Label: ETOPOSIDE capsule

- NDC Code(s): 0378-3266-94
- Packager: Mylan Pharmaceuticals Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated April 19, 2016
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- BOXED WARNING (What is this?)
-
DESCRIPTION
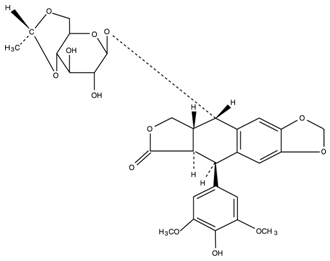
Etoposide (also commonly known as VP-16) is a semisynthetic derivative of podophyllotoxin used in the treatment of certain neoplastic diseases. It is 4’-Demethylepipodophyllotoxin 9-[4,6-O-(R)-ethylidene-β-D-glucopyranoside]. It is very soluble in methanol and chloroform, slightly soluble in ethanol, and sparingly soluble in water and ether. It is made more miscible with water by means of organic solvents. It has a molecular weight of 588.56 and a molecular formula of C29H32O13.
Etoposide may be administered either intravenously or orally. Etoposide capsules, USP are available as 50 mg opaque dark pink, oblong capsules. Each liquid filled, soft gelatin capsule contains 50 mg of etoposide, USP in a vehicle consisting of citric acid anhydrous, glycerol and polyethylene glycol. The soft gelatin capsules contain anidrisorb, gelatin and glycerol with the following dye system: red iron oxide and titanium dioxide; the capsules are printed with edible black ink containing FD&C Blue No. 1 Aluminum Lake, FD&C Red No. 40 Aluminum Lake, hypromellose and propylene glycol.
The structural formula is:
-
CLINICAL PHARMACOLOGY
Etoposide has been shown to cause metaphase arrest in chick fibroblasts. Its main effect, however, appears to be at the G2 portion of the cell cycle in mammalian cells. Two different dose dependent responses are seen. At high concentrations (10 mcg/mL or more), lysis of cells entering mitosis is observed. At low concentrations (0.3 mcg/mL to 10 mcg/mL), cells are inhibited from entering prophase. It does not interfere with microtubular assembly. The predominant macromolecular effect of etoposide appears to be the induction of DNA strand breaks by an interaction with DNA topoisomerase II or the formation of free radicals.
Pharmacokinetics
On intravenous administration, the disposition of etoposide is best described as a biphasic process with a distribution half-life of about 1.5 hours and terminal elimination half-life ranging from 4 to 11 hours. Total body clearance values range from 33 mL/min to 48 mL/min or 16 mL/min/m2 to 36 mL/min/m2 and, like the terminal elimination half-life, are independent of dose over a range 100 mg/m2 to 600 mg/m2. Over the same dose range, the areas under the plasma concentration vs. time curves (AUC) and the maximum plasma concentration (Cmax) values increase linearly with dose. Etoposide does not accumulate in the plasma following daily administration of 100 mg/m2 for 4 to 5 days.
The mean volumes of distribution at steady-state fall in the range of 18 to 29 liters or 7 L/m2 to 17 L/m2. Etoposide enters the CSF poorly. Although it is detectable in CSF and intracerebral tumors, the concentrations are lower than in extracerebral tumors and in plasma. Etoposide concentrations are higher in normal lung than in lung metastases and are similar in primary tumors and normal tissues of the myometrium. In vitro, etoposide is highly protein bound (97%) to human plasma proteins. An inverse relationship between plasma albumin levels and etoposide renal clearance is found in children. In a study determining the effect of other therapeutic agents on the in vitro binding of 14C-etoposide to human serum proteins, only phenylbutazone, sodium salicylate and aspirin displaced protein bound etoposide at concentrations achieved in vivo.
Etoposide binding ratio correlates directly with serum albumin in patients with cancer and in normal volunteers. The unbound fraction of etoposide significantly correlated with bilirubin in a population of cancer patients. Data have suggested a significant inverse correlation between serum albumin concentration and free fraction of etoposide (see PRECAUTIONS).
After intravenous administration of 14C-etoposide (100 mg/m2 to 124 mg/m2), mean recovery of radioactivity in the urine was 56% of the dose at 120 hours, 45% of which was excreted as etoposide; fecal recovery of radioactivity was 44% of the dose at 120 hours.
