Label: TARCEVA- erlotinib hydrochloride tablet



-
Contains inactivated NDC Code(s)
NDC Code(s): 54868-5290-0, 54868-5447-0, 54868-5474-0 - Packager: Physicians Total Care, Inc.
- This is a repackaged label.
- Source NDC Code(s): 50242-062, 50242-063, 50242-064
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated January 5, 2012
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TARCEVA safely and effectively. See full prescribing information for TARCEVA. TARCEVA (erlotinib hydrochloride) tablet for oral use Initial U.S. Approval: 2004
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
TARCEVA is a tyrosine kinase inhibitor indicated for the treatment of:
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
-
Tablets: 25 mg, 100 mg and 150 mg. (3)
CONTRAINDICATIONS
None
(4)WARNINGS AND PRECAUTIONS
-
Interstitial Lung Disease (ILD)-like events, including fatalities have been infrequently reported. Interrupt TARCEVA if acute onset of new or progressive unexplained pulmonary symptoms, such as dyspnea, cough and fever occur. Discontinue TARCEVA if ILD is diagnosed. (5.1)
-
Monitor patients with hepatic impairment closely. Interrupt or discontinue TARCEVA if changes in liver function are severe (5.2)
-
Cases of hepatic failure and hepatorenal syndrome (including fatalities) have been reported. Monitor periodic liver function testing. Interrupt or discontinue TARCEVA if liver function changes are severe. (5.3)
-
Cases of acute renal failure (including fatalities), and renal insufficiency have been reported. Interrupt TARCEVA in the event of dehydration. Monitor renal function and electrolytes in patients at risk of dehydration. (5.4)
-
Myocardial infarction/ischemia has been reported, including fatalities, in patients with pancreatic cancer. (5.5)
-
Cerebrovascular accidents, including a fatality, have been reported in patients with pancreatic cancer. (5.6)
-
Microangiopathic Hemolytic Anemia with thrombocytopenia has been reported in patients with pancreatic cancer. (5.7)
-
Women should be advised to avoid pregnancy while on TARCEVA. Treatment should only be continued if the potential benefit to the mother outweighs the risk to the fetus. (5.8)
-
International Normalized Ratio (INR) elevations and bleeding events, some associated with concomitant warfarin administration have been reported. Monitor patients taking warfarin or other coumarin-derivative anticoagulants. (5.9)
ADVERSE REACTIONS
The most common adverse reactions (>50%) in NSCLC are rash, diarrhea, anorexia and fatigue. ( 6.1)
The most common adverse reactions (>50%) in pancreatic cancer are fatigue, rash, nausea and anorexia. ( 6.2)
To report SUSPECTED ADVERSE REACTIONS, contact OSI Pharmaceuticals Inc. at 1-800-572-1932
or FDA at 1-800-FDA-1088 or www.fda.gov/medwatchDRUG INTERACTIONS
- CYP3A4 inhibitors may increase erlotinib plasma concentrations. (7)
- CYP3A4 inducers may decrease erlotinib plasma concentrations. (7)
- CYP1A2 inducers may decrease erlotinib plasma concentrations. (7)
- Erlotinib solubility is pH dependent. Drugs that alter the pH of the upper GI tract may alter the solubility of erlotinib and hence its absorption. (7)
- Cigarette smoking decreases erlotinib plasma concentrations (7)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 1/2012
-
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
RECENT MAJOR CHANGES
1. INDICATIONS AND USAGE
1.1 Non-Small Cell Lung Cancer (NSCLC)
1.2 Pancreatic Cancer
2. DOSAGE AND ADMINISTRATION
2.1 Recommended Dose - NSCLC
2.2 Recommended Dose - Pancreatic Cancer
2.3 Dose Modifications
3. DOSAGE FORMS AND STRENGTHS
4. CONTRAINDICATIONS
5. WARNINGS AND PRECAUTIONS
5.1 Pulmonary Toxicity
5.2 Patients with Hepatic Impairment
5.3 Hepatotoxicity
5.4 Renal Failure
5.5 Myocardial infarction/ischemia
5.6 Cerebrovascular accident
5.7 Microangiopathic Hemolytic Anemia with Thrombocytopenia
5.8 Use in Pregnancy
5.9 Elevated International Normalized Ratio and Potential Bleeding
6. ADVERSE REACTIONS
6.1 Non-Small Cell Lung Cancer
6.2 Pancreatic Cancer
6.3 NSCLC and Pancreatic Cancer Indications
7. DRUG INTERACTIONS
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Gender
8.7 Race
8.8 Patients with Hepatic Impairment
8.9 Patients with Renal Impairment
10. OVERDOSAGE
11. DESCRIPTION
12. CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13. NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14. CLINICAL STUDIES
14.1 Non-Small Cell Lung Cancer NSCLC – TARCEVA Administered as a Single Agent
14.2 NSCLC - TARCEVA Administered Concurrently with Chemotherapy
14.3 Pancreatic Cancer - TARCEVA Administered Concurrently with Gemcitabine
16. HOW SUPPLIED/STORAGE AND HANDLING
17. PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1. INDICATIONS AND USAGE
Enter section text here
1.1 Non-Small Cell Lung Cancer (NSCLC)
TARCEVA monotherapy is indicated for the treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of at least one prior chemotherapy regimen [see Clinical Studies (14.1)].
Results from two, multicenter, placebo-controlled, randomized, Phase 3 trials conducted in first-line patients with locally advanced or metastatic NSCLC showed no clinical benefit with the concurrent administration of TARCEVA with platinum-based chemotherapy [carboplatin and paclitaxel or gemcitabine and cisplatin] and its use is not recommended in that setting [see Clinical Studies (14.3)].
1.2 Pancreatic Cancer
TARCEVA in combination with gemcitabine is indicated for the first-line treatment of patients with locally advanced, unresectable or metastatic pancreatic cancer [see Clinical Studies (14.3)].
-
2. DOSAGE AND ADMINISTRATION
Enter section text here
2.1 Recommended Dose - NSCLC
The recommended daily dose of TARCEVA for non-small cell lung cancer is 150 mg taken at least one hour before or two hours after the ingestion of food. Treatment should continue until disease progression or unacceptable toxicity occurs. There is no evidence that treatment beyond progression is beneficial.
2.2 Recommended Dose - Pancreatic Cancer
The recommended daily dose of TARCEVA for pancreatic cancer is 100 mg taken at least one hour before or two hours after the ingestion of food, in combination with gemcitabine (see the gemcitabine package insert). Treatment should continue until disease progression or unacceptable toxicity occurs.
2.3 Dose Modifications
In patients who develop an acute onset of new or progressive pulmonary symptoms, such as dyspnea, cough or fever, treatment with TARCEVA should be interrupted pending diagnostic evaluation. If Interstitial Lung Disease (ILD) is diagnosed, TARCEVA should be discontinued and appropriate treatment instituted as necessary [see Warnings and Precautions (5.1)].
