SYLVANT- siltuximab injection, powder, lyophilized, for solution
Janssen Biotech, Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use SYLVANT® safely and effectively. See full prescribing information for SYLVANT.
SYLVANT (siltuximab) for injection, for intravenous use Initial U.S. Approval: 2014 RECENT MAJOR CHANGESINDICATIONS AND USAGESYLVANT is an interleukin-6 (IL-6) antagonist indicated for the treatment of patients with multicentric Castleman's disease (MCD) who are human immunodeficiency virus (HIV) negative and human herpesvirus-8 (HHV-8) negative. (1) Limitations of Use SYLVANT was not studied in patients with MCD who are HIV positive or HHV-8 positive because SYLVANT did not bind to virally produced IL-6 in a nonclinical study. DOSAGE AND ADMINISTRATIONFor intravenous infusion only. Administer as an 11 mg/kg dose given over 1 hour by intravenous infusion every 3 weeks. (2) DOSAGE FORMS AND STRENGTHSCONTRAINDICATIONSSevere hypersensitivity reaction to siltuximab or any of the excipients in SYLVANT. (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSThe most common adverse reactions (>10% of patients) were rash, pruritus, upper respiratory tract infection, increased weight, and hyperuricemia. (6) To report SUSPECTED ADVERSE REACTIONS, contact Janssen Biotech, Inc. at 1-800-526-7736 (1-800-JANSSEN) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. (6) See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 5/2018 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
SYLVANT is indicated for the treatment of patients with multicentric Castleman's disease (MCD) who are human immunodeficiency virus (HIV) negative and human herpesvirus-8 (HHV-8) negative.
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
Administer SYLVANT 11 mg/kg over 1 hour as an intravenous infusion every 3 weeks until treatment failure.
Perform hematology laboratory tests prior to each dose of SYLVANT therapy for the first 12 months and every 3 dosing cycles thereafter. If treatment criteria outlined in Table 1 are not met, consider delaying treatment with SYLVANT. Do not reduce dose.
| Laboratory parameter | Requirements before first SYLVANT administration | Retreatment criteria |
|---|---|---|
|
||
| Absolute Neutrophil Count | ≥1.0 × 109/L | ≥1.0 × 109/L |
| Platelet count | ≥75 × 109/L | ≥50 × 109/L |
| Hemoglobin* | <17 g/dL | <17 g/dL |
Do not administer SYLVANT to patients with severe infections until the infection resolves.
Discontinue SYLVANT in patients with severe infusion related reactions, anaphylaxis, severe allergic reactions, or cytokine release syndromes. Do not reinstitute treatment.
2.2 Instructions for Preparation and Administration
Use aseptic technique for reconstitution and preparation of dosing solution.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
1. Calculate the dose (mg), total volume (mL) of reconstituted SYLVANT solution required and the number of vials needed. A 21-gauge 1½ inch needle is recommended for preparation. Infusion bags (250 mL) must contain Dextrose 5% in Water and must be made of polyvinyl chloride (PVC), or polyolefin (PO), or polypropylene (PP), or polyethylene (PE). Alternatively PE bottles may be used.
2. Allow the vial(s) of SYLVANT to come to room temperature over approximately 30 minutes. SYLVANT should remain at room temperature for the duration of the preparation.
3. Aseptically reconstitute each SYLVANT vial as instructed in Table 2.
| Strength | Amount of Sterile Water for Injection, USP required for reconstitution | Post-reconstitution concentration |
|---|---|---|
| 100 mg vial | 5.2 mL | 20 mg/mL |
| 400 mg vial | 20 mL | 20 mg/mL |
Gently swirl the reconstituted vials to aid the dissolution of the lyophilized powder. DO NOT SHAKE or SWIRL VIGOROUSLY. Do not remove the contents until all of the solids have been completely dissolved. The lyophilized powder should dissolve in less than 60 minutes.
Once reconstituted, and prior to further dilution, inspect the vials for particulates and discoloration. Do not use if particles or solution discoloration are present or if visibly opaque. The reconstituted product should be kept for no more than two hours prior to addition into the infusion bag.
