Label: BIVALIRUDIN injection, powder, lyophilized, for solution


- NDC Code(s): 71288-427-10, 71288-427-11
- Packager: Meitheal Pharmaceuticals Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated June 8, 2021
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BIVALIRUDIN FOR INJECTION safely and effectively. See full prescribing information for BIVALIRUDIN FOR INJECTION.
BIVALIRUDIN for injection, for intravenous use
Initial U.S. Approval: 2000
INDICATIONS AND USAGE
Bivalirudin for Injection is a direct thrombin inhibitor indicated for use as an anticoagulant in patients undergoing percutaneous coronary intervention (PCI) including patients with heparin-induced thrombocytopenia (HIT) or heparin-induced thrombocytopenia and thrombosis syndrome (HITTS). (1)
DOSAGE AND ADMINISTRATION
- The recommended dosage is a 0.75 mg/kg intravenous bolus dose followed immediately by a 1.75 mg/kg/h intravenous infusion for the duration of the procedure. Five minutes after the bolus dose has been administered, an activated clotting time (ACT) should be performed and an additional bolus dose of 0.3 mg/kg should be given if needed.
- Extending duration of infusion post-procedure up to 4 hours should be considered in patients with ST segment elevation MI (STEMI). (2.1)
DOSAGE FORMS AND STRENGTHS
For Injection: 250 mg of bivalirudin as a lyophilized powder in a single-dose vial for reconstitution. (3)
WARNINGS AND PRECAUTIONS
- Bleeding Events: Bivalirudin increases the risk of bleeding. (5.1, 6.1, 12.2)
- Acute Stent Thrombosis: Increased incidence of acute stent thrombosis in STEMI patients undergoing primary PCI. (2.1, 5.2)
- Thrombotic Risk with Coronary Artery Brachytherapy: An increased risk of thrombus formation, including fatal outcomes, in gamma brachytherapy. (5.3)
ADVERSE REACTIONS
Most common adverse reaction (>2%) was bleeding. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Meitheal Pharmaceuticals, Inc. at 1-844-824-8426 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Heparin, warfarin, thrombolytics, or GPIs: Increased major bleeding risk with concomitant use. (7)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 10/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dose Adjustment in Renal Impairment
2.3 Instructions for Preparation and Administration
2.4 Storage after Reconstitution
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Bleeding Events
5.2 Acute Stent Thrombosis in Patients with STEMI Undergoing PCI
5.3 Thrombotic Risk with Coronary Artery Brachytherapy
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Bivalirudin for injection has been studied only in patients receiving concomitant aspirin.
The recommended dose of bivalirudin for injection is an intravenous bolus dose of 0.75 mg/kg, followed immediately by an infusion of 1.75 mg/kg/h for the duration of the procedure. Five minutes after the bolus dose has been administered, an activated clotting time (ACT) should be performed and an additional bolus of 0.3 mg/kg should be given if needed.
Extended duration of infusion following PCI at 1.75 mg/kg/h for up to 4 hours post-procedure should be considered in patients with ST segment elevation MI (STEMI).
2.2 Dose Adjustment in Renal Impairment
Maintenance Infusion
In patients with creatinine clearance less than 30 mL/min (by Cockcroft Gault equation), reduce the infusion rate to 1 mg/kg/h. Monitor anticoagulant status in patients with renal impairment.
In patients on hemodialysis, reduce the infusion rate to 0.25 mg/kg/h [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
2.3 Instructions for Preparation and Administration
Bivalirudin for injection is intended for intravenous bolus injection and continuous infusion after reconstitution and dilution.
Preparation Instructions for Bolus Injection and Continuous Infusion
- To each 250 mg vial, add 5 mL of Sterile Water for Injection, USP.
- Gently swirl until all material is dissolved.
- Withdraw and discard 5 mL from a 50 mL infusion bag containing 5% Dextrose in Water or 0.9% Sodium Chloride for Injection.
- Add the contents of the reconstituted vial to the infusion bag containing 5% Dextrose in Water or 0.9% Sodium Chloride for Injection to yield a final concentration of 5 mg per mL (e.g., 1 vial in 50 mL; 2 vials in 100 mL; 5 vials in 250 mL).
- Adjust the dose to be administered according to the patient's weight (see Table 1).