In children, approximately 55% of the dose is excreted in the urine as etoposide in 24 hours. The mean renal clearance of etoposide is 7 mL/min/m2 to 10 mL/min/m2 or about 35% of the total body clearance over a dose range of 80 mg/m2 to 600 mg/m2. Etoposide, therefore, is cleared by both renal and nonrenal processes, i.e., metabolism and biliary excretion. The effect of renal disease on plasma etoposide clearance is not known.
Biliary excretion of unchanged drug and/or metabolites is an important route of etoposide elimination as fecal recovery of radioactivity is 44% of the intravenous dose. The hydroxy acid metabolite [4’-demethylepipodophyllic acid-9-(4,6-0-(R)-ethylidene-ß-D-glucopyranoside)], formed by opening of the lactone ring, is found in the urine of adults and children. It is also present in human plasma, presumably as the trans isomer. Glucoronide and/or sulfate conjugates of etoposide are also excreted in human urine. Only 8% or less of an intravenous dose is excreted in the urine as radiolabeled metabolites of 14C-etoposide. In addition, 0-demethylation of the dimethoxyphenol ring occurs through the CYP450 3A4 isoenzyme pathway to produce the corresponding catechol.
After either intravenous infusion or oral capsule administration, the Cmax and AUC values exhibit marked intra- and inter-subject variability. This results in variability in the estimates of the absolute oral bioavailability of etoposide oral capsules.
Cmax and AUC values for orally administered etoposide capsules consistently fall in the same range as the Cmax and AUC values for an intravenous dose of one-half the size of the oral dose. The overall mean value of oral capsule bioavailability is approximately 50% (range 25% to 75%). The bioavailability of etoposide capsules appears to be linear up to a dose of at least 250 mg/m2.
There is no evidence of a first-pass effect for etoposide. For example, no correlation exists between the absolute oral bioavailability of etoposide capsules and nonrenal clearance. No evidence exists for any other differences in etoposide metabolism and excretion after administration of oral capsules as compared to intravenous infusion.
In adults, the total body clearance of etoposide is correlated with creatinine clearance, serum albumin concentration and nonrenal clearance. Patients with impaired renal function receiving etoposide have exhibited reduced total body clearance, increased AUC and a lower volume of distribution at steady-state (see PRECAUTIONS). Use of cisplatin therapy is associated with reduced total body clearance. In children, elevated serum SGPT levels are associated with reduced drug total body clearance. Prior use of cisplatin may also result in a decrease of etoposide total body clearance in children.
Although some minor differences in pharmacokinetic parameters between age and gender have been observed, these differences were not considered clinically significant.
- INDICATIONS AND USAGE
- CONTRAINDICATIONS
-
WARNINGS
Patients being treated with etoposide must be frequently observed for myelosuppression both during and after therapy. Myelosuppression resulting in death has been reported. Dose-limiting bone marrow suppression is the most significant toxicity associated with etoposide therapy. Therefore, the following studies should be obtained at the start of therapy and prior to each subsequent cycle of etoposide: platelet count, hemoglobin, white blood cell count and differential. The occurrence of a platelet count below 50,000/mm3 or an absolute neutrophil count below 500/mm3 is an indication to withhold further therapy until the blood counts have sufficiently recovered.
Pregnancy
Etoposide can cause fetal harm when administered to a pregnant woman. Etoposide has been shown to be teratogenic in mice and rats.
In rats, an intravenous etoposide dose of 0.4 mg/kg/day (about 1/20th of the human dose on a mg/m2 basis) during organogenesis caused maternal toxicity, embryotoxicity, and teratogenicity (skeletal abnormalities, exencephaly, encephalocele and anophthalmia); higher doses of 1.2 mg/kg/day and 3.6 mg/kg/day (about 1/7th and 1/2 of human dose on a mg/m2 basis) resulted in 90% and 100% embryonic resorptions. In mice, a single 1.0 mg/kg (1/16th of human dose on a mg/m2 basis) dose of etoposide administered intraperitoneally on days 6, 7 or 8 of gestation caused embryotoxicity, cranial abnormalities, and major skeletal malformations. An I.P. dose of 1.5 mg/kg (about 1/10th of human dose on a mg/m2 basis) on day 7 of gestation caused an increase in the incidence of intrauterine death and fetal malformations and a significant decrease in the average fetal body weight.
