleeding or report any fractures.
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
LIVMARLI® (liv-MAR-lee)
(maralixibat)
oral solutionThis Patient Information has been approved by the U.S. Food and Drug Administration Revised: 03/2024 What is LIVMARLI? - LIVMARLI is a prescription medicine used to treat:
- cholestatic pruritus (itch) in patients 3 months of age and older with Alagille syndrome (ALGS).
- cholestatic pruritus (itch) in patients 5 years of age and older with progressive familial intrahepatic cholestasis (PFIC).
- LIVMARLI is not for use in PFIC type 2 patients who have a severe defect in the bile salt export pump (BSEP) protein.
- It is not known if LIVMARLI is safe and effective in children with ALGS who are under 3 months of age.
- It is not known if LIVMARLI is safe and effective in children with PFIC who are under 5 years of age.
- It is not known if LIVMARLI is safe and effective in adults 65 years of age and older.
Before taking LIVMARLI, tell your healthcare provider about all of your medical conditions, including if you: - are pregnant or plan to become pregnant. It is not known if LIVMARLI will harm your unborn baby. Tell your healthcare provider right away if you think that you are pregnant.
- are breastfeeding or plan to breastfeed. It is not known if LIVMARLI passes into your breast milk. Talk with your healthcare provider about the best way to feed your baby if you take LIVMARLI.
Tell your healthcare provider about all medicines that you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. LIVMARLI may affect the way some other medicines work, and some other medicines may affect the way LIVMARLI works. How should you take LIVMARLI? Read the Instructions for Use that comes with LIVMARLI for information about the right way to prepare and take LIVMARLI. - Before you take LIVMARLI for the first time, talk to your healthcare provider or pharmacist about how to measure the prescribed dose.
- Take LIVMARLI exactly as your healthcare provider tells you to.
- Your healthcare provider may start you on a low dose of LIVMARLI and then increase the dose, especially if you have not taken LIVMARLI.
- Do not change your dose of LIVMARLI unless your healthcare provider tells you to.
- LIVMARLI is taken by mouth, 1 or 2 times each day (if 2 times per day, take in the morning and evening), 30 minutes before a meal.
- If you miss a dose of LIVMARLI and you take LIVMARLI 1-time a day:
- If it is 12 hours or less from the time you usually take LIVMARLI, take the missed dose as soon as possible. Then take your next dose at the usual time.
- If it is more than 12 hours from the time you usually take LIVMARLI, do not take the missed dose. Take your next dose at the usual time.
- If you miss a dose of LIVMARLI and you take LIVMARLI 2-times a day:
- If it is 6 hours or less from the time you usually take LIVMARLI, take the missed dose as soon as possible. Then take your next dose at the usual time.
- If it is more than 6 hours from the time you usually take LIVMARLI, do not take the missed dose. Take your next dose at the usual time.
- If you take a medicine that lowers cholesterol by binding bile acids, such as cholestyramine, colesevelam, or colestipol, take it at least 4 hours before or 4 hours after you take LIVMARLI. Ask your healthcare provider if you are not sure if you take these medicines.
- If you take too much LIVMARLI, call your healthcare provider or go to the nearest emergency room right away.
- Throw away any remaining LIVMARLI 100 days after first opening the bottle.
What are the possible side effects of LIVMARLI? LIVMARLI can cause serious side effects, including: -
Liver injury. Changes in certain liver tests are common in patients with ALGS and in patients with PFIC but may worsen during treatment with LIVMARLI. These changes may be a sign of liver injury and can be serious or may lead to liver transplant or death. Your healthcare provider should do blood tests and physical exams before starting and during treatment with LIVMARLI to check your liver function. Tell your healthcare provider right away if you get any signs or symptoms of liver problems, including:
- nausea or vomiting
- your skin or the white part of your eye turns yellow
- dark or brown urine
- pain on the right side of your stomach (abdomen)
- fullness, bloating, or fluid in your stomach area (ascites)
- loss of appetite
- bleeding or bruising more easily than normal, including vomiting blood
-
Stomach and intestinal (gastrointestinal) problems. LIVMARLI can cause stomach and intestinal problems, including diarrhea and stomach pain during treatment. Diarrhea can also cause the loss of too much body fluid (severe dehydration). Your healthcare provider should check your stool for blood and monitor you for too much body fluid loss.