4. Dilute the reconstituted SYLVANT solution dose to 250 mL with sterile Dextrose 5% in Water by withdrawing a volume equal to the total calculated volume of reconstituted SYLVANT from the Dextrose 5% in Water, 250 mL bag. Slowly add the total calculated volume (mL) of reconstituted SYLVANT solution to the Dextrose 5% in Water infusion bag. Gently invert the bag to mix the solution.
5. Administer the diluted SYLVANT solution in 5% Dextrose in Water 250 mL by intravenous infusion over a period of 1 hour using administration sets lined with PVC, or polyurethane (PU), or PE, containing a 0.2-micron inline polyethersulfone (PES) filter. The infusion should be completed within 4 hours of the dilution of the reconstituted solution to the infusion bag.
6. Do not infuse SYLVANT concomitantly in the same intravenous line with other agents.
7. SYLVANT does not contain preservatives. Do not store any unused portion of the reconstituted product or of the infusion solution. Waste material should be disposed of in accordance with local requirements.
3 DOSAGE FORMS AND STRENGTHS
SYLVANT (siltuximab) for injection is available as:
100 mg of lyophilized powder in a single-dose vial for intravenous infusion.
400 mg of lyophilized powder in a single-dose vial for intravenous infusion.
4 CONTRAINDICATIONS
Severe hypersensitivity reaction to siltuximab or any of the excipients in SYLVANT [see Warnings and Precautions (5.3)]. Hypersensitivity reactions, including anaphylactic reaction, hypersensitivity, and drug hypersensitivity have been reported in patients treated with siltuximab.
5 WARNINGS AND PRECAUTIONS
5.1 Concurrent Active Severe Infections
Do not administer SYLVANT to patients with severe infections until the infection resolves. SYLVANT may mask signs and symptoms of acute inflammation including suppression of fever and of acute Phase reactants such as C-reactive protein (CRP). Monitor patients receiving SYLVANT closely for infections. Institute prompt anti-infective therapy and do not administer further SYLVANT until the infection resolves.
5.2 Vaccinations
Do not administer live vaccines to patients receiving SYLVANT because IL-6 inhibition may interfere with the normal immune response to new antigens.
5.3 Infusion Related Reactions and Hypersensitivity
SYLVANT may cause infusion related reactions and anaphylaxis. Approximately 945 patients have been treated with SYLVANT in clinical trials. Of these, one patient experienced an anaphylactic reaction. Data from 254 patients treated with SYLVANT monotherapy forms the basis of the safety evaluation of infusion related reactions. Infusion related reactions were reported in 5.1% of these patients. Two (0.8%) were Grade 3 or higher, and 1 (0.4%) was serious; none were fatal. Symptoms of infusion reactions consisted of back pain, chest pain or discomfort, nausea and vomiting, flushing, erythema, and palpitations.
In long-term treatment of MCD patients with siltuximab at the recommended dosage of 11 mg/kg every 3 weeks, infusion related reactions or hypersensitivity reactions occurred at a frequency of 6.3% (1.3% for severe reactions).
Stop the infusion of SYLVANT if the patient develops signs of anaphylaxis. Discontinue further therapy with SYLVANT.
Stop the infusion if the patient develops a mild to moderate infusion reaction. If the reaction resolves, the SYLVANT infusion may be restarted at a lower infusion rate. Consider medication with antihistamines, acetaminophen, and corticosteroids. Discontinue SYLVANT if the patient does not tolerate the infusion following these interventions [see Adverse Reactions (6)].
Administer SYLVANT in a setting that provides resuscitation equipment, medication, and personnel trained to provide resuscitation.
5.4 Gastrointestinal Perforation
Gastrointestinal (GI) perforation has been reported in clinical trials although not in MCD trials. Use with caution in patients who may be at increased risk for GI perforation. Promptly evaluate patients presenting with symptoms that may be associated or suggestive of GI perforation.