Table 1: Dosing Table Weight
(kg)Using 5 mg per mL
ConcentrationBolus Infusion 0.75 mg/kg 1.75 mg/kg/h (mL) (mL/h) 43-47 7 16 48-52 7.5 17.5 53-57 8 19 58-62 9 21 63-67 10 23 68-72 10.5 24.5 73-77 11 26 78-82 12 28 83-87 13 30 88-92 13.5 31.5 93-97 14 33 98-102 15 35 103-107 16 37 108-112 16.5 38.5 113-117 17 40 118-122 18 42 123-127 19 44 128-132 19.5 45.5 133-137 20 47 138-142 21 49 143-147 22 51 148-152 22.5 52.5 Drug Compatibilities
No incompatibilities have been observed with administration sets.
Do not administer the drugs listed in Table 2 in the same intravenous line with Bivalirudin for injection.
Table 2: Drugs Not for Administration in the Same Intravenous Line with Bivalirudin for injection Alteplase Amiodarone HCl Amphotericin B Chlorpromazine HCl Diazepam Dobutamine Prochlorperazine Edisylate Reteplase Streptokinase Vancomycin HCl Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. Preparations of bivalirudin for injection containing particulate matter should not be used. Reconstituted material will be a clear to slightly opalescent, colorless to slightly yellow solution.
2.4 Storage after Reconstitution
Do not freeze reconstituted or diluted bivalirudin for injection. Reconstituted material may be stored at 2 to 8°C for up to 24 hours. Diluted bivalirudin for injection with a concentration of between 0.5 mg per mL and 5 mg per mL is stable at room temperature for up to 24 hours. Discard any unused portion of reconstituted solution remaining in the vial.
-
3 DOSAGE FORMS AND STRENGTHS
Bivalirudin for Injection: 250 mg of bivalirudin as a lyophilized powder in a single-dose vial for reconstitution. Each vial contains 250 mg of bivalirudin equivalent to an average of 275 mg bivalirudin trifluoroacetate*.
* The range of bivalirudin trifluoroacetate is 266 to 284 mg based on a range of trifluoroacetic acid composition of 1.2 to 2.6 equivalents.
-
4 CONTRAINDICATIONS
Bivalirudin is contraindicated in patients with:
- Active major bleeding;
- Hypersensitivity (e.g., anaphylaxis) to bivalirudin or its components [see Adverse Reactions (6.3)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Bleeding Events
Bivalirudin for injection increases the risk of bleeding [see Adverse Reactions (6.1)]. An unexplained fall in blood pressure or hematocrit should lead to serious consideration of a hemorrhagic event and cessation of bivalirudin for injection administration. Monitor patients receiving bivalirudin for injection for signs and symptoms of bleeding. Monitor patients with disease states associated with an increased risk of bleeding more frequently for bleeding.
5.2 Acute Stent Thrombosis in Patients with STEMI Undergoing PCI
Acute stent thrombosis (AST) (<4 hours) has been observed at a greater frequency in bivalirudin treated patients (1.2%, 36/2889) compared to heparin treated patients (0.2%, 6/2911) with STEMI undergoing primary PCI. Among patients who experienced an AST, one fatality (0.03%) occurred in an bivalirudin treated patient and one fatality (0.03%) in a heparin treated patient. These patients have been managed by Target Vessel Revascularization (TVR). Patients should remain for at least 24 hours in a facility capable of managing ischemic complications and should be carefully monitored following primary PCI for signs and symptoms consistent with myocardial ischemia.
5.3 Thrombotic Risk with Coronary Artery Brachytherapy
An increased risk of thrombus formation, including fatal outcomes, has been associated with the use of bivalirudin in gamma brachytherapy.
If a decision is made to use bivalirudin during brachytherapy procedures, maintain meticulous catheter technique, with frequent aspiration and flushing, paying special attention to minimizing conditions of stasis within the catheter or vessels [see Adverse Reactions (6.1)].
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In the BAT trials, 79 of the 2161 (3.7%) patients undergoing PCI for treatment of unstable angina and randomized to bivalirudin experienced major bleeding events which consisted of: intracranial bleeding, retroperitoneal bleeding, and clinically overt bleeding with a decrease in hemoglobin >3 g/dL or leading to a transfusion of >2 units of blood.