Women of childbearing potential should be advised to avoid becoming pregnant. If this drug is used during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be warned of the potential hazard to the fetus.
Etoposide should be considered a potential carcinogen in humans. The occurrence of acute leukemia with or without a preleukemic phase has been reported in rare instances in patients treated with etoposide alone or in association with other neoplastic agents. The risk of development of a preleukemic or leukemic syndrome is unclear. Carcinogenicity tests with etoposide have not been conducted in laboratory animals.
-
PRECAUTIONS
General
In all instances where the use of etoposide is considered for chemotherapy, the physician must evaluate the need and usefulness of the drug against the risk of adverse reactions. Most such adverse reactions are reversible if detected early. If severe reactions occur, the drug should be reduced in dosage or discontinued and appropriate corrective measures should be taken according to the clinical judgment of the physician. Reinstitution of etoposide therapy should be carried out with caution, and with adequate consideration of the further need for the drug and alertness as to possible recurrence of toxicity.
Patients with low serum albumin may be at an increased risk for etoposide associated toxicities.
Drug Interactions
High-dose cyclosporin A resulting in concentrations above 2000 ng/mL administered with oral etoposide has led to an 80% increase in etoposide exposure with a 38% decrease in total body clearance of etoposide compared to etoposide alone.
Laboratory Tests
Periodic complete blood counts should be done during the course of etoposide treatment. They should be performed prior to each cycle of therapy and at appropriate intervals during and after therapy. At least one determination should be done prior to each dose of etoposide.
Renal Impairment
In patients with impaired renal function, the following initial dose modification should be considered based on measured creatinine clearance:
Measured Creatinine Clearance
> 50 mL/min
15 to 50 mL/min
etoposide
100% of dose
75% of dose
Subsequent etoposide dosing should be based on patient tolerance and clinical effect.
Data are not available in patients with creatinine clearances < 15 mL/min and further dose reduction should be considered in these patients.
Carcinogenesis (see WARNINGS), Mutagenesis, Impairment of Fertility
Etoposide has been shown to be mutagenic in Ames assay.
Treatment of Swiss-Albino mice with 1.5 mg/kg I.P. of etoposide on day 7 of gestation increased the incidence of intrauterine death and fetal malformations as well as significantly decreased the average fetal body weight. Maternal weight gain was not affected.
Irreversible testicular atrophy was present in rats treated with etoposide intravenously for 30 days at 0.5 mg/kg/day (about 1/16th of the human dose on a mg/m2 basis).
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from etoposide, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric Use
Of more than 600 patients in four clinical studies in the NDA databases who received etoposide or etoposide phosphate in combination with other chemotherapeutic agents for the treatment of small cell lung cancer (SCLC), about one-third were older than 65 years. When advanced age was determined to be a prognostic factor for response or survival in these studies, comparisons between treatment groups were performed for the elderly subset. In the one study (etoposide in combination with cyclophosphamide and vincristine compared with cyclophosphamide and vincristine or cyclophosphamide, vincristine and doxorubicin) where age was a significant prognostic factor for survival, a survival benefit for elderly patients was observed for the etoposide regimen compared with the control regimens. No differences in myelosuppression were seen between elderly and younger patients in these studies except for an increased frequency of WHO Grade III or IV leukopenia among elderly patients in a study of etoposide phosphate or etoposide in combination with cisplatin. Elderly patients in this study also had more anorexia, mucositis, dehydration, somnolence and elevated BUN levels than younger patients.
In five single-agent studies of etoposide phosphate in patients with a variety of tumor types, 34% of patients were age 65 years or more. WHO Grade III or IV leukopenia, granulocytopenia and asthenia were more frequent among elderly patients.
Post-marketing experience also suggests that elderly patients may be more sensitive to some of the known adverse effects of etoposide, including myelosuppression, gastrointestinal effects, infectious complications and alopecia.
Although some minor differences in pharmacokinetic parameters between elderly and nonelderly patients have been observed, these differences were not considered clinically significant.
Etoposide and its metabolites are known to be substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function (see PRECAUTIONS: Renal Impairment for recommended dosing adjustments in patients with renal impairment).
-
ADVERSE REACTIONS
The following data on adverse reactions are based on both oral and intravenous administration of etoposide as a single agent, using several different dose schedules for treatment of a wide variety of malignancies.