6 ADVERSE REACTIONS
The following adverse reactions are also discussed in other sections of the labeling:
- Concurrent active severe infections [see Warnings and Precautions (5.1)]
- Infusion-related reactions and hypersensitivity [see Warnings and Precautions (5.3)]
- Gastrointestinal perforation [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Study 1, in MCD, was an international, multicenter, randomized Phase 2 study of every 3 week infusions comparing SYLVANT and best supportive care (BSC) to placebo and BSC. There were 53 patients randomized to the SYLVANT arm at a dosage of 11 mg/kg and 26 patients randomized to the placebo arm. Of the 26 placebo-treated patients, 13 patients subsequently crossed-over to receive SYLVANT. The median age was 48 years (range 20 to 78), 66% male, 48% Asian, 39% White, 4% Black or African American, 7% other. The patients randomized to SYLVANT received a median of 19 infusions (range 1 to 50) compared to patients randomized to placebo who received a median of 8 infusions (range 2 to 32). To control for disparate exposure between arms, Table 3 reports the per patient incidence of adverse reactions that occurred during the first 8 infusions. Adverse reactions that occurred >3% in the SYLVANT arm are presented.
The most common adverse reactions (> 10% compared to placebo) during treatment with SYLVANT in the MCD clinical trial were rash, pruritus, upper respiratory tract infection, increased weight, and hyperuricemia.
| Body System/Adverse Reactions | SYLVANT+BSC*
n=53 | Placebo+BSC n=26 |
||
|---|---|---|---|---|
| All Grades | Grades 3–4 | All Grades | Grades 3–4 | |
| Skin disorders | ||||
| Rash (rash, rash generalized, rash maculo-papular, rash popular and rash pruritic) | 15 (28%) | 1 (2%) | 3 (12%) | 0 |
| Pruritus | 15 (28%) | 0 | 2 (8%) | 0 |
| Skin hyperpigmentation | 2 (4%) | 0 | 0 | 0 |
| Eczema | 2 (4%) | 0 | 0 | 0 |
| Psoriasis | 2 (4%) | 0 | 0 | 0 |
| Dry skin | 2 (4%) | 0 | 0 | 0 |
| Infections | ||||
| Lower respiratory tract | 4 (8%) | 2 (4%) | 1 (4%) | 1 (4%) |
| Upper respiratory tract | 14 (26%) | 1 (2%) | 4 (15%) | 1 (4%) |
| Blood and lymphatic system disorders | ||||
| Thrombocytopenia | 5 (9%) | 2 (4%) | 1 (4%) | 1 (4%) |
| General disorders | ||||
| Edema (general and localized) | 14 (26%) | 4 (8%) | 7 (27%) | 0 |
| Gastrointestinal disorders | ||||
| Constipation | 4 (8%) | 0 | 1 (4%) | 0 |
| Metabolism | ||||
| Hypertriglyceridemia | 4 (8%) | 0 | 0 | 0 |
| Hypercholesterolemia | 2 (4%) | 0 | 0 | 0 |
| Hyperuricemia | 6 (11%) | 1 (2%) | 0 | 0 |
| Respiratory, thoracic and mediastinal disorders | ||||
| Oropharyngeal pain | 4 (8%) | 0 | 1 (4%) | 0 |
| Renal and urinary disorders | ||||
| Renal impairment | 4 (8%) | 0 | 0 | 0 |
| Nervous system disorders | ||||
| Headache | 4 (8%) | 0 | 1 (4%) | 0 |
| Investigations | ||||
| Weight increased | 10 (19%) | 1 (2%) | 0 | 0 |
| Vascular disorders | ||||
| Hypotension | 2 (4%) | 1 (2%)† | 0 | 0 |
Study CNTO328MCD2002 (referred to as Study 2) (NCT01400503) was an open label, long term extension study of patients with MCD treated on prior trials. The median duration of siltuximab treatment was 5.52 years (range: 0.8 to 10.8 years); more than 50% of patients received siltuximab treatment for ≥5 years. The rate of serious or Grade ≥3 adverse events did not increase over time as a function of cumulative exposure.