6.2 Immunogenicity
As with all peptides, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to bivalirudin in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In in vitro studies, bivalirudin exhibited no platelet aggregation response against sera from patients with a history of HIT/HITTS.
Among 494 subjects who received bivalirudin in clinical trials and were tested for antibodies, 2 subjects had treatment-emergent positive bivalirudin antibody tests. Neither subject demonstrated clinical evidence of allergic or anaphylactic reactions and repeat testing was not performed. Nine additional patients who had initial positive tests were negative on repeat testing.
6.3 Postmarketing Experience
Because postmarketing adverse reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following adverse reactions have been identified during post approval use of bivalirudin: fatal bleeding; hypersensitivity and allergic reactions including reports of anaphylaxis; lack of anticoagulant effect; thrombus formation during PCI with and without intracoronary brachytherapy, including reports of fatal outcomes; pulmonary hemorrhage; cardiac tamponade; and INR increased.
-
7 DRUG INTERACTIONS
In clinical trials in patients undergoing PCI/percutaneous transluminal coronary angioplasty (PTCA), co-administration of bivalirudin with heparin, warfarin, thrombolytics, or GPIs was associated with increased risks of major bleeding events compared to patients not receiving these concomitant medications.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no data available on use of bivalirudin in pregnant women to inform a drug-associated risk of adverse developmental outcomes. Reproduction studies in rats and rabbits administered subcutaneously doses up to 1.6 times and 3.2 times the maximum recommended human dose (MRHD) of 15 mg/kg/day based on body surface area (BSA) during organogenesis, respectively, revealed no evidence of fetal harm.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Data
Animal Data
Reproductive studies have been performed in rats at subcutaneous doses up to 150 mg/kg/day (1.6 times the maximum recommended human dose based on body surface area) and rabbits at subcutaneous doses up to 150 mg/kg/day (3.2 times the maximum recommended human dose based on body surface area). These studies revealed no harm to the fetus attributable to bivalirudin.
At 500 mg/kg/day (equivalent to 5.4 times the maximum recommended human dose based on body surface area) subcutaneously, litter sizes and live fetuses in rats were reduced. Fetal skeletal variations were also noted. Some of these changes could be attributed to maternal toxicity observed at high doses.
There is no study covering the peri-natal period because of the potential complications of drug-induced hemorrhage during delivery.
8.2 Lactation
Risk Summary
It is not known whether bivalirudin is present in human milk. No data are available on the effects on the breastfed child or on milk production.
Bivalirudin was administered to lactating rats in reproduction studies (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for bivalirudin and any potential adverse effects on the breastfed child from bivalirudin or from the underlying maternal condition.
Data
Animal Data
Reproduction studies conducted in lactating female rats dosed subcutaneously daily with bivalirudin at doses up to 150 mg/kg/day (1.6 times the maximum recommended human dose, based on body surface area) from day 2 through day 20 of lactation revealed no adverse developmental outcomes to the pups.
8.4 Pediatric Use
The safety and effectiveness of bivalirudin in pediatric patients have not been established.
8.5 Geriatric Use
In studies of patients undergoing PCI, 44% were ≥65 years of age and 12% of patients were ≥75 years old. Elderly patients experienced more bleeding events than younger patients.
8.6 Renal Impairment
The disposition of bivalirudin was studied in PTCA patients with mild, moderate and severe renal impairment. The clearance of bivalirudin was reduced approximately 21% in patients with moderate and severe renal impairment and was reduced approximately 70% in dialysis-dependent patients [see Clinical Pharmacology (12.3)]. Reduce the infusion dose of bivalirudin and monitor the anticoagulant status more frequently in patients with renal impairment creatinine clearance less than 30 mL/min (by Cockcroft Gault equation) [see Dosage and Administration (2.2)].
