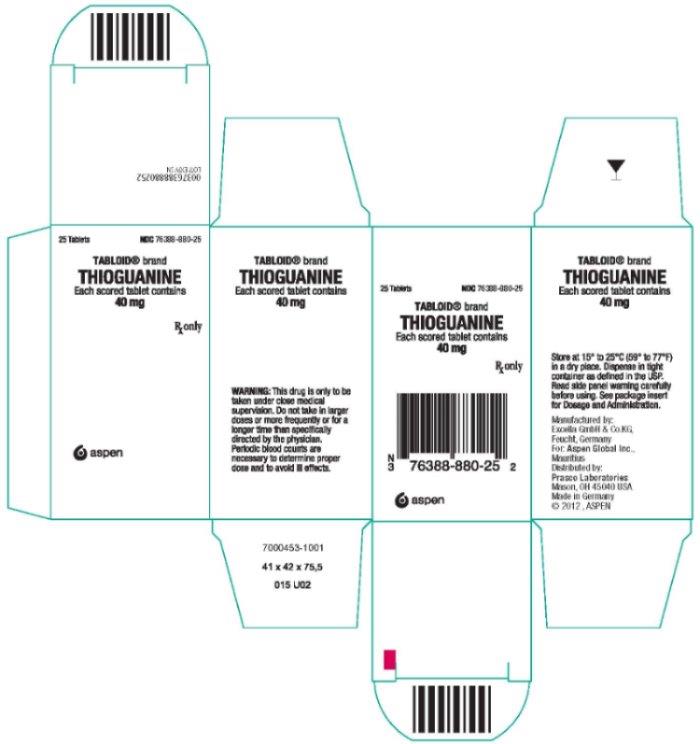
Label: TABLOID- thioguanine tablet


- NDC Code(s): 76388-880-25
- Packager: Aspen Global Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated August 3, 2022
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- CAUTION
-
DESCRIPTION
TABLOID brand Thioguanine was synthesized and developed by Hitchings, Elion, and associates at the Wellcome Research Laboratories. It is one of a large series of purine analogues which interfere with nucleic acid biosynthesis, and has been found active against selected human neoplastic diseases.
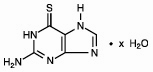
Thioguanine, known chemically as 2-amino-1,7-dihydro-6H-purine-6-thione, is an analogue of the nucleic acid constituent guanine, and is closely related structurally and functionally to PURINETHOL® (mercaptopurine). Its structural formula is:
TABLOID brand Thioguanine is available in tablets for oral administration. Each scored tablet contains 40 mg thioguanine and the inactive ingredients acacia, lactose monohydrate, magnesium stearate, potato starch, and stearic acid.
-
CLINICAL PHARMACOLOGY
Clinical studies have shown that the absorption of an oral dose of thioguanine in humans is incomplete and variable, averaging approximately 30% of the administered dose (range: 14% to 46%). Following oral administration of 35S-6-thioguanine, total plasma radioactivity reached a maximum at 8 hours and declined slowly thereafter. Parent drug represented only a very small fraction of the total plasma radioactivity at any time, being virtually undetectable throughout the period of measurements.
The oral administration of radiolabeled thioguanine revealed only trace quantities of parent drug in the urine. However, a methylated metabolite, 2-amino-6-methylthiopurine (MTG), appeared very early, rose to a maximum 6 to 8 hours after drug administration, and was still being excreted after 12 to 22 hours.
Radiolabeled sulfate appeared somewhat later than MTG but was the principal metabolite after 8 hours. Thiouric acid and some unidentified products were found in the urine in small amounts. Intravenous administration of 35S-6-thioguanine disclosed a median plasma half-disappearance time of 80 minutes (range: 25 to 240 minutes) when the compound was given in single doses of 65 to 300 mg/m2. Although initial plasma levels of thioguanine did correlate with the dose level, there was no correlation between the plasma half-disappearance time and the dose.