Other important adverse reactions reported in MCD clinical studies, all of which were very common, were:
Infections and infestations: nasopharyngitis, urinary tract infection
Blood and lymphatic system disorders: neutropenia
Nervous system disorders: dizziness
Vascular disorders: hypertension
Gastrointestinal disorders: nausea, abdominal pain, vomiting, diarrhea, gastroesophageal reflux disease, mouth ulceration
Immunogenicity
Immunogenicity data are highly dependent on the sensitivity and specificity of the test methods used. Additionally, the observed incidence of a positive result in a test method may be influenced by several factors, including sample handling, timing of sample collection, drug interference, concomitant medication and the underlying disease. Therefore, comparison of the incidence of antibodies to SYLVANT with the incidence of antibodies to other products may be misleading. The clinical significance of anti-siltuximab antibodies following treatment with SYLVANT is not known.
The immunogenicity of siltuximab has been evaluated using antigen-bridging enzyme immunoassay (EIA) and electrochemiluminescence-based immunoassay (ECLIA) methods. A total of 432 patients across the clinical studies were evaluated at multiple time points for anti-therapeutic antibody (ATA) responses to siltuximab after treatment with SYLVANT. Following SYLVANT dosing, 0/243 (0%) patients tested positive for anti-siltuximab antibodies by EIA and 4/189 (2%) patients tested positive by ECLIA. Further immunogenicity analyses were conducted for all positive samples from the 4 patients with detectable anti-siltuximab antibodies. None of these patients had neutralizing antibodies.
7 DRUG INTERACTIONS
7.1 Cytochrome P450 Substrates
Cytochrome P450s in the liver are down regulated by infection and inflammation stimuli including cytokines such as IL-6. Inhibition of IL-6 signaling in patients treated with SYLVANT may restore CYP450 activities to higher levels leading to increased metabolism of drugs that are CYP450 substrates compared to metabolism prior to treatment with SYLVANT.
Upon initiation or discontinuation of SYLVANT, in patients being treated with CYP450 substrates with a narrow therapeutic index, perform therapeutic monitoring of effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) as needed and adjust dose. The effect of SYLVANT on CYP450 enzyme activity can persist for several weeks after stopping therapy. Exercise caution when SYLVANT is co-administered with CYP3A4 substrate drugs where a decrease in effectiveness would be undesirable (e.g., oral contraceptives, lovastatin, atorvastatin).
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C
Risk-Summary
There are no adequate or well-controlled studies in pregnant women. In animal reproduction studies, administration of a human antibody to IL-6 to pregnant cynomolgus monkeys caused decreases in globulin levels in pregnant animals and in the offspring. Siltuximab crossed the placenta in monkeys. Infants born to pregnant women treated with SYLVANT may be at increased risk of infection, and caution is advised in the administration of live vaccines to these infants. SYLVANT should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Advise patients of childbearing potential to avoid pregnancy. Women of childbearing potential should use contraception during and for 3 months after treatment.
Animal Data
In an embryo-fetal development study, siltuximab doses of 9.2 or 46 mg/kg/week were administered intravenously to pregnant monkeys during gestation days (GD) 20 to 118, which includes the period of organogenesis. Fetuses were evaluated on GD 140, approximately 25 days prior to the natural birth. Exposures at the low and high dose after the 25th administration were approximately 3 and 7 times respectively the exposure in humans at the recommended dose of 11 mg/kg. There was no siltuximab-related maternal or fetal toxicity. However, siltuximab crossed the placenta at both doses and when measured on GD 140, fetal serum concentrations of siltuximab were similar to maternal concentrations. In a combined embryofetal and pre- and post-natal development study, cynomolgus monkeys were intravenously administered doses of 10 or 50 mg/kg/week of a human antibody to IL-6 from GD 20 to natural delivery (GD 167). The offspring was evaluated up to 7 months after birth for developmental effects. No maternal or infant toxicity was observed; however, globulin levels were decreased in pregnant animals (GD 34 through lactation day 30) and in the offspring (lactation days 30–120) at both doses.
8.3 Nursing Mothers
It is not known whether siltuximab is excreted in human milk or absorbed systemically after ingestion. Because many drugs and immunoglobulins are excreted in human milk, and because of the potential for adverse reactions in nursing infants from SYLVANT, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
The safety and efficacy of SYLVANT have not been established in pediatric patients.