Diarrhea can usually be managed with loperamide. Patients with severe diarrhea who are unresponsive to loperamide or who become dehydrated may require dose reduction or temporary interruption of therapy. Patients with severe skin reactions may also require dose reduction or temporary interruption of therapy.
When dose reduction is necessary, the TARCEVA dose should be reduced in 50 mg decrements.
In patients who are taking TARCEVA with a strong CYP3A4 inhibitor such as, but not limited to, atazanavir, clarithromycin, indinavir, itraconazole, ketoconazole, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, troleandomycin (TAO), voriconazole, or grapefruit or grapefruit juice, a dose reduction should be considered if severe adverse reactions occur. Similarly, in patients who are taking TARCEVA with an inhibitor of both CYP3A4 and CYP1A2 like ciprofloxacin, a dose reduction of TARCEVA should be considered if severe adverse reactions occur. [see Drug Interactions (7)].
Pre-treatment with the CYP3A4 inducer rifampicin decreased erlotinib AUC by about 2/3 to 4/5. Use of alternative treatments lacking CYP3A4 inducing activity is strongly recommended. If an alternative treatment is unavailable, an increase in the dose of TARCEVA should be considered as tolerated at two week intervals while monitoring the patient’s safety. The maximum dose of TARCEVA studied in combination with rifampicin is 450 mg. If the TARCEVA dose is adjusted upward, the dose will need to be reduced immediately to the indicated starting dose upon discontinuation of rifampicin or other inducers. Other CYP3A4 inducers include, but are not limited to rifabutin, rifapentine, phenytoin, carbamazepine, phenobarbital and St. John's Wort. These too should be avoided if possible [see Drug Interactions (7)].
Cigarette smoking has been shown to reduce erlotinib exposure. Patients should be advised to stop smoking. If a patient continues to smoke, a cautious increase in the dose of TARCEVA, not exceeding 300 mg may be considered, while monitoring the patient’s safety. However, efficacy and long-term safety (> 14 days) of a dose higher than the recommended starting doses have not been established in patients who continue to smoke cigarettes. If the TARCEVA dose is adjusted upward, the dose should be reduced immediately to the indicated starting dose upon cessation of smoking [see Clinical Pharmacology (12.3)].
Erlotinib is eliminated by hepatic metabolism and biliary excretion. Although erlotinib exposure was similar in patients with moderately impaired hepatic function (Child-Pugh B), patients with hepatic impairment (total bilirubin > ULN or Child-Pugh A, B and C) should be closely monitored during therapy with TARCEVA [see WARNINGS and PRECAUTIONS (5.2 )]. Treatment with TARCEVA should be used with extra caution in patients with total bilirubin > 3 x ULN. TARCEVA dosing should be interrupted or discontinued if changes in liver function are severe such as doubling of total bilirubin and/or tripling of transaminases in the setting of pretreatment values outside normal range. In the setting of worsening liver function tests, before they become severe, dose interruption and/or dose reduction with frequent liver function test monitoring should be considered. TARCEVA dosing should be interrupted or discontinued if total bilirubin is >3 x ULN and/or transaminases are >5 x ULN in the setting of normal pretreatment values [see Warnings and Precautions (5.2 , 5.3 ), Adverse Reactions (6.3) and Use in Specific Populations (8.6 )].
-
3. DOSAGE FORMS AND STRENGTHS
25 mg tablets
White film-coated tablets for daily oral administration. Round, biconvex face and straight sides, white film-coated, printed in orange with a “T” and “25” on one side and plain on the other side.
100 mg tablets
White film-coated tablets for daily oral administration. Round, biconvex face and straight sides, white film-coated, printed in gray with “T” and “100” on one side and plain on the other side.
150 mg tablets
White film-coated tablets for daily oral administration. Round, biconvex face and straight sides, white film-coated, printed in maroon with “T” and “150” on one side and plain on the other side.
- 4. CONTRAINDICATIONS
-
5. WARNINGS AND PRECAUTIONS
Enter section text here
5.1 Pulmonary Toxicity
There have been infrequent reports of serious Interstitial Lung Disease (ILD)-like events, including fatalities, in patients receiving TARCEVA for treatment of NSCLC, pancreatic cancer or other advanced solid tumors. In the randomized single-agent NSCLC study [see CLINICAL STUDIES (14.1)], the incidence of ILD-like events (0.8%) was the same in both the placebo and TARCEVA groups. In the pancreatic cancer study - in combination with gemcitabine – [see Clinical Studies (14.3)], the incidence of ILD-like events was 2.5% in the TARCEVA plus gemcitabine group vs. 0.4% in the placebo plus gemcitabine group.
The overall incidence of ILD-like events in approximately 4900 TARCEVA-treated patients from all studies (including uncontrolled studies and studies with concurrent chemotherapy) was approximately 0.7%. Reported diagnoses in patients suspected of having ILD-like events included pneumonitis, radiation pneumonitis, hypersensitivity pneumonitis, interstitial pneumonia, interstitial lung disease, obliterative bronchiolitis, pulmonary fibrosis, Acute Respiratory Distress Syndrome and lung infiltration. Symptoms started from 5 days to more than 9 months (median 39 days) after initiating TARCEVA therapy. In the lung cancer trials most of the cases were associated with confounding or contributing factors such as concomitant/prior chemotherapy, prior radiotherapy, pre-existing parenchymal lung disease, metastatic lung disease, or pulmonary infections.
In the event of an acute onset of new or progressive unexplained pulmonary symptoms such as dyspnea, cough, and fever, TARCEVA therapy should be interrupted pending diagnostic evaluation. If ILD is diagnosed, TARCEVA should be discontinued and appropriate treatment instituted as needed [see Dosage and Administration (2.3 )].
5.2 Patients with Hepatic Impairment
In a pharmacokinetic study in patients with moderate hepatic impairment (Child-Pugh B) associated with significant liver tumor burden, 10 out of 15 patients died on treatment or within 30 days of the last TARCEVA dose. One patient died from hepatorenal syndrome, 1 patient died from rapidly progressing liver failure and the remaining 8 patients died from progressive disease. Six out of the 10 patients who died had baseline total bilirubin > 3 x ULN suggesting severe hepatic impairment. Treatment with TARCEVA should be used with extra caution in patients with total bilirubin > 3 x ULN. Patients with hepatic impairment (total bilirubin > ULN or Child-Pugh A, B and C) should be closely monitored during therapy with TARCEVA. TARCEVA dosing should be interrupted or discontinued if changes in liver function are severe such as doubling of total bilirubin and/or tripling of transaminases in the setting of pretreatment values outside normal range [see Clinical Pharmacology (12.3) and Dosage and Administration (2.3 )].