Thioguanine is incorporated into the DNA and the RNA of human bone marrow cells. Studies with intravenous 35S-6-thioguanine have shown that the amount of thioguanine incorporated into nucleic acids is more than 100 times higher after 5 daily doses than after a single dose. With the 5-dose schedule, from one-half to virtually all of the guanine in the residual DNA was replaced by thioguanine. Tissue distribution studies of 35S-6-thioguanine in mice showed only traces of radioactivity in brain after oral administration. No measurements have been made of thioguanine concentrations in human cerebrospinal fluid (CSF), but observations on tissue distribution in animals, together with the lack of CNS penetration by the closely related compound, mercaptopurine, suggest that thioguanine does not reach therapeutic concentrations in the CSF.
Monitoring of plasma levels of thioguanine during therapy is of questionable value. There is technical difficulty in determining plasma concentrations, which are seldom greater than 1 to 2 mcg/mL after a therapeutic oral dose. More significantly, thioguanine enters rapidly into the anabolic and catabolic pathways for purines, and the active intracellular metabolites have appreciably longer half-lives than the parent drug. The biochemical effects of a single dose of thioguanine are evident long after the parent drug has disappeared from plasma. Because of this rapid metabolism of thioguanine to active intracellular derivatives, hemodialysis would not be expected to appreciably reduce toxicity of the drug.
Thioguanine competes with hypoxanthine and guanine for the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRTase) and is itself converted to 6-thioguanylic acid (TGMP). This nucleotide reaches high intracellular concentrations at therapeutic doses. TGMP interferes at several points with the synthesis of guanine nucleotides. It inhibits de novo purine biosynthesis by pseudo-feedback inhibition of glutamine-5-phosphoribosylpyrophosphate amidotransferase—the first enzyme unique to the de novo pathway for purine ribonucleotide synthesis. TGMP also inhibits the conversion of inosinic acid (IMP) to xanthylic acid (XMP) by competition for the enzyme IMP dehydrogenase. At one time TGMP was felt to be a significant inhibitor of ATP:GMP phosphotransferase (guanylate kinase), but recent results have shown this not to be so.
Thioguanylic acid is further converted to the di- and tri-phosphates, thioguanosine diphosphate (TGDP) and thioguanosine triphosphate (TGTP) (as well as their 2′-deoxyribosyl analogues) by the same enzymes which metabolize guanine nucleotides. Thioguanine nucleotides are incorporated into both the RNA and the DNA by phosphodiester linkages and it has been argued that incorporation of such fraudulent bases contributes to the cytotoxicity of thioguanine.
Thus, thioguanine has multiple metabolic effects and at present it is not possible to designate one major site of action. Its tumor inhibitory properties may be due to one or more of its effects on (a) feedback inhibition of de novo purine synthesis; (b) inhibition of purine nucleotide interconversions; or (c) incorporation into the DNA and the RNA. The net consequence of its actions is a sequential blockade of the synthesis and utilization of the purine nucleotides.
The catabolism of thioguanine and its metabolites is complex and shows significant differences between humans and the mouse. In both humans and mice, after oral administration of 35S-6-thioguanine, urine contains virtually no detectable intact thioguanine. While deamination and subsequent oxidation to thiouric acid occurs only to a small extent in humans, it is the main pathway in mice. The product of deamination by guanase, 6-thioxanthine is inactive, having negligible antitumor activity. This pathway of thioguanine inactivation is not dependent on the action of xanthine oxidase, and an inhibitor of that enzyme (such as allopurinol) will not block the detoxification of thioguanine even though the inactive 6-thioxanthine is normally further oxidized by xanthine oxidase to thiouric acid before it is eliminated. In humans, methylation of thioguanine is much more extensive than in the mouse. The product of methylation, 2-amino-6-methylthiopurine, is also substantially less active and less toxic than thioguanine and its formation is likewise unaffected by the presence of allopurinol. Appreciable amounts of inorganic sulfate are also found in both murine and human urine, presumably arising from further metabolism of the methylated derivatives.