Tell your healthcare provider right away if you have any new or worsening signs or symptoms of stomach and intestinal problems including:
- diarrhea
- more frequent bowel movements than usual
- stools that are black, tarry, sticky, or have blood or mucous
- severe stomach-area pain or tenderness
Tell your healthcare provider if you have any signs and symptoms of a loss of too much body fluid including: - vomiting
- diarrhea
- urinating less often than usual
- headache
- dizziness
-
A condition called Fat Soluble Vitamin (FSV) Deficiency caused by low levels of certain vitamins (vitamin A, D, E, and K) stored in body fat. FSV deficiency is common in patients with ALGS and in patients with PFIC but may worsen during treatment with LIVMARLI. Your healthcare provider should do blood tests before starting and during treatment with LIVMARLI.
Other common side effects of FSV deficiency reported during treatment with LIVMARLI were bone fractures and bleeding.
These are not all of the possible side effects of LIVMARLI. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store LIVMARLI? - Store unopened LIVMARLI at room temperature between 68°F and 77°F (20°C and 25°C).
- After opening the LIVMARLI bottle, store below 30°C (86°F).
General information about the safe and effective use of LIVMARLI. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use LIVMARLI for a condition for which it was not prescribed. Do not give LIVMARLI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about LIVMARLI that is written for health professionals. What are the ingredients in LIVMARLI? Active ingredients: maralixibat chloride. Inactive ingredients: edetate disodium, grape flavor, propylene glycol, purified water, and sucralose. Manufactured for: Mirum Pharmaceuticals, Inc., 950 Tower Lane, Suite 1050, Foster City, CA 94404 For more information, go to www.LIVMARLI.com or call 1-855-MRM-4YOU - LIVMARLI is a prescription medicine used to treat:
-
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
LIVMARLI® [liv-MAR-lee]
(maralixibat)
oral solutionThis Instructions for Use contains information on how to take LIVMARLI. Read this Instructions for Use before you start taking LIVMARLI for the first time and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. Follow your healthcare provider's instructions for the dose of LIVMARLI to give. Ask your healthcare provider or pharmacist if you have questions about how to prepare or give the prescribed dose of LIVMARLI. Important information about measuring LIVMARLI - Before you give LIVMARLI for the first time, talk to your healthcare provider or pharmacist about how to correctly measure your prescribed dose.
- You may be given 1 or more dosing dispensers of different sizes as shown in Table 1 below. Always use the correct dosing dispenser size provided with LIVMARLI based on your current prescribed dose.
Table 1 Dose (mL) Dosing dispenser size (mL) 0.1 to 0.5 0.5 0.6 to 1 1 1.25 to 3 3 - Your prescribed dose may change over time. Use the table above to choose the correct dosing dispenser size for your prescribed dose.
- If you do not have the correct dosing dispenser size for your prescribed dose, contact your healthcare provider or pharmacist.
- The dosing dispenser may be used for 100 days if cleaned correctly (see Section C). After 100 days, replace the dosing dispenser with a new one. A new replacement dosing dispenser may be used within the 100 days if necessary.
- Do not use a household teaspoon or any other dosing device to measure the dose.
- Do not open more than 1 bottle at a time.
- Do not give more than the prescribed dose.
- Do not use LIVMARLI after 100 days of first opening the bottle or after the throw away (discard) date listed on the pharmacy label (see Section D).
- When you start a new bottle of LIVMARLI, use a new dosing dispenser.
Storage information - Store unopened LIVMARLI at room temperature, between 68°F and 77°F (20°C and 25°C).
- After opening the LIVMARLI bottle, store below 86°F (30°C).
- Store the dosing dispenser in a clean, dry place when not in use.