8.5 Geriatric Use
Of the patients treated with SYLVANT monotherapy in clinical studies 127 (35%) were 65 years and older. No overall differences in safety profile were observed between these patients and younger patients, and other reported clinical experience has not identified differences in the safety profile between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. Clinical studies did not include sufficient numbers of patients aged 65 years and older to determine the effect of age on efficacy in MCD population.
8.6 Patients with Renal Impairment
Based on a population pharmacokinetic analysis using data from clinical trials in patients, no significant difference in siltuximab clearance was observed in patients with pre-existing renal impairment (creatinine clearance (CLCr) ≥ 15 mL/min) compared to patients with baseline normal renal function (CLCr ≥ 90 mL/min). No initial dosage adjustment is necessary for patients with CLCr ≥ 15 mL/min. The potential effect of end stage renal disease on siltuximab pharmacokinetics cannot be determined [see Clinical Pharmacology (12.3)].
8.7 Patients with Hepatic Impairment
Based on a population pharmacokinetic analysis using data from clinical trials in patients, no significant difference in siltuximab clearance was observed in patients with pre-existing mild to moderate hepatic impairment (Child-Pugh Class A and B, respectively) compared to patients with baseline normal hepatic function. No initial dosage adjustment is necessary for patients with mild to moderate hepatic impairment. Patients with baseline severe hepatic impairment (Child-Pugh Class C) were not included in clinical trials [see Clinical Pharmacology (12.3)].
11 DESCRIPTION
Siltuximab is a human-mouse chimeric monoclonal antibody that binds human interleukin-6 (IL-6) and is produced by Chinese hamster ovary cells.
SYLVANT (siltuximab) for injection is supplied as a sterile, white, preservative free, lyophilized powder in single-dose vials.
Each SYLVANT 100 mg single-dose vial contains 100 mg siltuximab, 3.7 mg L-histidine (from L-histidine and L-histidine monohydrochloride monohydrate), 0.8 mg polysorbate 80, and 169 mg sucrose.
Each SYLVANT 400 mg single-dose vial contains 400 mg siltuximab, 14.9 mg L-histidine (from L-histidine and L-histidine monohydrochloride monohydrate), 3.2 mg polysorbate 80, and 677 mg sucrose.
Following reconstitution with Sterile Water for Injection, USP (per section 2.2), the resulting pH is approximately 5.2. The resulting solution contains 20 mg/mL siltuximab to be administered by intravenous infusion following dilution [see Dosage and Administration (2.2)].
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Siltuximab binds human IL-6 and prevents the binding of IL-6 to both soluble and membrane-bound IL-6 receptors. IL-6 has been shown to be involved in diverse normal physiologic processes such as induction of immunoglobulin secretion. Overproduction of IL-6 has been linked to systemic manifestations in patients with MCD.
12.2 Pharmacodynamics
Cardiac Electrophysiology: The effect of multiple doses of SYLVANT (15 mg/kg every 3 weeks for 4 cycles) on the QTc interval was evaluated in an open label, single arm study in 30 patients with Monoclonal Gammopathy of Undetermined Significance, Smoldering Multiple Myeloma, or Indolent Multiple Myeloma. No large changes in the mean QTc interval (i.e., > 20 ms) were detected in the study.
Measurement of IL-6 concentrations in serum or plasma during treatment should not be used as pharmacodynamic marker, as siltuximab-neutralized antibody-IL-6 complexes interfere with current immunological-based IL-6 quantification methods.
12.3 Pharmacokinetics
The pharmacokinetics of siltuximab were evaluated in patients with multicentric Castleman's disease and hematological and non-hematological malignancies. The serum siltuximab pharmacokinetics are adequately described by a linear two-compartment intravenous model with first-order elimination.
Distribution
Following SYLVANT administration (11 mg/kg, once every 3 weeks as 1-hour intravenous infusion) in patients with multicentric Castleman's disease, the maximum serum siltuximab concentration (Cmax) occurred close to the end of infusion. At steady state, the serum mean Cmax value for siltuximab is 332 mcg/mL (42% CV), and the serum mean predose trough value is 84 mcg/mL (78% CV).