In some animal tumors, resistance to the effect of thioguanine correlates with the loss of HGPRTase activity and the resulting inability to convert thioguanine to thioguanylic acid. However, other resistance mechanisms, such as increased catabolism of TGMP by a nonspecific phosphatase, may be operative. Although not invariable, it is usual to find cross-resistance between thioguanine and its close analogue, PURINETHOL (mercaptopurine).
Metabolism and Genetic Polymorphism
Several published studies indicate that patients with reduced TPMT or NUDT15 activity receiving usual doses of mercaptopurine, accumulate excessive cellular concentrations of active 6-TGNs, and are at higher risk for severe myelosuppression.In a study of 1028 children with ALL, the approximate tolerated mercaptopurine dosage range for patients with TPMT and/or NUDT15 deficiency on mercaptopurine maintenance therapy (as a percentage of the planned dosage) was as follows: heterozygous for either TPMT or NUDT15, 50-90%; heterozygous for both TPMT and NUDT15, 30-50%; homozygous for either TPMT or NUDT15, 5-10%.Approximately 0.3% (1:300) of patients of European or African ancestry have two loss-of-function alleles of the TPMT gene and have little or no TPMT activity (homozygous deficient or poor metabolizers), and approximately 10% of patients have one loss-of-function TPMT allele leading to intermediate TPMT activity (heterozygous deficient or intermediate metabolizers). The TPMT*2, TPMT*3A, and TPMT*3C alleles account for about 95% of individuals with reduced levels of TPMT activity. NUDT15 deficiency is detected in <1% of patients of European or African ancestry. Among patients of East Asian ancestry (i.e., Chinese, Japanese, Vietnamese), 2% have two loss-of-function alleles of the NUDT15 gene, and approximately 21% have one loss-of-function allele. The p.R139C variant of NUDT15 (present on the *2 and *3 alleles) is the most commonly observed, but other less common loss-of-function NUDT15 alleles have been observed.
Consider all clinical information when interpreting results from phenotypic testing used to determine the level of thiopurine nucleotides or TPMT activity in erythrocytes, since some coadministered drugs can influence measurement of TPMT activity in blood, and blood from recent transfusions will misrepresent a patient’s actual TPMT activity.
-
INDICATIONS AND USAGE
a) Acute Nonlymphocytic Leukemias
TABLOID brand Thioguanine is indicated for remission induction and remission consolidation treatment of acute nonlymphocytic leukemias. However, it is not recommended for use during maintenance therapy or similar long-term continuous treatments due to the high risk of liver toxicity (see WARNINGS and ADVERSE REACTIONS).
The response to this agent depends upon the age of the patient (younger patients faring better than older) and whether thioguanine is used in previously treated or previously untreated patients. Reliance upon thioguanine alone is seldom justified for initial remission induction of acute nonlymphocytic leukemias because combination chemotherapy including thioguanine results in more frequent remission induction and longer duration of remission than thioguanine alone.
b) Other Neoplasms
TABLOID brand Thioguanine is not effective in chronic lymphocytic leukemia, Hodgkin’s lymphoma, multiple myeloma, or solid tumors. Although thioguanine is one of several agents with activity in the treatment of the chronic phase of chronic myelogenous leukemia, more objective responses are observed with MYLERAN® (busulfan), and therefore busulfan is usually regarded as the preferred drug.
- CONTRAINDICATIONS
-
WARNINGS
SINCE DRUGS USED IN CANCER CHEMOTHERAPY ARE POTENTIALLY HAZARDOUS, IT IS RECOMMENDED THAT ONLY PHYSICIANS EXPERIENCED WITH THE RISKS OF THIOGUANINE AND KNOWLEDGEABLE IN THE NATURAL HISTORY OF ACUTE NONLYMPHOCYTIC LEUKEMIAS ADMINISTER THIS DRUG.