-
10 OVERDOSAGE
Cases of overdose of up to 10 times the recommended bolus or continuous infusion dose of bivalirudin have been reported in clinical trials and in postmarketing reports. A number of the reported overdoses were due to failure to adjust the infusion dose of bivalirudin in persons with renal dysfunction including persons on hemodialysis [see Dosage and Administration (2.2)]. Bleeding, as well as deaths due to hemorrhage, have been observed in some reports of overdose. In cases of suspected overdosage, discontinue bivalirudin immediately and monitor the patient closely for signs of bleeding. There is no known antidote to bivalirudin. Bivalirudin is hemodialyzable [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
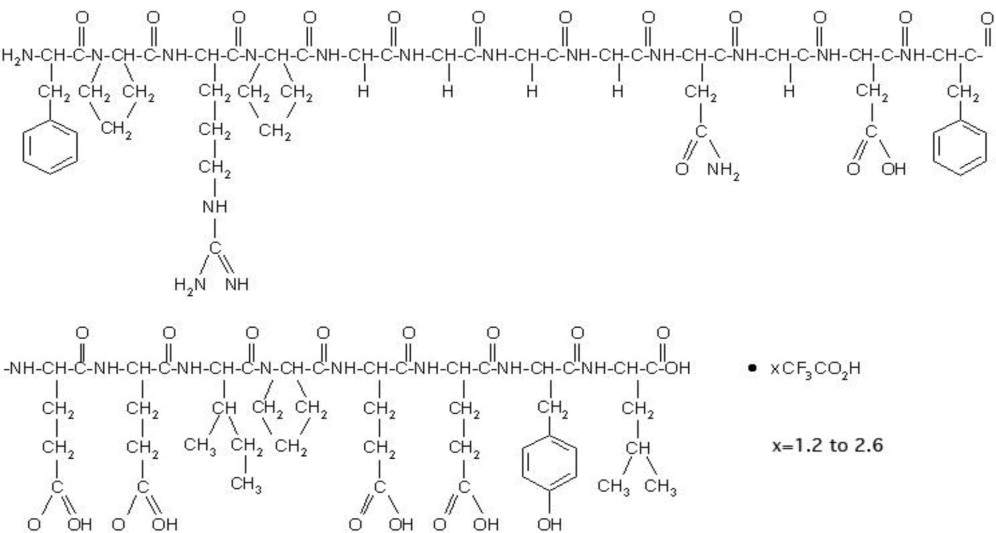
Bivalirudin for Injection contains bivalirudin which is a specific and reversible direct thrombin inhibitor. Bivalirudin is a synthetic, 20 amino acid peptide, with the chemical name of D-phenylalanyl-L-prolyl-L-arginyl-L-prolyl-glycyl-glycyl-glycyl-glycyl-L-asparagyl-glycyl-L-aspartyl-L-phenylalanyl-L-glutamyl-L-glutamyl-L-isoleucyl-L-prolyl-L-glutamyl-L-glutamyl-L-tyrosyl-L-leucine. The active pharmaceutical ingredient is in the form of bivalirudin trifluoroacetate as a white to off-white powder. The chemical name for bivalirudin trifluoroacetate is D-phenylalanyl-L-prolyl-L-arginyl-L-prolyl-glycyl-glycyl-glycyl-glycyl-L-asparagyl-glycyl-L-aspartyl-L-phenylalanyl-L-glutamyl-L-glutamyl-L-isoleucyl-L-prolyl-L-glutamyl-L-glutamyl-L-tyrosyl-L-leucine trifluoroacetate (Figure 1). The molecular weight of bivalirudin is 2180 daltons (anhydrous free base peptide).
Figure 1: Structural formula for bivalirudin
Bivalirudin for Injection is supplied as a sterile white lyophilized cake, in single-dose vials. Each vial contains 250 mg bivalirudin, equivalent to an average of 275 mg of bivalirudin trifluoroacetate*, 125 mg mannitol, and sodium hydroxide to adjust the pH to 5-6 (equivalent of approximately 12.5 mg sodium). When reconstituted with Sterile Water for Injection, the product yields a clear to opalescent, colorless to slightly yellow solution, pH 5-6.
* The range of bivalirudin trifluoroacetate is 266 to 284 mg based on a range of trifluoroacetic acid composition of 1.2 to 2.6 equivalents.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Bivalirudin directly inhibits thrombin by specifically binding both to the catalytic site and to the anion-binding exosite of circulating and clot-bound thrombin. Thrombin is a serine proteinase that plays a central role in the thrombotic process, acting to cleave fibrinogen into fibrin monomers and to activate Factor XIII to Factor XIIIa, allowing fibrin to develop a covalently cross-linked framework which stabilizes the thrombus; thrombin also activates Factors V and VIII, promoting further thrombin generation, and activates platelets, stimulating aggregation and granule release. The binding of bivalirudin to thrombin is reversible as thrombin slowly cleaves the bivalirudin-Arg3-Pro4 bond, resulting in recovery of thrombin active site functions.