5.3 Hepatotoxicity
Cases of hepatic failure and hepatorenal syndrome (including fatalities) have been reported during use of TARCEVA, particularly in patients with baseline hepatic impairment. Therefore, periodic liver function testing (transaminases, bilirubin, and alkaline phosphatase) is recommended. In the setting of worsening liver function tests, dose interruption and/or dose reduction with frequent liver function test monitoring should be considered. TARCEVA dosing should be interrupted or discontinued if total bilirubin is >3 x ULN and/or transaminases are >5 x ULN in the setting of normal pretreatment values [see Adverse Reactions (6.3) and Dosage and Administration (2.3 )].
5.4 Renal Failure
Cases of hepatorenal syndrome, acute renal failure (including fatalities), and renal insufficiency have been reported. Some were secondary to baseline hepatic impairment while others were associated with severe dehydration due to diarrhea, vomiting, and/or anorexia or concurrent chemotherapy use. In the event of dehydration, particularly in patients with contributing risk factors for renal failure (eg, pre-existing renal disease, medical conditions or medications that may lead to renal disease, or other predisposing conditions including advanced age), TARCEVA therapy should be interrupted and appropriate measures should be taken to intensively rehydrate the patient. Periodic monitoring of renal function and serum electrolytes is recommended in patients at risk of dehydration [see Adverse Reactions (6.3) and Dosage and Administration (2.3 )].
5.5 Myocardial infarction/ischemia
In the pancreatic carcinoma trial, six patients (incidence of 2.3%) in the TARCEVA/gemcitabine group developed myocardial infarction/ischemia. One of these patients died due to myocardial infarction. In comparison, 3 patients in the placebo/gemcitabine group developed myocardial infarction (incidence 1.2%) and one died due to myocardial infarction.
5.6 Cerebrovascular accident
In the pancreatic carcinoma trial, six patients in the TARCEVA/gemcitabine group developed cerebrovascular accidents (incidence: 2.3%). One of these was hemorrhagic and was the only fatal event. In comparison, in the placebo/gemcitabine group there were no cerebrovascular accidents.
5.7 Microangiopathic Hemolytic Anemia with Thrombocytopenia
In the pancreatic carcinoma trial, two patients in the TARCEVA/gemcitabine group developed microangiopathic hemolytic anemia with thrombocytopenia (incidence: 0.8%). Both patients received TARCEVA and gemcitabine concurrently. In comparison, in the placebo/gemcitabine group there were no cases of microangiopathic hemolytic anemia with thrombocytopenia.
5.8 Use in Pregnancy
Pregnancy Category D
Women of childbearing potential should avoid becoming pregnant while being treated with TARCEVA. Erlotinib administered to rabbits during organogenesis at doses that result in plasma drug concentrations of approximately 3 times those in humans (AUCs at 150 mg daily dose) was associated with embryo/fetal lethality and abortion. When erlotinib was administered to female rats prior to mating and through the first week of pregnancy, at doses 0.3 or 0.7 times the clinical dose of 150 mg, on a mg/m2 basis, there was an increase in early resorptions that resulted in a decrease in the number of live fetuses. [see Use in Specific Populations (8.1)]
5.9 Elevated International Normalized Ratio and Potential Bleeding
International Normalized Ratio (INR) elevations and infrequent reports of bleeding events including gastrointestinal and non-gastrointestinal bleedings have been reported in clinical studies, some associated with concomitant warfarin administration. Patients taking warfarin or other coumarin-derivative anticoagulants should be monitored regularly for changes in prothrombin time or INR. [see Adverse Reactions (6.3)].
-
6. ADVERSE REACTIONS
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Safety evaluation of TARCEVA is based on 856 cancer patients who received TARCEVA as monotherapy, 308 patients who received TARCEVA 100 or 150 mg plus gemcitabine, and 1228 patients who received TARCEVA concurrently with other chemotherapies.
There have been reports of serious events, including fatalities, in patients receiving TARCEVA for treatment of NSCLC, pancreatic cancer or other advanced solid tumors [see Warnings and Precautions (5) and Dosage and Administration (2.3 )].
6.1 Non-Small Cell Lung Cancer
Adverse events, regardless of causality, that occurred in at least 10% of patients treated with single-agent TARCEVA at 150 mg and at least 3% more often than in the placebo group in the randomized trial of patients with NSCLC are summarized by NCI-CTC (version 2.0) Grade in Table 1.
The most common adverse reactions in patients receiving single-agent TARCEVA 150 mg were rash and diarrhea. Grade 3/4 rash and diarrhea occurred in 9% and 6%, respectively, in TARCEVA-treated patients. Rash and diarrhea each resulted in study discontinuation in 1% of TARCEVA-treated patients. Six percent and 1% of patients needed dose reduction for rash and diarrhea, respectively. The median time to onset of rash was 8 days, and the median time to onset of diarrhea was 12 days.
Table 1: Adverse Reactions Occurring More Frequently (≥ 3%) in the Single Agent TARCEVA Group than in the Placebo Group and in ≥10% of Patients in the TARCEVA Group. TARCEVA 150 mg
N = 485Placebo
N = 242NCI-CTC Grade Any Grade Grade 3 Grade 4 Any Grade Grade 3 Grade 4 MedDRA Preferred Term % % % % % % Rash 75 8 <1 17 0 0 Diarrhea 54 6 <1 18 <1 0 Anorexia 52 8 1 38 5 <1 Fatigue 52 14 4 45 16 4 Dyspnea 41 17 11 35 15 11 Cough 33 4 0 29 2 0 Nausea 33 3 0 24 2 0 Infection 24 4 0 15 2 0 Vomiting 23 2 <1 19 2 0 Stomatitis 17 <1 0 3 0 0 Pruritus 13 <1 0 5 0 0 Dry skin 12 0 0 4 0 0 Conjunctivitis 12 <1 0 2 <1 0 Keratoconjunctivitis sicca 12 0 0 3 0 0 Abdominal pain 11 2 <1 7 1 <1 Liver function test abnormalities (including elevated alanine aminotransferase (ALT), aspartate aminotransferase (AST) and bilirubin) were observed in patients receiving single-agent TARCEVA 150 mg. These elevations were mainly transient or associated with liver metastases. Grade 2 (>2.5 – 5.0 x ULN) ALT elevations occurred in 4% and <1% of TARCEVA and placebo treated patients, respectively. Grade 3 (>5.0 – 20.0 x ULN) elevations were not observed in TARCEVA-treated patients. TARCEVA dosing should be interrupted or discontinued if changes in liver function are severe. [see Dosage and Administration (2.3)]
6.2 Pancreatic Cancer
Adverse events, regardless of causality, that occurred in at least 10% of patients treated with TARCEVA 100 mg plus gemcitabine in the randomized trial of patients with pancreatic cancer are summarized by NCI-CTC (version 2.0) Grade in Table 2.