THIOGUANINE IS NOT RECOMMENDED FOR MAINTENANCE THERAPY OR SIMILAR LONG-TERM CONTINUOUS TREATMENTS DUE TO THE HIGH RISK OF LIVER TOXICITY ASSOCIATED WITH VASCULAR ENDOTHELIAL DAMAGE (see DOSAGE AND ADMINISTRATION and ADVERSE REACTIONS). This liver toxicity has been observed in a high proportion of children receiving thioguanine as part of maintenance therapy for acute lymphoblastic leukemia and in other conditions associated with continuous use of thioguanine. This liver toxicity is particularly prevalent in males. Liver toxicity usually presents as the clinical syndrome of hepatic veno-occlusive disease (hyperbilirubinemia, tender hepatomegaly, weight gain due to fluid retention, and ascites) or with signs of portal hypertension (splenomegaly, thrombocytopenia, and oesophageal varices). Histopathological features associated with this toxicity include hepatoportal sclerosis, nodular regenerative hyperplasia, peliosis hepatitis, and periportal fibrosis.
Thioguanine therapy should be discontinued in patients with evidence of liver toxicity as reversal of signs and symptoms of liver toxicity have been reported upon withdrawal.
Patients must be carefully monitored (see PRECAUTIONS, Laboratory Tests). Early indications of liver toxicity are signs associated with portal hypertension such as thrombocytopenia out of proportion with neutropenia and splenomegaly. Elevations of liver enzymes have also been reported in association with liver toxicity but do not always occur.
The most consistent, dose-related toxicity is bone marrow suppression. This may be manifested by anemia, leukopenia, thrombocytopenia, or any combination of these. Any one of these findings may also reflect progression of the underlying disease. Since thioguanine may have a delayed effect, it is important to withdraw the medication temporarily at the first sign of an abnormally large fall in any of the formed elements of the blood.
Evaluate patients with repeated severe myelosuppression for thiopurine S-methyltransferase (TPMT) or nucleotide diphosphatase (NUDT15) deficiency. TPMT genotyping or phenotyping (red blood cell TPMT activity) and NUDT15 genotyping can identify patients who have reduced activity of these enzymes. Patients with homozygous TPMT or NUDT15 deficiency require substantial dosage reductions. Bone marrow suppression could be exacerbated by coadministration with drugs that inhibit TPMT, such as olsalazine, mesalazine, or sulphasalazine.
It is recommended that evaluation of the hemoglobin concentration or hematocrit, total white blood cell count and differential count, and quantitative platelet count be obtained frequently while the patient is on thioguanine therapy. In cases where the cause of fluctuations in the formed elements in the peripheral blood is obscure, bone marrow examination may be useful for the evaluation of marrow status. The decision to increase, decrease, continue, or discontinue a given dosage of thioguanine must be based not only on the absolute hematologic values, but also upon the rapidity with which changes are occurring. In many instances, particularly during the induction phase of acute leukemia, complete blood counts will need to be done more frequently in order to evaluate the effect of the therapy. The dosage of thioguanine may need to be reduced when this agent is combined with other drugs whose primary toxicity is myelosuppression.
Myelosuppression is often unavoidable during the induction phase of adult acute nonlymphocytic leukemias if remission induction is to be successful. Whether or not this demands modification or cessation of dosage depends both upon the response of the underlying disease and a careful consideration of supportive facilities (granulocyte and platelet transfusions) which may be available.
Life-threatening infections and bleeding have been observed as consequences of thioguanine-induced granulocytopenia and thrombocytopenia.
The effect of thioguanine on the immunocompetence of patients is unknown.
Pregnancy
Drugs such as thioguanine are potential mutagens and teratogens. Thioguanine may cause fetal harm when administered to a pregnant woman. Thioguanine has been shown to be teratogenic in rats when given in doses 5 times the human dose. When given to the rat on the 4th and 5th days of gestation, 13% of surviving placentas did not contain fetuses, and 19% of offspring were malformed or stunted. The malformations noted included generalized edema, cranial defects, and general skeletal hypoplasia, hydrocephalus, ventral hernia, situs inversus, and incomplete development of the limbs. There are no adequate and well-controlled studies in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking the drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant.