Keep LIVMARLI and all medicines out of the reach of children. You will receive LIVMARLI oral solution Each package of LIVMARLI contains: LIVMARLI (9.5 mg/mL):
Dosing dispensers, provided separately by your pharmacist: 
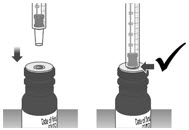
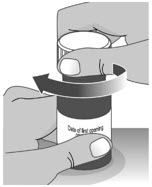
Note: Dosing dispenser sizes shown are for example only. Section A: Prepare the bottle 1. Remove the LIVMARLI bottle from the box (See Figure A). Note: The LIVMARLI bottle may not be in a box. 2. Write the date (bottle open date) on the LIVMARLI bottle (See Figure B). Note: The date of first opening or a throw away (discard) date may already be written on the bottle label by your pharmacist. 3. Remove the plastic seal from the bottle (See Figure C). Note: Your pharmacist may have already removed the plastic seal from the bottle. Section B: Prepare and give LIVMARLI Step 1: Draw the dose 1.1: Open the bottle by pushing down firmly on the child-resistant cap and turning the cap to the left (counter-clockwise) (See Figure D).
Do not throw away the child-resistant cap.1.2: If using a new dosing dispenser, remove the dosing dispenser from the wrapper (See Figure E). Throw away (dispose of) the wrapper in household trash.
If using a used dosing dispenser, make sure the dosing dispenser has been cleaned (See Section C).
If there is a cap on the dosing dispenser, remove it and throw away (dispose of) the cap into the household trash (See Figure F).
Important:- Make sure you use the correct dosing dispenser size for your prescribed dose (See Table 1).
- After 100 days, replace with a new dosing dispenser provided. A new replacement dosing dispenser may be used within the 100 days if necessary.
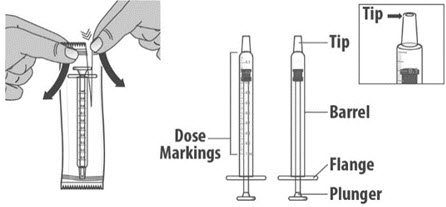
- Check the dosing dispenser for any damage to the barrel, plunger or tip (See Figure G). If you cannot see the dosage marking or if it becomes hard to move the plunger, replace with a new dosing dispenser.
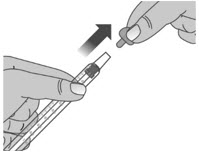
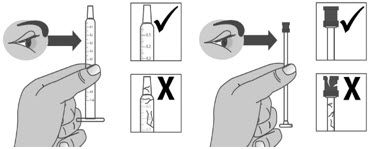
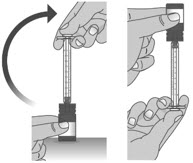
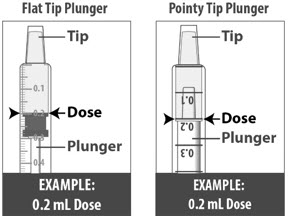
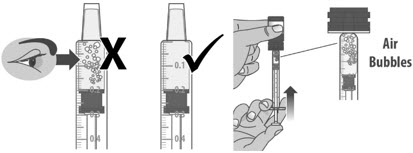
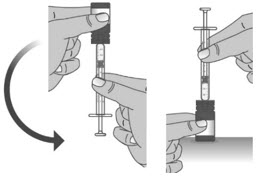
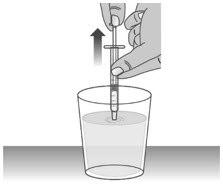
1.3: Push the plunger down fully to remove air from the dosing dispenser (See Figure H). 1.4: Make sure the cap is removed from bottle and insert the tip of the dosing dispenser into the bottle. The tip of the dosing dispenser should fit securely into the hole of the bottle (See Figure I). 1.5: Keep the dosing dispenser in place and turn the bottle upside down (See Figure J). 1.6: Pull back the plunger slowly until the top of the plunger is even with the marking on the barrel of the dosing dispenser for your prescribed dose of LIVMARLI (See Figure K).
See Figure L on how to align the plunger with your prescribed dose.