The most common adverse reactions in pancreatic cancer patients receiving TARCEVA 100 mg plus gemcitabine were fatigue, rash, nausea, anorexia and diarrhea. In the TARCEVA plus gemcitabine arm, Grade 3/4 rash and diarrhea were each reported in 5% of TARCEVA plus gemcitabine-treated patients. The median time to onset of rash and diarrhea was 10 days and 15 days, respectively. Rash and diarrhea each resulted in dose reductions in 2% of patients, and resulted in study discontinuation in up to 1% of patients receiving TARCEVA plus gemcitabine. The 150 mg cohort was associated with a higher rate of certain class-specific adverse reactions including rash and required more frequent dose reduction or interruption.
Table 2: Adverse Reactions Occurring in ≥ 10% of TARCEVA-treated Pancreatic Cancer Patients: 100 mg cohort *Includes all MedDRA preferred terms in the Infections and Infestations System Organ Class TARCEVA + Gemcitabine
1000 mg/m2 IV
N=259Placebo + Gemcitabine
1000 mg/m2 IV
N=256NCI-CTC Grade Any Grade Grade 3 Grade 4 Any Grade Grade 3 Grade 4 MedDRA Preferred Term % % % % % % Fatigue 73 14 2 70 13 2 Rash 69 5 0 30 1 0 Nausea 60 7 0 58 7 0 Anorexia 52 6 <1 52 5 <1 Diarrhea 48 5 <1 36 2 0 Abdominal pain 46 9 <1 45 12 <1 Vomiting 42 7 <1 41 4 <1 Weight decreased 39 2 0 29 <1 0 Infection* 39 13 3 30 9 2 Edema 37 3 <1 36 2 <1 Pyrexia 36 3 0 30 4 0 Constipation 31 3 1 34 5 1 Bone pain 25 4 <1 23 2 0 Dyspnea 24 5 <1 23 5 0 Stomatitis 22 <1 0 12 0 0 Myalgia 21 1 0 20 <1 0 Depression 19 2 0 14 <1 0 Dyspepsia 17 <1 0 13 <1 0 Cough 16 0 0 11 0 0 Dizziness 15 <1 0 13 0 <1 Headache 15 <1 0 10 0 0 Insomnia 15 <1 0 16 <1 0 Alopecia 14 0 0 11 0 0 Anxiety 13 1 0 11 <1 0 Neuropathy 13 1 <1 10 <1 0 Flatulence 13 0 0 9 <1 0 Rigors 12 0 0 9 0 0 In the pancreatic carcinoma trial, 10 patients in the TARCEVA/gemcitabine group developed deep venous thrombosis (incidence: 3.9%). In comparison, 3 patients in the placebo/gemcitabine group developed deep venous thrombosis (incidence 1.2%). The overall incidence of grade 3 or 4 thrombotic events, including deep venous thrombosis, was similar in the two treatment arms: 11% for TARCEVA plus gemcitabine and 9% for placebo plus gemcitabine.
No differences in Grade 3 or Grade 4 hematologic laboratory toxicities were detected between the TARCEVA plus gemcitabine group compared to the placebo plus gemcitabine group.
Severe adverse events (≥grade 3 NCI-CTC) in the TARCEVA plus gemcitabine group with incidences < 5% included syncope, arrhythmias, ileus, pancreatitis, hemolytic anemia including microangiopathic hemolytic anemia with thrombocytopenia, myocardial infarction/ischemia, cerebrovascular accidents including cerebral hemorrhage, and renal insufficiency [see Warnings and Precautions (5)].
Liver function test abnormalities (including elevated alanine aminotransferase (ALT), aspartate aminotransferase (AST) and bilirubin) have been observed following the administration of TARCEVA plus gemcitabine in patients with pancreatic cancer. Table 3 displays the most severe NCI-CTC grade of liver function abnormalities that developed. TARCEVA dosing should be interrupted or discontinued if changes in liver function are severe [see Dosage and Administration (2.3)].
Table 3: Liver Function Test Abnormalities (most severe NCI-CTC grade) in Pancreatic Cancer Patients: 100 mg Cohort TARCEVA + Gemcitabine 1000 mg/m2 IV
N = 259Placebo + Gemcitabine 1000 mg/m2 IV
N = 256NCI-CTC Grade Grade 2 Grade 3 Grade 4 Grade 2 Grade 3 Grade 4 Bilirubin 17 % 10% <1% 11% 10% 3% ALT 31% 13% <1% 22% 9% 0% AST 24% 10% <1% 19% 9% 0% 6.3 NSCLC and Pancreatic Cancer Indications
During the NSCLC and the combination pancreatic cancer trials, infrequent cases of gastrointestinal bleeding have been reported, some associated with concomitant warfarin or NSAID administration. [see Warnings and Precautions (5.9)]. These adverse events were reported as peptic ulcer bleeding (gastritis, gastroduodenal ulcers), hematemesis, hematochezia, melena and hemorrhage from possible colitis. Cases of acute renal failure or renal insufficiency, including fatalities, with or without hypokalemia have been reported. [see Warnings and Precautions (5.4)]. Cases of Grade 1 epistaxis were also reported in both the single-agent NSCLC and the pancreatic cancer clinical trials.
NCI-CTC Grade 3 conjunctivitis and keratitis have been reported infrequently in patients receiving TARCEVA therapy in the NSCLC and pancreatic cancer clinical trials. Corneal ulcerations may also occur. [see Patient Counseling Information (17)].
Hair and nail disorders including alopecia, hirsutism, eyelash/eyebrow changes, paronychia and brittle and loose nails have been reported in clinical trials and during post-marketing use of TARCEVA.
In patients who develop skin rash, the appearance of the rash is typically erythematous and maculopapular and it may resemble acne with follicular pustules, but is histopathologically different. This skin reaction commonly occurs on the face, upper chest and back, but may be more generalized or severe (NCI-CTC Grade 3 or 4) with desquamation. Associated symptoms may include itching, tenderness and/or burning. Dry skin with or without digital skin fissures may occur.
Hepatic failure has been reported in patients treated with single-agent TARCEVA or TARCEVA combined with chemotherapy in clinical studies and during post-marketing use of TARCEVA [see Warnings and Precautions (5.3 )]; it is not possible to reliably estimate the frequency or establish a causal relationship to TARCEVA treatment.
In general, no notable differences in the safety of TARCEVA monotherapy or in combination with gemcitabine could be discerned between females or males and between patients younger or older than the age of 65 years [see Use in Specific Populations (8.4)]. The safety of TARCEVA appears similar in Caucasian and Asian patients.