-
PRECAUTIONS
General
Although the primary toxicity of thioguanine is myelosuppression, other toxicities have occasionally been observed, particularly when thioguanine is used in combination with other cancer chemotherapeutic agents.
A few cases of jaundice have been reported in patients with leukemia receiving thioguanine. Among these were 2 adult male patients and 4 pediatric patients with acute myelogenous leukemia and an adult male with acute lymphocytic leukemia who developed hepatic veno-occlusive disease while receiving chemotherapy for their leukemia. Six patients had received cytarabine prior to treatment with thioguanine, and some were receiving other chemotherapy in addition to thioguanine when they became symptomatic. While hepatic veno-occlusive disease has not been reported in patients treated with thioguanine alone, it is recommended that thioguanine be withheld if there is evidence of toxic hepatitis or biliary stasis, and that appropriate clinical and laboratory investigations be initiated to establish the etiology of the hepatic dysfunction. Deterioration in liver function studies during thioguanine therapy should prompt discontinuation of treatment and a search for an explanation of the hepatotoxicity.
Administration of live vaccines to immunocompromised patients should be avoided.
Information for Patients
Patients should be informed that the major toxicities of thioguanine are related to myelosuppression, hepatotoxicity, and gastrointestinal toxicity. Patients should never be allowed to take the drug without medical supervision and should be advised to consult their physician if they experience fever, sore throat, jaundice, nausea, vomiting, signs of local infection, bleeding from any site, or symptoms suggestive of anemia. Women of childbearing potential should be advised to avoid becoming pregnant.
Laboratory Tests
Consider testing for TPMT and NUDT15 deficiency in patients who experience severe bone marrow toxicities or repeated episodes of myelosuppression. (see WARNINGS).
It is advisable to monitor liver function tests (serum transaminases, alkaline phosphatase, bilirubin) at weekly intervals when first beginning therapy and at monthly intervals thereafter. It may be advisable to perform liver function tests more frequently in patients with known pre-existing liver disease or in patients who are receiving thioguanine and other hepatotoxic drugs. Patients should be instructed to discontinue thioguanine immediately if clinical jaundice is detected (see WARNINGS).
Drug Interactions
There is usually complete cross-resistance between PURINETHOL (mercaptopurine) and TABLOID brand Thioguanine.
As there is in vitro evidence that aminosalicylate derivatives (e.g., olsalazine, mesalazine, or sulphasalazine) inhibit the TPMT enzyme, they should be administered with caution to patients receiving concurrent thioguanine therapy (see WARNINGS).
Carcinogenesis, Mutagenesis, Impairment of Fertility
In view of its action on cellular DNA, thioguanine is potentially mutagenic and carcinogenic, and consideration should be given to the theoretical risk of carcinogenesis when thioguanine is administered (see WARNINGS).
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because of the potential for tumorigenicity shown for thioguanine, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric Use
Clinical studies of thioguanine did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact Aspen Global Inc. Toll-Free at 1-855-800-8165 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
The most frequent adverse reaction to thioguanine is myelosuppression. The induction of complete remission of acute myelogenous leukemia usually requires combination chemotherapy in dosages which produce marrow hypoplasia. Since consolidation and maintenance of remission are also effected by multiple-drug regimens whose component agents cause myelosuppression, pancytopenia is observed in nearly all patients. Dosages and schedules must be adjusted to prevent life-threatening cytopenias whenever these adverse reactions are observed.Hyperuricemia frequently occurs in patients receiving thioguanine as a consequence of rapid cell lysis accompanying the antineoplastic effect. Adverse effects can be minimized by increased hydration, urine alkalinization, and the prophylactic administration of a xanthine oxidase inhibitor such as ZYLOPRIM® (allopurinol). Unlike PURINETHOL (mercaptopurine) and IMURAN® (azathioprine), thioguanine may be continued in the usual dosage when allopurinol is used conjointly to inhibit uric acid formation.