With the once every 3 week dosing regimen, siltuximab steady state is achieved by the sixth infusion, and siltuximab accumulates approximately 1.7-fold relative to a single dose. Following multiple dosing, siltuximab showed approximately dose proportional pharmacokinetics over the dose range of 2.8 to 11 mg/kg.
Based on population pharmacokinetic analysis, the central volume of distribution in a male subject with body weight of 70 kg is 4.5 L (20% CV).
Elimination
Based on the population pharmacokinetic analysis, the clearance of siltuximab in patients is 0.23 L/day (51% CV). Based on population pharmacokinetic analysis (n=378), body weight was identified as the only statistically significant covariate for siltuximab clearance. Therefore, the body weight based dosing is appropriate.
The mean terminal half-life (t1/2) for siltuximab in patients after the first intravenous infusion of 11 mg/kg is 20.6 days (range: 14.2 to 29.7 days).
Specific Populations
Age and Gender
Based on population pharmacokinetic analysis, age [range: 18 to 85 years (n=378)] and gender [female (n=175), male (n=203)] do not affect exposure of siltuximab.
Renal Impairment
A population pharmacokinetic analysis (based on pre-existing renal function) was carried out with data from 377 patients enrolled in clinical trials, including 176 with normal renal function (CLCr ≥ 90 mL/min), 122 with mild renal impairment (CLCr 60 to <90 mL/min), 75 with moderate renal impairment (CLCr 30 to <60 mL/min), and 3 with severe renal impairment (CLCr 15 to 29 mL/min). The apparent clearance of siltuximab was similar in patients with pre-existing mild, moderate and severe renal impairment (CLCr 15 to <90 mL/min) compared to patients with normal renal function. The potential effect of end stage renal disease on siltuximab pharmacokinetics cannot be determined as clinical and pharmacokinetic data are available from only one patient.
Hepatic Impairment
A population pharmacokinetic analysis (based on pre-existing hepatic function) was carried out with data from 377 patients enrolled in clinical trials, including 302 with normal hepatic function, 72 with mild hepatic impairment (Child-Pugh A), and 3 with moderate hepatic impairment (Child-Pugh B). The apparent clearance of siltuximab was similar in patients with pre-existing mild and moderate hepatic impairment (Child-Pugh Class A and B) compared to patients with normal hepatic function. The potential effect of severe hepatic impairment on siltuximab pharmacokinetics cannot be determined as clinical and pharmacokinetic data are not available.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or genotoxicity studies have been conducted with siltuximab.
Two fertility studies were conducted. In one study, drug-treated male mice were mated with untreated females and in the second study drug-treated female mice were mated with untreated males. A murine analog of siltuximab was administered subcutaneously at doses up to 100 mg/kg/week for a total of 7 doses in both studies. There was no effect on male or female fertility parameters. In addition, siltuximab did not produce any toxicity in the reproductive organs in cynomolgus monkeys in the 6-month repeat-dose toxicology study at doses up to 46 mg/kg (approximately 7 times) the systemic exposure in patients at the recommended dose.
14 CLINICAL STUDIES
Study 1
Study CNTO328MCD2001 (referred to as Study 1) (NCT01024036) was a Phase 2, multinational, randomized (2:1) double blind, placebo controlled study to evaluate the clinical efficacy and safety of SYLVANT for the treatment of patients with MCD. In this study 53 patients were randomized to Best Supportive Care (BSC) and SYLVANT at a dose of 11 mg/kg every 3 weeks and 26 patients were randomized to BSC and placebo. The median age was 48 years (range 20 to 78), 66% male, 48% Asian, 39% White, 4% Black or African American, 7% other. The histological subtype of MCD was similar in both treatment arms, with 33% hyaline vascular subtype, 23% plasmacytic subtype and 44% mixed subtype. Treatment was continued until treatment failure (defined as disease progression based on increase in symptoms, radiologic progression or deterioration in performance status) or unacceptable toxicity.