-
7. DRUG INTERACTIONS
Erlotinib is metabolized predominantly by CYP3A4, and inhibitors of CYP3A4 would be expected to increase exposure. Co-treatment with the potent CYP3A4 inhibitor ketoconazole increases erlotinib AUC by 2/3. When TARCEVA was co-administered with ciprofloxacin, an inhibitor of both CYP3A4 and CYP1A2, the erlotinib exposure [AUC] and maximum concentration [Cmax] increased by 39% and 17% respectively. Caution should be used when administering or taking TARCEVA with ketoconazole and other strong CYP3A4 inhibitors such as, but not limited to, atazanavir, clarithromycin, indinavir, itraconazole, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, troleandomycin (TAO), voriconazole and grapefruit or grapefruit juice. [see Dosage and Administration (2.3 )].
Pre-treatment with the CYP3A4 inducer rifampicin for 7 days prior to TARCEVA decreased erlotinib AUC by about 2/3 to 4/5, which is equivalent to a dose of about 30 to 50 mg in NSCLC patients. In a separate study, treatment with rifampicin for 11 days, with co-administration of a single 450 mg dose of TARCEVA on day 8 resulted in a mean erlotinib exposure (AUC) that was 57.6% of that observed following a single 150 mg TARCEVA dose in the absence of rifampicin treatment [see Dose Modifications (2.3 )]. Use of alternative treatments lacking CYP3A4 inducing activity is strongly recommended. If an alternative treatment is unavailable, adjusting the starting dose should be considered. If the TARCEVA dose is adjusted upward, the dose will need to be reduced immediately to the indicated starting dose upon discontinuation of rifampicin or other inducers. Other CYP3A4 inducers include, but are not limited to, rifabutin, rifapentine, phenytoin, carbamazepine, phenobarbital and St. John's Wort [see Dosage and Administration (2.3 )].
Cigarette smoking has been shown to reduce erlotinib AUC. Patients should be advised to stop smoking; however, if they continue to smoke, a cautious increase in the dose of TARCEVA may be considered, while monitoring the patient’s safety. If the TARCEVA dose is adjusted upward, the dose should be reduced immediately to the indicated starting dose upon cessation of smoking. [see Dosage and Administration(2.3 ) and Clinical Pharmacology(12.3)].
Pretreatment and co-administration of TARCEVA decreased the AUC of CYP3A4 substrate, midazolam, by 24%. The mechanism is not clear.
In a study, there were no significant effects of gemcitabine on the pharmacokinetics of erlotinib nor were there significant effects of erlotinib on the pharmacokinetics of gemcitabine.
Drugs that alter the pH of the upper GI tract may alter the solubility of erlotinib and reduce its bioavailability. Co-administration of TARCEVA with omeprazole, a proton pump inhibitor, decreased the erlotinib AUC by 46%. Increasing the dose of TARCEVA when co-administered with such agents is not likely to compensate for the loss of exposure. Since proton pump inhibitors affect pH of the upper GI tract for an extended period, separation of doses may not eliminate the interaction. The concomitant use of proton pump inhibitors with TARCEVA should be avoided if possible. The use of antacids may be considered in place of histamine 2 receptor blockers (H2 blockers) or proton pump inhibitors in patients receiving TARCEVA. However, no clinical study has been conducted to evaluate the effect of antacids on erlotinib pharmacokinetics. If an antacid is necessary, the antacid dose and the TARCEVA dose should be separated by several hours [see Clinical Pharmacology (12.3)].
-
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category D [See Warnings and Precautions (5.8)]
Erlotinib has been shown to cause maternal toxicity with associated embryo/fetal lethality and abortion in rabbits when given at doses that result in plasma drug concentrations of approximately 3 times those in humans (AUCs at 150 mg daily dose). When given during the period of organogenesis to achieve plasma drug concentrations approximately equal to those in humans, based on AUC, there was no increased incidence of embryo/fetal lethality or abortion in rabbits or rats. However, female rats treated with 30 mg/m2/day or 60 mg/m2/day (0.3 or 0.7 times the clinical dose, on a mg/m2 basis) of erlotinib prior to mating through the first week of pregnancy had an increase in early resorptions that resulted in a decrease in the number of live fetuses.
No teratogenic effects were observed in rabbits or rats dosed with erlotinib during organogenesis at doses up to 600 mg/m2/day in the rabbit (3 times the plasma drug concentration seen in humans at 150 mg/day) and up to 60 mg/m2/day in the rat (0.7 times the clinical dose of 150 mg/day on a mg/m2 basis).
There are no adequate and well-controlled studies in pregnant women using TARCEVA. Women of childbearing potential should be advised to avoid pregnancy while on TARCEVA. Adequate contraceptive methods should be used during therapy, and for at least 2 weeks after completing therapy. Treatment should only be continued in pregnant women if the potential benefit to the mother outweighs the risk to the fetus. If TARCEVA is used during pregnancy, the patient should be apprised of the potential hazard to the fetus or potential risk for loss of the pregnancy [see Warnings and Precautions (5.8)].
8.3 Nursing Mothers
It is not known whether erlotinib is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from TARCEVA, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
The safety and effectiveness of TARCEVA in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of patients participating in the randomized NSCLC trial, 62% were less than 65 years of age, and 38% of patients were aged 65 years or older. The survival benefit was maintained across both age groups [HR = 0.75 (95% CI: 0.6, 0.9) in patients less than 65 years of age, and HR = 0.79 (95% CI: 0.6, 1.0) in patients who were 65 years or older].
In the pancreatic cancer study, 53% of patients were younger than 65 years of age and 47% were 65 years of age or older. There were no clinically relevant differences between the age groups [HR = 0.78 (95% CI: 0.6, 1.0) in patients less than 65 years of age, and HR = 0.94 (95% CI: 0.7, 1.2) in patients who were 65 years or older]. No meaningful differences in safety or pharmacokinetics were observed between younger and older patients in either study. Therefore, no dosage adjustments are recommended in elderly patients.
8.6 Gender
Of the total number of patients participating in the randomized NSCLC trial, 65% were males and 35% females. There were no clinically relevant differences in safety and efficacy based on gender [HR = 0.76 (95% CI: 0.6, 0.9) in males and HR = 0.80 (95% CI: 0.6, 1.1) in females].
In the pancreatic cancer study, 52% of patients were males and 48% females. There were no clinically relevant differences in safety and efficacy based on gender [HR = 0.74 (95% CI: 0.6, 0.9) in males and HR = 1.0 (95% CI: 0.8, 1.3) in females].
8.7 Race
In the randomized NSCLC trial, 78% of all patients were Caucasian and 12% were Asian. There were no clinically relevant differences in safety and efficacy based on race [HR = 0.79 (95% CI: 0.6, 1.0) in Caucasians and HR = 0.61 (95% CI: 0.4, 1.0) in Asians].
In the pancreatic cancer study, 88% of all patients were Caucasian and 7% were Asian. There were no clinically relevant differences in safety and efficacy based on race [HR = 0.88 (95% CI: 0.7, 1.1) in Caucasians and HR = 0.61 (95% CI: 0.3, 1.3) in Asians].