In in vitro studies, bivalirudin inhibited both soluble (free) and clot-bound thrombin, was not neutralized by products of the platelet release reaction, and prolonged the activated partial thromboplastin time (aPTT), thrombin time (TT), and prothrombin time (PT) of normal human plasma in a concentration-dependent manner. The clinical relevance of these findings is unknown.
12.2 Pharmacodynamics
In healthy volunteers and patients (with ≥70% vessel occlusion undergoing routine PTCA), bivalirudin exhibited dose- and concentration-dependent anticoagulant activity as evidenced by prolongation of the ACT, aPTT, PT, and TT. Intravenous administration of bivalirudin produces an immediate anticoagulant effect. Coagulation times return to baseline approximately 1 hour following cessation of bivalirudin administration. Bivalirudin also increases INR. Therefore INR measurements made in bivalirudin treated patients may not be useful for determining the appropriate dose of warfarin.
In 291 patients with ≥70% vessel occlusion undergoing routine PTCA, a positive correlation was observed between the dose of bivalirudin and the proportion of patients achieving ACT values of 300 sec or 350 sec. At a bivalirudin dose of 1 mg/kg IV bolus plus 2.5 mg/kg/h IV infusion for 4 hours, followed by 0.2 mg/kg/h, all patients reached maximal ACT values >300 sec.
12.3 Pharmacokinetics
Bivalirudin exhibits linear pharmacokinetics following IV administration to patients undergoing PTCA. In these patients, a mean steady state bivalirudin concentration of 12.3 ± 1.7 mcg/mL is achieved following an IV bolus of 1 mg/kg and a 4-hour 2.5 mg/kg/h IV infusion.
Distribution
Bivalirudin does not bind to plasma proteins (other than thrombin) or to red blood cells.
Elimination
Bivalirudin has a half-life of 25 minutes in PTCA patients with normal renal function. The total body clearance of bivalirudin in PTCA patients with normal renal function is 3.4 mL/min/kg.
Excretion
Bivalirudin undergoes glomerular filtration. Tubular secretion and tubular reabsorption are also implicated in the excretion of bivalirudin, although the extent is unknown.
Specific Populations
Patients with Renal Impairment
Total body clearance was similar for PTCA patients with normal renal function and with mild renal impairment. Clearance was reduced by 21% in patients with moderate and severe renal impairment with a half-life of 34 and 57 minutes, respectively. In dialysis patients, clearance was reduced by 70%, with a half-life of 3.5 hours. Approximately 25% bivalirudin is cleared by hemodialysis.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term studies in animals have been performed to evaluate the carcinogenic potential of bivalirudin. Bivalirudin displayed no genotoxic potential in the in vitro bacterial cell reverse mutation assay (Ames test), the in vitro Chinese hamster ovary cell forward gene mutation test (CHO/HGPRT), the in vitro human lymphocyte chromosomal aberration assay, the in vitro rat hepatocyte unscheduled DNA synthesis (UDS) assay, and the in vivo rat micronucleus assay. Fertility and general reproductive performance in rats were unaffected by subcutaneous doses of bivalirudin up to 150 mg/kg/day, about 1.6 times the dose on a body surface area basis (mg/m2) of a 50 kg person given the maximum recommended dose of 15 mg/kg/day.
-
14 CLINICAL STUDIES
Bivalirudin Angioplasty Trial (BAT)
In the BAT studies, patients with unstable angina undergoing PCI were randomized 1:1 to a 1 mg/kg bolus of bivalirudin and then 2.5 mg/kg/h for four hours and then 0.2 mg/kg/h for 14-20 hours or to 175 IU/kg bolus of heparin followed by an 18-24 hour infusion of 15 IU/kg/h infusion. Additional heparin but not bivalirudin could be administered for ACT <350 seconds. The studies were designed to demonstrate the superiority of bivalirudin to heparin on the occurrence of any of the following during hospitalization up to seven days of death, MI, abrupt closure of dilated vessel, or clinical deterioration requiring revascularization or placement of an aortic balloon pump.