The major efficacy outcome of the study was durable tumor and symptomatic response, defined as tumor response (PR and CR based on modified International Working Group response criteria for malignant lymphoma) assessed by independent review and complete resolution or stabilization of MCD symptoms. Thirty-four MCD related signs and symptoms prospectively identified were collected and graded according to the NCI-CTCAE v 4, by investigators. A durable response was defined as tumor and symptomatic response that persisted for a minimum of 18 weeks without treatment failure. The durable tumor and symptomatic response in the SYLVANT arm was 34% compared to 0% in the placebo arm (95% CI: 11.1, 54.8; p=0.0012).
Other analyses included tumor response, time to treatment failure and an increase in hemoglobin of 1.5 g/dL or more, in patients who were anemic at time of study entry, at week 13. The results are summarized in Table 4.
| Efficacy Endpoint | SYLVANT n=53 | Placebo n=26 | p-value* |
|---|---|---|---|
| Durable tumor and symptomatic response (independent review) | 34% | 0 | 0.0012 |
| Tumor response | 38% | 4% | <0.05 |
| Median time to treatment failure (days) | NR† | 134 | <0.05 |
| ≥1.5 g/dL increase in hemoglobin | 61% (19/31) | 0% (0/11) | <0.05 |
A consistent treatment effect was confirmed on subgroup analysis for all parameters evaluated with the exception of the hyaline vascular histological subtype. There were no patients with hyaline vascular histology who demonstrated a durable tumor and symptomatic response. However, activity was suggested in this subtype based on change in hemoglobin and median time to treatment failure.
At the time of the analysis, overall survival data were not mature. One year survival rate was 100% in the SYLVANT arm and 92% in the placebo arm.
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Instruct the patient of the risks of SYLVANT treatment.
Infections
Inform patients that SYLVANT may lower their resistance to infections. Instruct the patient of the importance of contacting their doctor immediately when symptoms suggesting infection appear in order to assure rapid evaluation and appropriate treatment.
Vaccination
Inform the patient that they should discuss the recommended vaccinations prior to treatment with SYLVANT.
Allergic Reactions
Advise patients to seek immediate medical attention if they experience any symptoms of serious allergic reactions during the infusion. Signs include: difficulty breathing, chest tightness, wheezing, severe dizziness or light-headedness, swelling of the lips or skin rash.
Manufactured by:
Janssen Biotech, Inc., Horsham, PA 19044
U.S. License No. 1864
At: Cilag AG, Schaffhausen, Switzerland
Product of the Netherlands
© Janssen Biotech, Inc. 2015
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 05/2018 | |
|
PATIENT INFORMATION |
||
|
What is SYLVANT? SYLVANT is a prescription medicine used to treat people with multicentric Castleman's disease (MCD) who do not have human immunodeficiency virus (HIV) and human herpesvirus-8 (HHV-8) infection. It is not known if SYLVANT is safe and effective in children. |
||
|
Who should not receive SYLVANT? Do not receive SYLVANT if you have had a severe allergic reaction to siltuximab or any of the ingredients in SYLVANT. See the end of this leaflet for a complete list of ingredients in SYLVANT. |
||
|
Before you receive SYLVANT, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. |
||
|
How will I receive SYLVANT?