8.8 Patients with Hepatic Impairment
Patients with hepatic impairment (total bilirubin > ULN or Child Pugh A, B and C) should be closely monitored during therapy with TARCEVA. Treatment with TARCEVA should be used with extra caution in patients with total bilirubin > 3 x ULN [see Warnings (5.2 ), Adverse Reactions (6.3), and Dosage and Administration (2.3 )].
In vitro and in vivo evidence suggest that erlotinib is cleared primarily by the liver. However, erlotinib exposure was similar in patients with moderately impaired hepatic function (Child-Pugh B) compared with patients with adequate hepatic function including patients with primary liver cancer or hepatic metastases [see Dosage and Administration (2.3 ) and Clinical Pharmacology (12.3)].
-
10. OVERDOSAGE
Single oral doses of TARCEVA up to 1,000 mg in healthy subjects and weekly doses up to 1,600 mg in cancer patients have been tolerated. Repeated twice-daily doses of 200 mg single-agent TARCEVA in healthy subjects were poorly tolerated after only a few days of dosing. Based on the data from these studies, an unacceptable incidence of severe adverse events, such as diarrhea, rash, and liver transaminase elevation, may occur above the recommended dose [see Dosage and Administration (2)]. In case of suspected overdose, TARCEVA should be withheld and symptomatic treatment instituted.
-
11. DESCRIPTION
TARCEVA (erlotinib), a kinase inhibitor, is a quinazolinamine with the chemical name N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine. TARCEVA contains erlotinib as the hydrochloride salt that has the following structural formula:
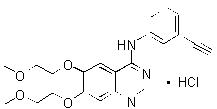
Erlotinib hydrochloride has the molecular formula C22H23N3O4.HCl and a molecular weight of 429.90. The molecule has a pKa of 5.42 at 25oC. Erlotinib hydrochloride is very slightly soluble in water, slightly soluble in methanol and practically insoluble in acetonitrile, acetone, ethyl acetate and hexane.
Aqueous solubility of erlotinib hydrochloride is dependent on pH with increased solubility at a pH of less than 5 due to protonation of the secondary amine. Over the pH range of 1.4 to 9.6, maximal solubility of approximately 0.4 mg/mL occurs at a pH of approximately 2.
TARCEVA tablets for oral administration are available in three dosage strengths containing erlotinib hydrochloride (27.3 mg, 109.3 mg and 163.9 mg) equivalent to 25 mg, 100 mg and 150 mg erlotinib and the following inactive ingredients: lactose monohydrate, hypromellose, hydroxypropyl cellulose, magnesium stearate, microcrystalline cellulose, sodium starch glycolate, sodium lauryl sulfate and titanium dioxide. The tablets also contain trace amounts of color additives, including FD&C Yellow #6 (25 mg only) for product identification.
-
12. CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism of clinical antitumor action of erlotinib is not fully characterized. Erlotinib inhibits the intracellular phosphorylation of tyrosine kinase associated with the epidermal growth factor receptor (EGFR). Specificity of inhibition with regard to other tyrosine kinase receptors has not been fully characterized. EGFR is expressed on the cell surface of normal cells and cancer cells.
12.3 Pharmacokinetics
Absorption and Distribution:
Erlotinib is about 60% absorbed after oral administration and its bioavailability is substantially increased by food to almost 100%. Peak plasma levels occur 4 hours after dosing. The solubility of erlotinib is pH dependent. Erlotinib solubility decreases as pH increases. Co-administration of TARCEVA with omeprazole, a proton pump inhibitor, decreased the erlotinib exposure [AUC] and maximum concentration [Cmax] by 46% and 61% respectively [see Drug Interactions (7)].
Following absorption, erlotinib is approximately 93% protein bound to plasma albumin and alpha-1 acid glycoprotein (AAG). Erlotinib has an apparent volume of distribution of 232 liters.
Metabolism and Excretion:
A population pharmacokinetic analysis in 591 patients receiving single-agent TARCEVA showed a median half-life of 36.2 hours. Time to reach steady state plasma concentration would therefore be 7 – 8 days. No significant relationships of clearance to covariates of patient age, body weight or gender were observed. Smokers had a 24% higher rate of erlotinib clearance.
A second population pharmacokinetic analysis was conducted that incorporated erlotinib data from 204 pancreatic cancer patients who received erlotinib plus gemcitabine. This analysis demonstrated that covariates affecting erlotinib clearance in patients from the pancreatic study were very similar to those seen in the prior single-agent pharmacokinetic analysis. No new covariate effects were identified. Co-administration of gemcitabine had no effect on erlotinib plasma clearance.
In vitro assays of cytochrome P450 metabolism showed that erlotinib is metabolized primarily by CYP3A4 and to a lesser extent by CYP1A2, and the extrahepatic isoform CYP1A1. Following a 100 mg oral dose, 91% of the dose was recovered: 83% in feces (1% of the dose as intact parent) and 8% in urine (0.3% of the dose as intact parent).
Cigarette smoking reduces erlotinib exposure. In the Phase 3 NSCLC trial, current smokers achieved erlotinib steady-state trough plasma concentrations which was approximately 2-fold less than the former smokers or patients who had never smoked. This effect was accompanied by a 24% increase in apparent erlotinib plasma clearance. In a separate study which evaluated the single-dose pharmacokinetics of erlotinib in healthy volunteers, current smokers cleared the drug faster than former smokers or volunteers who had never smoked. The AUC0-infinity in smokers was about 1/3 to 1/2 of that in never/former smokers. In another study which was conducted in NSCLC patients (N=35) who were current smokers, pharmacokinetic analyses at steady-state indicated a dose-proportional increase in erlotinib exposure when the TARCEVA dose was increased from 150 mg to 300 mg. However, the exact dose to be recommended for patients who currently smoke is unknown [see Drug Interactions (7) and Patient Counseling Information (17)].
Special Populations:
Patients with Hepatic Impairment
Patients with hepatic impairment (total bilirubin > ULN or Child Pugh A, B and C) should be closely monitored during therapy with TARCEVA. Treatment with TARCEVA should be used with extra caution in patients with total bilirubin > 3 x ULN [see Warnings and Precautions (5.2 ), Adverse Reactions (6.3), and Dosage and Administration (2.3 )].
In vitro and in vivo evidence suggest that erlotinib is cleared primarily by the liver. However, erlotinib exposure was similar in patients with moderately impaired hepatic function (Child-Pugh B) compared with patients with adequate hepatic function including patients with primary liver cancer or hepatic metastases.
Patients with Renal Impairment
Less than 9% of a single dose is excreted in the urine. No clinical studies have been conducted in patients with compromised renal function.
-
13. NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Erlotinib has not been tested for carcinogenicity.
Erlotinib has been tested for genotoxicity in a series of in vitro assays (bacterial mutation, human lymphocyte chromosome aberration, and mammalian cell mutation) and an in vivo mouse bone marrow micronucleus test and did not cause genetic damage.