Less frequent adverse reactions include nausea, vomiting, anorexia, and stomatitis. Intestinal necrosis and perforation have been reported in patients who received multiple-drug chemotherapy including thioguanine.Hepatic Effects
Liver toxicity associated with vascular endothelial damage has been reported when thioguanine is used in maintenance or similar long-term continuous therapy which is not recommended (see WARNINGS and DOSAGE AND ADMINISTRATION). This usually presents as the clinical syndrome of hepatic veno-occlusive disease (hyperbilirubinemia, tender hepatomegaly, weight gain due to fluid retention, and ascites) or signs and symptoms of portal hypertension (splenomegaly, thrombocytopenia, and esophageal varices). Elevation of liver transaminases, alkaline phosphatase, and gamma glutamyl transferase and jaundice may also occur. Histopathological features associated with this toxicity include hepatoportal sclerosis, nodular regenerative hyperplasia, peliosis hepatitis, and periportal fibrosis.
Liver toxicity during short-term cyclical therapy presents as veno-occlusive disease. Reversal of signs and symptoms of this liver toxicity has been reported upon withdrawal of short-term or long-term continuous therapy.
Centrilobular hepatic necrosis has been reported in a few cases; however, the reports are confounded by the use of high doses of thioguanine, other chemotherapeutic agents, and oral contraceptives and chronic alcohol abuse.
-
OVERDOSAGE
Signs and symptoms of overdosage may be immediate, such as nausea, vomiting, malaise, hypotension, and diaphoresis; or delayed, such as myelosuppression and azotemia. It is not known whether thioguanine is dialyzable. Hemodialysis is thought to be of marginal use due to the rapid intracellular incorporation of thioguanine into active metabolites with long persistence. The oral LD50 of thioguanine was determined to be 823 mg/kg ± 50.73 mg/kg and 740 mg/kg ± 45.24 mg/kg for male and female rats, respectively. Symptoms of overdosage may occur after a single dose of as little as 2.0 to 3.0 mg/kg thioguanine. As much as 35 mg/kg has been given in a single oral dose with reversible myelosuppression observed. There is no known pharmacologic antagonist of thioguanine. The drug should be discontinued immediately if unintended toxicity occurs during treatment. Severe hematologic toxicity may require supportive therapy with platelet transfusions for bleeding, and granulocyte transfusions and antibiotics if sepsis is documented. If a patient is seen immediately following an accidental overdosage of the drug, it may be useful to induce emesis.
-
DOSAGE AND ADMINISTRATION
TABLOID brand Thioguanine is administered orally. The dosage which will be tolerated and effective varies according to the stage and type of neoplastic process being treated. Because the usual therapies for adult and pediatric acute nonlymphocytic leukemias involve the use of thioguanine with other agents in combination, physicians responsible for administering these therapies should be experienced in the use of cancer chemotherapy and in the chosen protocol.
Patients with homozygous deficiency of either TPMT or NUDT15 enzyme typically require 10% or less of the standard thioguanine dosage. Reduce initial dosage in patients who are known to have homozygous TPMT or NUDT15 deficiency. Most patients with heterozygous TPMT or NUDT15 deficiency tolerate recommended thioguanine doses, but some require dose reduction based on toxicities. Patients who are heterozygous for both TPMT and NUDT15 may require more substantial dosage reductions. Reduce the dosage based on tolerability.
Ninety-six (59%) of 163 pediatric patients with previously untreated acute nonlymphocytic leukemia obtained complete remission with a multiple-drug protocol including thioguanine, prednisone, cytarabine, cyclophosphamide, and vincristine. Remission was maintained with daily thioguanine, 4-day pulses of cytarabine and cyclophosphamide, and a single dose of vincristine every 28 days. The median duration of remission was 11.5 months.