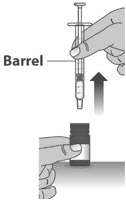
Note: The medicine should appear colorless to light yellow and clear. If it is not, do not use the medicine and contact your pharmacist.Note: Your dose may be different than the dose shown in the figures. 1.7: Check the dosing dispenser for air bubbles. If you see any air bubbles, fully push the plunger so that the medicine flows back into the bottle and withdraw the prescribed dose (See Figure M). 1.8: When you have measured the correct dose, leave the dosing dispenser in the bottle, and turn the bottle right side up (See Figure N). 1.9: Carefully remove the dosing dispenser from the bottle by pulling straight up on the barrel of the dosing dispenser (See Figure O).
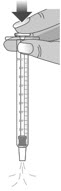
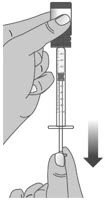
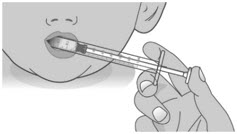
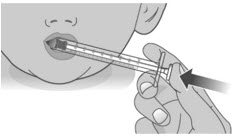
Do not push the dosing dispenser plunger during this step.Step 2: Give the dose Note: LIVMARLI should be taken while sitting up or standing. After taking LIVMARLI wait a few minutes before lying down. 2.1: Place the tip of the dosing dispenser against the inside of the cheek (See Figure P) and slowly push the plunger all the way in to give the entire dose of LIVMARLI (See Figure Q). 2.2: Swallow the dose.
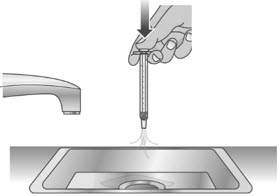
If you are not sure the entire dose was swallowed, do not give another dose. Wait until the next scheduled dose.2.3: Place the child-resistant cap back on the bottle and turn the cap to the right (clockwise) (See Figure R). Step 1: Rinse dosing dispenser 1.1: Fill a cup with water (See Figure S). 1.2: Clean the dosing dispenser by pulling back on the plunger slowly to fill the dosing dispenser with water from the cup (See Figure T). 1.3: Over a sink, push the water out of the dosing dispenser (See Figure U). Repeat several times to make sure that all of the LIVMARLI has been removed. Step 2: Dry the dosing dispenser 2.1: Remove the plunger from the barrel of the dosing dispenser by pulling the plunger and barrel away from each other (See Figure V). 2.2: Shake off excess water (See Figure W). 2.3: Place the plunger and barrel on a clean, dry paper towel to air dry. Store the dosing dispenser in a clean, dry place until your next dose (See Figure X). Before you give the next dose, put the dosing dispenser back together by pushing the plunger into the barrel (See Figure Y).
- Throw away (dispose of) the bottle of LIVMARLI following the steps below 100 days after first opened or after the throw away (discard) date listed on the pharmacy label, even if there is still medicine in it.
- Mix medicine with a substance such as dirt, cat litter, or used coffee grounds.
- Place the mixture in a container such as a sealed plastic bag.
- Delete all personal information on the prescription label of the empty medicine bottle, then throw away (dispose of) in the household trash or recycle the empty bottle.
- Throw away (dispose of) used dosing dispensers in the household trash.
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Revised:03/2023 LB00004v5 - PRINCIPAL DISPLAY PANEL - 30 mL Bottle Label
-
INGREDIENTS AND APPEARANCE
LIVMARLI
maralixibat chloride solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:79378-110 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength maralixibat chloride (UNII: V78M04F0XC) (maralixibat - UNII:UYB6UOF69L) maralixibat 9.5 mg in 1 mL Inactive Ingredients Ingredient Name Strength Propylene Glycol (UNII: 6DC9Q167V3) Sucralose (UNII: 96K6UQ3ZD4) Edetate Disodium (UNII: 7FLD91C86K) Water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:79378-110-01 1 in 1 CARTON 09/29/2021 1 30 mL in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA214662 09/29/2021 Labeler - Mirum Pharmaceuticals Inc. (116902386)