Erlotinib did not impair fertility in either male or female rats.
-
14. CLINICAL STUDIES
14.1 Non-Small Cell Lung Cancer NSCLC – TARCEVA Administered as a Single Agent
The efficacy and safety of single-agent TARCEVA was assessed in a randomized, double blind, placebo-controlled trial in 731 patients with locally advanced or metastatic NSCLC after failure of at least one chemotherapy regimen. Patients were randomized 2:1 to receive TARCEVA 150 mg or placebo (488 Tarceva, 243 placebo) orally once daily until disease progression or unacceptable toxicity. Study endpoints included overall survival, response rate, and progression-free survival (PFS). Duration of response was also examined. The primary endpoint was survival. The study was conducted in 17 countries.
Table 4 summarizes the demographic and disease characteristics of the study population. Demographic characteristics were well balanced between the two treatment groups. About two-thirds of the patients were male. Approximately one-fourth had a baseline ECOG performance status (PS) of 2, and 9% had a baseline ECOG PS of 3. Fifty percent of the patients had received only one prior regimen of chemotherapy. About three quarters of these patients were known to have smoked at some time.
Table 4: Demographic and Disease Characteristics * Stratification factor as documented at baseline; distribution differs slightly from values reported at time of randomization. TARCEVA
(N = 488)Placebo
(N = 243)Characteristics n (%) n (%) Gender Female 173 (35) 83 (34) Male 315 (65) 160 (66) Age (years) < 65 299 (61) 153 (63) ≥ 65 189 (39) 90 (37) Race Caucasian 379 (78) 188 (77) Black 18 (4) 12 (5) Asian 63 (13) 28 (12) Other 28 (6) 15 (6) ECOG Performance Status at Baseline* 0 64 (13) 34 (14) 1 256 (52) 132 (54) 2 126 (26) 56 (23) 3 42 (9) 21 (9) Weight Loss in Previous 6 Months < 5% 320 (66) 166 (68) 5 – 10% 96 (20) 36 (15) > 10% 52 (11) 29 (12) Unknown 20 (4) 12 (5) Smoking History Never Smoked 104 (21) 42 (17) Current or Ex-smoker 358 (73) 187 (77) Unknown 26 (5) 14 (6) Histological Classification Adenocarcinoma 246 (50) 119 (49) Squamous 144 (30) 78 (32) Undifferentiated Large Cell 41 (8) 23 (9) Mixed Non-Small Cell 11 (2) 2 (<1) Other 46 (9) 21 (9) Time from Initial Diagnosis to Randomization (Months) < 6 63 (13) 34 (14) 6 – 12 157 (32) 85 (35) > 12 268 (55) 124 (51) Best Response to Prior Therapy at Baseline* CR/PR 196 (40) 96 (40) PD 101 (21) 51 (21) SD 191 (39) 96 (40) Number of Prior Regimens at Baseline* 1 243 (50) 121 (50) 2 238 (49) 119 (49) 3 7 (1) 3 (1) Exposure to Prior Platinum at Baseline* Yes 454 (93) 224 (92) No 34 (7) 19 (8) The results of the study are shown in Table 5.
Table 5: Efficacy Results - *
- Two-sided Log-Rank test stratified by ECOG performance status, number of prior regimens, prior platinum, best response to prior chemotherapy.
TARCEVA Placebo Hazard
Ratio ()95% CI p-value Survival Median
6.7 moMedian
4.7 mo0.73 0.61 – 0.86 <0.001 (*) 1-year Survival 31.2% 21.5% Progression-Free Survival Median
9.9 wkMedian
7.9 wk0.59 0.50 – 0.70 <0.001 (*) Tumor Response (CR+PR) 8.9% 0.9% <0.001 () Response Duration Median
34.3 wkMedian 15.9 wk Survival was evaluated in the intent-to-treat population. Figure 1 depicts the Kaplan-Meier curves for overall survival. The primary survival and PFS analyses were two-sided Log-Rank tests stratified by ECOG performance status, number of prior regimens, prior platinum, best response to prior chemotherapy.
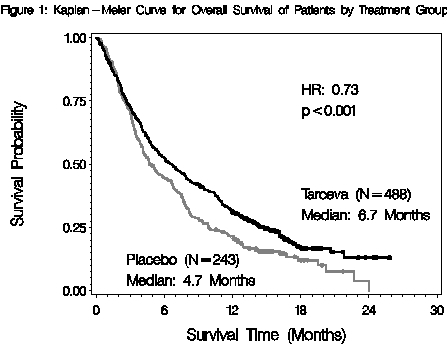
Note: HR is from Cox regression model with the following covariates: ECOG performance status, number of prior regimens, prior platinum, best response to prior chemotherapy. P-value is from two-sided Log-Rank test stratified by ECOG performance status, number of prior regimens, prior platinum, best response to prior chemotherapy.
14.2 NSCLC - TARCEVA Administered Concurrently with Chemotherapy
Results from two, multicenter, placebo-controlled, randomized, trials in over 1000 patients conducted in first-line patients with locally advanced or metastatic NSCLC showed no clinical benefit with the concurrent administration of TARCEVA with platinum-based chemotherapy [carboplatin and paclitaxel (TARCEVA, N = 526) or gemcitabine and cisplatin (TARCEVA, N = 580)].
14.3 Pancreatic Cancer - TARCEVA Administered Concurrently with Gemcitabine
The efficacy and safety of TARCEVA in combination with gemcitabine as a first-line treatment was assessed in a randomized, double blind, placebo-controlled trial in 569 patients with locally advanced, unresectable or metastatic pancreatic cancer. Patients were randomized 1:1 to receive TARCEVA (100 mg or 150 mg) or placebo once daily on a continuous schedule plus gemcitabine IV (1000 mg/m2, Cycle 1 - Days 1, 8, 15, 22, 29, 36 and 43 of an 8 week cycle; Cycle 2 and subsequent cycles - Days 1, 8 and 15 of a 4 week cycle [the approved dose and schedule for pancreatic cancer, see the gemcitabine package insert]). TARCEVA or placebo was taken orally once daily until disease progression or unacceptable toxicity. The primary endpoint was survival. Secondary endpoints included response rate, and progression-free survival (PFS). Duration of response was also examined. The study was conducted in 18 countries. A total of 285 patients were randomized to receive gemcitabine plus TARCEVA (261 patients in the 100 mg cohort and 24 patients in the 150 mg cohort) and 284 patients were randomized to receive gemcitabine plus placebo (260 patients in the 100 mg cohort and 24 patients in the 150 mg cohort). Too few patients were treated in the 150 mg cohort to draw conclusions.
Table 6 summarizes the demographic and disease characteristics of the study population that was randomized to receive 100 mg of TARCEVA plus gemcitabine or placebo plus gemcitabine. Baseline demographic and disease characteristics of the patients were similar between the 2 treatment groups, except for a slightly larger proportion of females in the TARCEVA arm (51%) compared with the placebo arm (44%). The median time from initial diagnosis to randomization was approximately 1.0 month. Most patients presented with metastatic disease at study entry as the initial manifestation of pancreatic cancer.