Hematologic Toxicity
Myelosuppression is dose related and dose limiting, with granulocyte nadirs occurring 7 to 14 days after drug administration and platelet nadirs occurring 9 to 16 days after drug administration. Bone marrow recovery is usually complete by day 20, and no cumulative toxicity has been reported. Fever and infection have also been reported in patients with neutropenia. Death associated with myelosuppression has been reported.
The occurrence of acute leukemia with or without a preleukemic phase has been reported rarely in patients treated with etoposide in association with other antineoplastic agents (see WARNINGS).
Gastrointestinal Toxicity
Nausea and vomiting are the major gastrointestinal toxicities. The severity of such nausea and vomiting is generally mild to moderate with treatment discontinuation required in 1% of patients. Nausea and vomiting can usually be controlled with standard antiemetic therapy. Mild to severe mucositis/esophagitis may occur. Gastrointestinal toxicities are slightly more frequent after oral administration than after intravenous infusion.
Hypotension
Transient hypotension following rapid intravenous administration has been reported in 1% to 2% of patients. It has not been associated with cardiac toxicity or electrocardiographic changes. No delayed hypotension has been noted. To prevent this rare occurrence, it is recommended that etoposide be administered by slow intravenous infusion over a 30- to 60-minute period. If hypotension occurs, it usually responds to cessation of the infusion and administration of fluids or other supportive therapy as appropriate. When restarting the infusion, a slower administration rate should be used.
Allergic Reactions
Anaphylactic-like reactions characterized by chills, fever, tachycardia, bronchospasm, dyspnea and/or hypotension have been reported to occur in 0.7% to 2% of patients receiving intravenous etoposide and in less than 1% of the patients treated with the oral capsules. These reactions have usually responded promptly to the cessation of the infusion and administration of pressor agents, corticosteroids, antihistamines or volume expanders as appropriate; however, the reactions can be fatal. Hypertension and/or flushing have also been reported. Blood pressure usually normalizes within a few hours after cessation of the infusion. Anaphylactic-like reactions have occurred during the initial infusion of etoposide.
Facial/tongue swelling, coughing, diaphoresis, cyanosis, tightness in throat, laryngospasm, back pain and/or loss of consciousness have sometimes occurred in association with the above reactions. In addition, an apparent hypersensitivity-associated apnea has been reported rarely.
Rash, urticaria, and/or pruritus have infrequently been reported at recommended doses. At investigational doses, a generalized pruritic erythematous maculopapular rash, consistent with perivasculitis, has been reported.
Alopecia
Reversible alopecia, sometimes progressing to total baldness, was observed in up to 66% of patients.
Other Toxicities
The following adverse reactions have been infrequently reported: abdominal pain, aftertaste, constipation, dysphagia, asthenia, fatigue, malaise, somnolence, transient cortical blindness, optic neuritis, interstitial pneumonitis/pulmonary fibrosis, fever, seizure (occasionally associated with allergic reactions), Stevens-Johnson Syndrome, and toxic epidermal necrolysis, pigmentation, and a single report of radiation recall dermatitis.
Hepatic toxicity, generally in patients receiving higher doses of the drug than those recommended, has been reported with etoposide. Metabolic acidosis has also been reported in patients receiving higher doses.
The incidences of adverse reactions in the table that follows are derived from multiple data bases from studies in 2,081 patients when etoposide was used either orally or by injection as a single agent.
ADVERSE DRUG EFFECT
PERCENT RANGE OF REPORTED INCIDENCE
Hematologic toxicity
Leukopenia (less than 1,000 WBC/mm3)
Leukopenia (less than 4,000 WBC/mm3)
Thrombocytopenia (less than 50,000 platelets/mm3)
Thrombocytopenia (less than 100,000 platelets/mm3)
Anemia
3 to 17
60 to 91
1 to 20
22 to 41
0 to 33
Gastrointestinal toxicity
Nausea and vomiting
Abdominal pain
Anorexia
Diarrhea
Stomatitis
Hepatic
31 to 43
0 to 2
10 to 13
1 to 13
1 to 6
0 to 3
Alopecia
Peripheral neurotoxicity
Hypotension
Allergic reaction
8 to 66
1 to 2
1 to 2
1 to 2
- OVERDOSAGE
-
DOSAGE AND ADMINISTRATION
Etoposide Capsules
In small cell lung cancer, the recommended dose of etoposide capsules is two times the IV dose rounded to the nearest 50 mg (i.e., Two times 35 mg/m2/day for 4 days to 50 mg/m2/day for 5 days).