Fifty-three percent of previously untreated adults with acute nonlymphocytic leukemias attained remission following use of the combination of thioguanine and cytarabine according to a protocol developed at The Memorial Sloan-Kettering Cancer Center. A median duration of remission of 8.8 months was achieved with the multiple-drug maintenance regimen which included thioguanine.
On those occasions when single-agent chemotherapy with thioguanine may be appropriate, the usual initial dosage for pediatric patients and adults is approximately 2 mg/kg of body weight per day. If, after 4 weeks on this dosage, there is no clinical improvement and no leukocyte or platelet depression, the dosage may be cautiously increased to 3 mg/kg/day. The total daily dose may be given at one time.
The dosage of thioguanine used does not depend on whether or not the patient is receiving ZYLOPRIM (allopurinol); this is in contradistinction to the dosage reduction which is mandatory when PURINETHOL (mercaptopurine) or IMURAN (azathioprine) is given simultaneously with allopurinol.
Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published.1-8
There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
- HOW SUPPLIED
-
REFERENCES
1. ONS Clinical Practice Committee. Cancer Chemotherapy Guidelines and Recommendations for Practice. Pittsburgh, PA: Oncology Nursing Society; 1999:32-41.
2. Recommendations for the safe handling of parenteral antineoplastic drugs. Washington, DC: Division of Safety, Clinical Center Pharmacy Department and Cancer Nursing Services, National Institutes of Health and Human Services, 1992, US Dept of Health and Human Services, Public Health Service publication NIH 92-2621.
3. AMA Council on Scientific Affairs. Guidelines for handling parenteral antineoplastics. JAMA. 1985;253:1590-1591.
4. National Study Commission on Cytotoxic Exposure. Recommendations for handling cytotoxic agents. 1987. Available from Louis P. Jeffrey, Chairman, National Study Commission on Cytotoxic Exposure. Massachusetts College of Pharmacy and Allied Health Sciences, 179 Longwood Avenue, Boston, MA 02115.
5. Clinical Oncological Society of Australia. Guidelines and recommendations for safe handling of antineoplastic agents. Med J Australia. 1983;1:426-428.
6. Jones RB, Frank R, Mass T. Safe handling of chemotherapeutic agents: a report from the Mount Sinai Medical Center. CA-A Cancer J for Clin. 1983;33:258-263.
7. American Society of Hospital Pharmacists. ASHP technical assistance bulletin on handling cytotoxic and hazardous drugs. Am J Hosp Pharm. 1990;47:1033-1049.
8. Controlling Occupational Exposure to Hazardous Drugs. (OSHA Work-Practice Guidelines.) Am J Health-Syst Pharm. 1996;53:1669-1685.Manufactured by: Excella GmbH & Co.KG, Feucht, Germany
For: Aspen Global Inc., Mauritius
Distributed by: Prasco Laboratories
Mason, OH 45040 USA
Made in Germany
©2018, Aspen Global Inc. All rights reserved.
Rev. 05/2020
7000296-1003
- Principal Display Panel
-
INGREDIENTS AND APPEARANCE
TABLOID
thioguanine tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:76388-880 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength THIOGUANINE (UNII: FTK8U1GZNX) (THIOGUANINE ANHYDROUS - UNII:WIX31ZPX66) THIOGUANINE 40 mg Inactive Ingredients Ingredient Name Strength ACACIA (UNII: 5C5403N26O) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, POTATO (UNII: 8I089SAH3T) STEARIC ACID (UNII: 4ELV7Z65AP) Product Characteristics Color white (off-white) Score 2 pieces Shape ROUND Size 9mm Flavor Imprint Code T40 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:76388-880-25 1 in 1 CARTON 03/12/2013 03/31/2025 1 25 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA012429 03/12/2013 03/31/2025 Labeler - Aspen Global Inc. (850491625)