Table 6: Demographic and Disease Characteristics: 100 mg Cohort *Unknown includes responses of 'Unknown' and missing.
**Stratification factor as documented at baseline; distribution differs slightly from values reported at time of randomization.TARCEVA+ Gemcitabine
(N=261)Placebo + Gemcitabine
(N=260)Characteristics n (%) n (%) Gender Female 134 (51) 114 (44) Male 127 (49) 146 (56) Age (Years) <65 136 (52) 138 (53) ≥65 125 (48) 122 (47) Race Caucasian 225 (86) 231 (89) Black 8 (3) 5 (2) Asian 20 (8) 14 (5) Other 8 (3) 10 (3) ECOG Performance Status* 0 82 (31) 83 (32) 1 134 (51) 132 (51) 2 44 (17) 45 (17) Unknown* 1 (<1) 0 (0) Disease Status at Baseline** Locally Advanced 61 (23) 63 (24) Distant Metastasis 200 (77) 197 (76) The results of the study are shown in Table 7.
Table 7: Efficacy Results: 100 mg Cohort - *
- Two-sided Log-Rank test stratified by ECOG performance status and extent of disease.
TARCEVA + Gemcitabine Placebo+ Gemcitabine Hazard
Ratio ()95% CI p-value Survival Median
6.4 mo
250 deathsMedian
6.0 mo
254 deaths0.81 0.68 – 0.97 0.028 (*) 1-year Survival 23.8% 19.4% Progression-Free Survival Median
3.8 mo
225 eventsMedian
3.5 mo
232 events0.76 0.64 – 0.92 0.006 (*) Tumor Response (CR+PR) 8.6% 7.9% 0.87 () Response Duration Median
23.9 wkMedian
23.3 wkSurvival was evaluated in the intent-to-treat population. Figure 2 depicts the Kaplan-Meier curves for overall survival in the 100 mg cohort. The primary survival and PFS analyses were two-sided Log-Rank tests stratified by ECOG performance status and extent of disease.
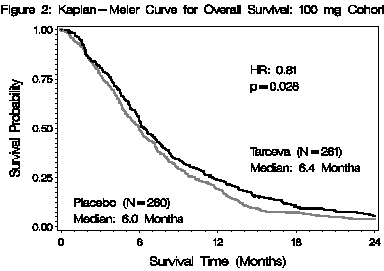
Note: HR is from Cox regression model with the following covariates: ECOG performance status and extent of disease. P-value is from two-sided Log-Rank test stratified by ECOG performance status and extent of disease.
-
16. HOW SUPPLIED/STORAGE AND HANDLING
25 mg Tablets
Round, biconvex face and straight sides, white film-coated, printed in orange with a “T” and “25” on one side and plain on the other side; supplied in:
Bottles of 30
NDC 54868-5290-0
100 mg Tablets
Round, biconvex face and straight sides, white film-coated, printed in gray with “T” and “100” on one side and plain on the other side; supplied in:
Bottles of 30
NDC 54868-5474-0
150 mg Tablets
Round, biconvex face and straight sides, white film-coated, printed in maroon with “T” and “150” on one side and plain on the other side; supplied in:
Bottles of 30
NDC 54868-5447-0
Store at 25°C (77°F); excursions permitted to 15° – 30°C (59° – 86°F). See USP Controlled Room Temperature.
-
17. PATIENT COUNSELING INFORMATION
If the following signs or symptoms occur, patients should be advised to seek medical advice promptly [see Warnings and Precautions (5), Adverse Reactions (6) and Dosage and Administration(2.3)].
- Onset or worsening of skin rash
- Severe or persistent diarrhea, nausea, anorexia, or vomiting
- Onset or worsening of unexplained shortness of breath or cough
- Eye irritation
Given that skin reactions are anticipated when taking TARCEVA, proactive intervention may include alcohol-free emollient cream and sunscreen and the management of rash should be discussed with the patient. This may include topical corticosteroids or antibiotics with anti-inflammatory properties. These approaches were used in the NSCLC and pancreatic pivotal clinical trials. Acne preparations with drying properties may aggravate the dry skin and erythema. Treatment of rash has not been formally studied and should be based on rash severity.
Women of childbearing potential should be advised to avoid becoming pregnant while taking TARCEVA [see Warnings and Precautions (5.8) and Use in Specific Populations (8.1)].
Smokers should be advised to stop smoking while taking TARCEVA as plasma concentrations of erlotinib are reduced due to the effect of cigarette smoking [see Clinical Pharmacology (12.3 )].
Manufactured for:
OSI Pharmaceuticals Inc., Melville, NY 11747
Manufactured by:
Schwarz Pharma Manufacturing, Seymour, IN 47274
Distributed by:
Genentech USA, Inc., 1 DNA Way, South San Francisco, CA 94080-4990
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - PACKAGE LABEL
-
INGREDIENTS AND APPEARANCE
TARCEVA
erlotinib hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54868-5290(NDC:50242-062) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ERLOTINIB HYDROCHLORIDE (UNII: DA87705X9K) (ERLOTINIB - UNII:J4T82NDH7E) ERLOTINIB 25 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) HYPROMELLOSES (UNII: 3NXW29V3WO) HYDROXYPROPYL CELLULOSE (UNII: RFW2ET671P) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) Product Characteristics Color white Score no score Shape ROUND Size 6mm Flavor Imprint Code T;25 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54868-5290-0 30 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021743 07/01/2005 TARCEVA
erlotinib hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54868-5474(NDC:50242-063) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ERLOTINIB HYDROCHLORIDE (UNII: DA87705X9K) (ERLOTINIB - UNII:J4T82NDH7E) ERLOTINIB 100 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) HYPROMELLOSES (UNII: 3NXW29V3WO) HYDROXYPROPYL CELLULOSE (UNII: RFW2ET671P) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color white Score no score Shape ROUND Size 9mm Flavor Imprint Code T;100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54868-5474-0 30 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021743 11/21/2005 TARCEVA
erlotinib hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54868-5447(NDC:50242-064) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ERLOTINIB HYDROCHLORIDE (UNII: DA87705X9K) (ERLOTINIB - UNII:J4T82NDH7E) ERLOTINIB 150 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) HYPROMELLOSES (UNII: 3NXW29V3WO) HYDROXYPROPYL CELLULOSE (UNII: RFW2ET671P) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color white Score no score Shape ROUND Size 10mm Flavor Imprint Code T;150 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54868-5447-0 30 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021743 10/13/2005 Labeler - Physicians Total Care, Inc. (194123980) Establishment Name Address ID/FEI Business Operations Physicians Total Care, Inc. 194123980 relabel, repack