|
||
|
What are the possible side effects of SYLVANT? SYLVANT may cause serious side effects, including:
|
||
|
|
|
|
The most common side effects of SYLVANT include: rash, itching, upper respiratory tract infection, swelling, weight gain, and increased blood level of uric acid. These are not all the possible side effects of SYLVANT. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
|
General information about the safe and effective use of SYLVANT Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your healthcare provider or pharmacist for information about SYLVANT that is written for health professionals. |
||
|
What are the ingredients in SYLVANT? Active ingredient: siltuximab Inactive ingredients: L-histidine and L-histidine monohydrochloride monohydrate, polysorbate 80, and sucrose Manufactured by: Janssen Biotech, Inc., Horsham, PA 19044 For more information, call 1-800-526-7736 or go to www.Sylvant.com. |
||
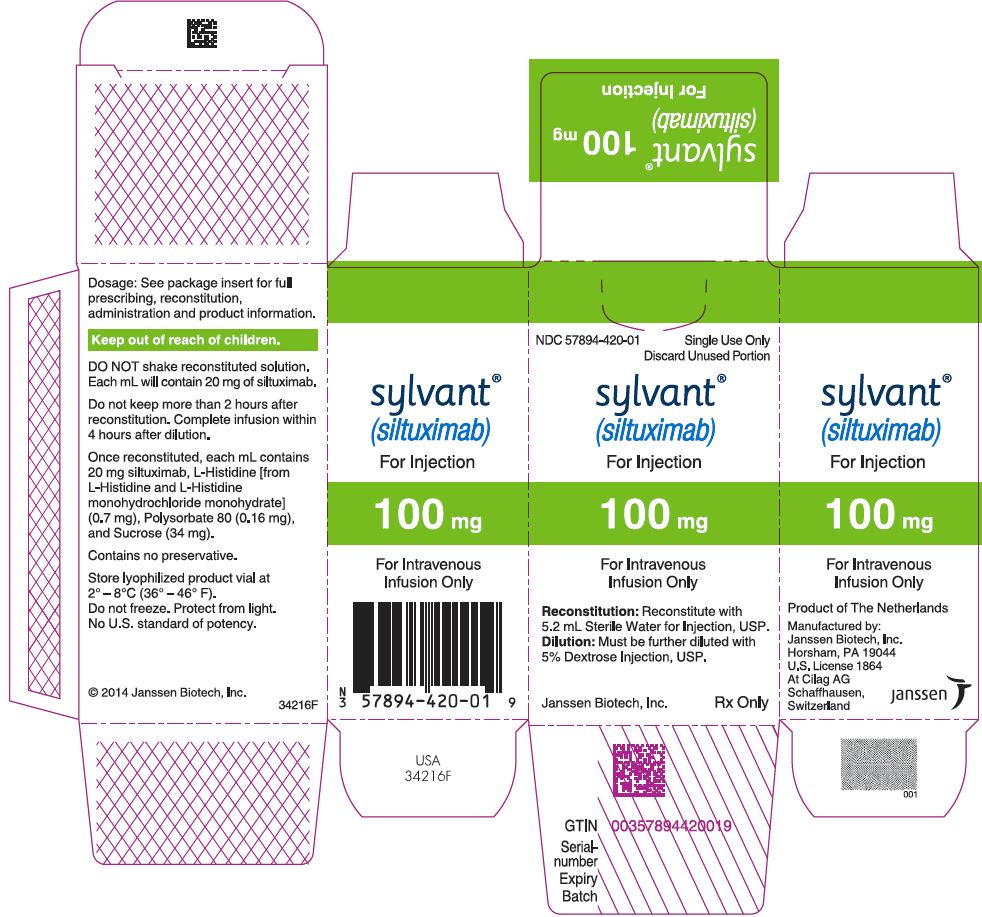
PRINCIPAL DISPLAY PANEL - 100 mg Vial Carton
NDC 57894-420-01
Single Use Only
Discard Unused Portion
sylvant®
(siltuximab)
For Injection
100 mg
For Intravenous
Infusion Only
Reconstitution: Reconstitute with
5.2 mL Sterile Water for Injection, USP.
Dilution: Must be further diluted with
5% Dextrose Injection, USP.
Janssen Biotech, Inc.
Rx Only
PRINCIPAL DISPLAY PANEL - 400 mg Vial Carton
NDC 57894-421-01
Single Use Only
Discard Unused Portion
sylvant®
(siltuximab)
For Injection
400 mg
For Intravenous Infusion Only
Reconstitution: Reconstitute with
20 mL Sterile Water for
Injection, USP.
Dilution: Must be further diluted with
5% Dextrose Injection, USP.
Janssen Biotech, Inc.
Rx Only

| SYLVANT
siltuximab injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| SYLVANT
siltuximab injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Janssen Biotech, Inc. (099091753) |
| Registrant - Janssen Pharmaceuticals, Inc. (063137772) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen Biologics BV | 409612918 | API MANUFACTURE(57894-420, 57894-421) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen Sciences Ireland UC | 986030167 | API MANUFACTURE(57894-420, 57894-421) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Cilag AG | 483237103 | MANUFACTURE(57894-420, 57894-421) | |