The 4312 subjects ranged in age from 29-90 (median 63) years. 68% were male, and 91% were Caucasian. Median weight was 80 kg (39-120 kg). 741 (17%) subjects had post-MI angina. Twenty-three percent of patients were treated with heparin within one hour prior to randomization.
The studies did not demonstrate that bivalirudin was statistically superior to heparin for reducing the risk of death, MI, abrupt closure of the dilated vessel, or clinical deterioration requiring revascularization or placement of an aortic balloon pump, but the occurrence of these events was similar in both treatment groups. Study outcomes are shown in Table 3.
Table 3: Incidences of In-hospital Endpoints in BAT Trial 1 A composite of death or MI or clinical deterioration of cardiac origin requiring revascularization or placement of an aortic balloon pump or angiographic evidence of abrupt vessel closure.
Endpoint BIVALIRUDIN
(n=2161)HEPARIN
(n=2151)Primary Endpoint1 7.9% 9.3% Death, MI, revascularization 6.2% 7.9% Death 0.2% 0.2% MI 3.3% 4.2% AT-BAT Trial (NCT# 00043940)
This was a single-arm open-label study in which 51 patients with heparin-induced thrombocytopenia (HIT) or heparin induced thrombocytopenia and thrombosis syndrome (HITTS) underwent PCI. The majority of patients achieved adequate ACT at the time of device activation and no major bleeding was reported. Evidence for the diagnosis of HIT/HITTS was based on a clinical history of a decrease of platelets in patients after heparin administration [new diagnosis or history of clinically suspected or objectively documented HIT/HITTS defined as either: 1) HIT: positive heparin-induced platelet aggregation (HIPA) or other functional assay where the platelet count has decreased to <100,000/mL (minimum 30% from prior to heparin), or has decreased to <150,000/mL (minimum 40% from prior to heparin), or has decreased as above within hours of receiving heparin in a patient with a recent, previous exposure to heparin; 2) HITTS: thrombocytopenia as above plus arterial or venous thrombosis diagnosed by physician examination/laboratory and/or appropriate imaging studies]. Patients ranged in age from 48 to 89 years (median 70); weight ranged from 42-123 kg (median 76); 50% were male and 50% were female. Bivalirudin was administered as either 1 mg/kg bolus followed by 2.5 mg/kg/h (high dose in 28 patients) or 0.75 mg/kg bolus followed by a 1.75 mg/kg/h infusion (lower dose in 25 patients) for up to 4 hours. Ninety-eight percent of patients received aspirin, 86% received clopidogrel and 19% received GPIs.
The median ACT values at the time of device activation were 379 sec (high dose) and 317 sec (lower dose). Following the procedure, 48 of the 51 patients (94%) had TIMI grade 3 flow and stenosis <50%. One patient died during a bradycardic episode 46 hours after successful PCI, another patient required surgical revascularization, and one patient experienced no flow requiring a temporary intra-aortic balloon.
Two of the fifty-one patients with the diagnosis of HIT/HITTS developed thrombocytopenia after receiving bivalirudin and GPIs.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Bivalirudin for Injection is supplied as follows:
NDC Bivalirudin for Injection Package Factor 71288-427-11 250 mg Single-Dose Vial 10 vials per carton Bivalirudin for Injection is a lyophilized powder. Each vial contains 250 mg of bivalirudin equivalent to an average of 275 mg of bivalirudin trifluoroacetate*.
* The range of bivalirudin trifluoroacetate is 266 to 284 mg based on a range of trifluoroacetic acid composition of 1.2 to 2.6 equivalents.
- 17 PATIENT COUNSELING INFORMATION
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
BIVALIRUDIN
bivalirudin injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:71288-427 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BIVALIRUDIN (UNII: TN9BEX005G) (BIVALIRUDIN - UNII:TN9BEX005G) BIVALIRUDIN 250 mg in 5 mL Inactive Ingredients Ingredient Name Strength mannitol (UNII: 3OWL53L36A) 125 mg in 5 mL sodium hydroxide (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:71288-427-11 10 in 1 CARTON 07/16/2018 1 NDC:71288-427-10 5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA091602 07/16/2018 Labeler - Meitheal Pharmaceuticals Inc. (080548348) Establishment Name Address ID/FEI Business Operations Nanjing King-Friend Biochemical Pharmaceutical Co., Ltd. 421297554 MANUFACTURE(71288-427)