The dosage should be modified to take into account the myelosuppressive effects of other drugs in the combination or the effects of prior x-ray therapy or chemotherapy which may have compromised bone marrow reserve.
Stability
Etoposide capsules must be stored under refrigeration 2° to 8°C (36° to 46°F). The capsules are stable for 36 months under such refrigeration conditions.
Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published1-8. There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
-
HOW SUPPLIED
Etoposide Capsules, USP are available containing 50 mg of etoposide, USP.
The 50 mg capsule is an opaque dark pink, soft gelatin capsule printed with E50 in black ink. They are available as follows:
NDC 0378-3266-94
20 Capsules - Unit DoseCapsules are to be stored under refrigeration, between 2° to 8°C (36° to 46°F).
Protect from freezing.
Dispense in a tight, light-resistant container as defined in the USP using a child-resistant closure.
-
REFERENCES
- 1.
- ONS Clinical Practice Committee. Cancer Chemotherapy Guidelines and Recommendations for Practice. Pittsburgh, PA: Oncology Nursing Society; 1999:32-41.
- 2.
- Recommendations for the safe handling of parenteral antineoplastic drugs. Washington, DC: Division of Safety, National Institutes of Health; 1983. US Dept of Health and Human Services, Public Health Service publication NIH 83-2621.
- 3.
- AMA Council on Scientific Affairs. Guidelines for handling parenteral antineoplastics. JAMA.1985; 253: 1590-1591.
- 4.
- National Study Commission on Cytotoxic Exposure. Recommendations for handling cytotoxic agents. 1987. Available from Louis P. Jeffrey, Chairman, National Study Commission on Cytotoxic Exposure. Massachusetts College of Pharmacy and Allied Health Sciences, 179 Longwood Avenue, Boston, MA 02115.
- 5.
- Clinical Oncological Society of Australia. Guidelines and recommendations for safe handling of antineoplastic agents. Med J Australia.1983; 1:426–428.
- 6.
- Jones RB, Frank R, Mass T. Safe handling of chemotherapeutic agents: a report from the Mount Sinai Medical Center. CA–A Cancer J for Clin.1983; 33:258–263.
- 7.
- American Society of Hospital Pharmacists, ASHP technical assistance bulletin on handling cytotoxic and hazardous drugs. Am J Hosp Pharm.1990; 47:1033–1049.
- 8.
- Controlling Occupational Exposure to Hazardous Drugs. (OSHA Work-Practice Guidelines.). Am J Health-SystPharm.1996; 53:1669-1685.
Manufactured for:
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.Manufactured by:
Catalent Germany Eberbach GmbH
D-69412 Eberbach/Baden
GermanyREVISED: 4/2016
ETOP:R3 -
PRINCIPAL DISPLAY PANEL - 50 mg
NDC 0378-3266-94
Etoposide
Capsules, USP
50 mg20 Capsules Unit Dose
Rx only 2 Blister Strips of 10 Capsules
Each capsule contains: Etoposide, USP 50 mg
Usual Dosage: See accompanying prescribing information.
This unit dose package is not child-resistant. Dispense in child-resistant containers.
Store under refrigeration, 2° to 8°C (36° to 46°F). Protect from freezing.
Manufactured for:
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505Made in Germany
M3266:94:2C:R5
Mylan.com
-
INGREDIENTS AND APPEARANCE
ETOPOSIDE
etoposide capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0378-3266 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ETOPOSIDE (UNII: 6PLQ3CP4P3) (ETOPOSIDE - UNII:6PLQ3CP4P3) ETOPOSIDE 50 mg Inactive Ingredients Ingredient Name Strength ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) GLYCERIN (UNII: PDC6A3C0OX) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) FERRIC OXIDE RED (UNII: 1K09F3G675) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C RED NO. 40 (UNII: WZB9127XOA) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) Product Characteristics Color PINK (dark pink opaque) Score no score Shape CAPSULE Size 23mm Flavor Imprint Code E50 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0378-3266-94 20 in 1 CARTON; Type 0: Not a Combination Product 10/22/2001 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA075635 10/22/2001 Labeler - Mylan Pharmaceuticals Inc. (059295980)