Label: MULTAQ- dronedarone tablet, film coated

- NDC Code(s): 55154-8104-2
- Packager: Cardinal Health 107, LLC
- This is a repackaged label.
- Source NDC Code(s): 0024-4142
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated January 5, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use MULTAQ safely and effectively. See full prescribing information for MULTAQ.
MULTAQ (dronedarone) tablets, for oral use
Initial U.S. Approval: 2009WARNING: INCREASED RISK OF DEATH, STROKE AND HEART FAILURE IN PATIENTS WITH DECOMPENSATED HEART FAILURE OR PERMANENT ATRIAL FIBRILLATION
See full prescribing information for complete boxed warning.
MULTAQ is contraindicated in patients with symptomatic heart failure with recent decompensation requiring hospitalization or NYHA Class IV heart failure. MULTAQ doubles the risk of death in these patients. (4, 5.1, 14.3)
MULTAQ is contraindicated in patients in atrial fibrillation (AF) who will not or cannot be cardioverted into normal sinus rhythm. In patients with permanent AF, MULTAQ doubles the risk of death, stroke, and hospitalization for heart failure. (4, 5.2, 14.4)
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
One tablet of 400 mg twice a day with morning and evening meals (2)
DOSAGE FORMS AND STRENGTHS
400 mg film-coated tablets (3)
CONTRAINDICATIONS
- •
- Permanent AF (patients in whom normal sinus rhythm will not or cannot be restored) (Boxed Warning, 4)
- •
- Recently decompensated heart failure requiring hospitalization or Class IV heart failure (Boxed Warning, 4)
- •
- Second or third-degree atrioventricular (AV) block or sick sinus syndrome (except when used in conjunction with a functioning pacemaker) (4)
- •
- Bradycardia <50 bpm (4)
- •
- Concomitant use of a strong CYP3A inhibitor (4)
- •
- Concomitant use of drugs or herbal products that prolong the QT interval and may induce torsade de pointes (4)
- •
- Liver or lung toxicity related to the previous use of amiodarone (4)
- •
- Severe hepatic impairment (4)
- •
- QTc Bazett interval ≥500 ms (4)
- •
- Pregnancy (4, 8.1) and Nursing mothers (4, 8.3)
- •
- Hypersensitivity to the active substance or to any of the excipients (4)
WARNINGS AND PRECAUTIONS
- •
- Determine cardiac rhythm at least once every 3 months. If AF is detected discontinue MULTAQ or cardiovert. (5.2)
- •
- Ensure appropriate antithrombotic therapy prior to and throughout MULTAQ use. (5.3)
- •
- Liver injury: If hepatic injury is suspected, discontinue MULTAQ. (5.5)
- •
- If pulmonary toxicity is confirmed, discontinue treatment. (5.6)
- •
- Hypokalemia and hypomagnesemia: Maintain potassium and magnesium levels within the normal range. (5.7)
- •
- Renal impairment: Monitor renal function periodically. (5.9)
- •
- Teratogen: Women of childbearing potential should use effective contraception while using MULTAQ. (5.10)
ADVERSE REACTIONS
Most common adverse reactions (≥2%) are diarrhea, nausea, abdominal pain, vomiting, and asthenia (6)
To report SUSPECTED ADVERSE REACTIONS, contact sanofi-aventis U.S. LLC at 1-800-633-1610 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Dronedarone is metabolized by CYP3A and is a moderate inhibitor of CYP3A and CYP2D6 and has potentially important pharmacodynamic interactions (7)
- •
- Class I or III antiarrhythmics: Contraindicated. (4, 7.1)
- •
- Digoxin: Consider discontinuation or halve dose of digoxin before treatment and monitor digoxin levels. (7.1, 7.3)
- •
- Calcium channel blockers (CCB): Initiate CCB with low dose and increase after ECG verification of tolerability. (7.1,7.2, 7.3)
- •
- Beta-blockers: May provoke excessive bradycardia. Initiate with low dose and increase after ECG verification of tolerability. (7.1, 7.3)
- •
- CYP3A inducers: Avoid concomitant use. (7.2)
- •
- Grapefruit juice: Avoid concomitant use. (7.2)
- •
- Statins: Avoid simvastatin doses greater than 10 mg daily. Follow label recommendations for concomitant use of other statins with a CYP3A and P-gp inhibitor like dronedarone. (7.3)
- •
- CYP3A substrates with a narrow therapeutic index (e.g., sirolimus and tacrolimus): Monitor and adjust dosage of concomitant drug as needed when used with MULTAQ. (7.3)
- •
- Warfarin: Monitor INR after initiating dronedarone in patients taking warfarin. (7.3)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: INCREASED RISK OF DEATH, STROKE AND HEART FAILURE IN PATIENTS WITH DECOMPENSATED HEART FAILURE OR PERMANENT ATRIAL FIBRILLATION
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Cardiovascular Death in NYHA Class IV or Decompensated Heart Failure
5.2 Cardiovascular Death and Heart Failure in Permanent AF
5.3 Increased Risk of Stroke in Permanent AF
5.4 New Onset or Worsening Heart Failure
5.5 Liver Injury
5.6 Pulmonary Toxicity
5.7 Hypokalemia and Hypomagnesemia with Potassium-Depleting Diuretics
5.8 QT Interval Prolongation
5.9 Renal Impairment and Failure
5.10 Women of Childbearing Potential
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Pharmacodynamic Interactions
7.2 Effects of Other Drugs on Dronedarone
7.3 Effects of Dronedarone on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.3 Developmental Toxicity
14 CLINICAL STUDIES
14.1 ATHENA
14.2 EURIDIS and ADONIS
14.3 ANDROMEDA
14.4 PALLAS
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: INCREASED RISK OF DEATH, STROKE AND HEART FAILURE IN PATIENTS WITH DECOMPENSATED HEART FAILURE OR PERMANENT ATRIAL FIBRILLATION
In patients with symptomatic heart failure and recent decompensation requiring hospitalization or NYHA Class IV heart failure, MULTAQ doubles the risk of death [see Clinical Studies (14.3)]. MULTAQ is contraindicated in patients with symptomatic heart failure with recent decompensation requiring hospitalization or NYHA Class IV heart failure [see Contraindications (4), Warnings and Precautions (5.1)].
In patients with permanent atrial fibrillation, MULTAQ doubles the risk of death, stroke and hospitalization for heart failure [see Clinical Studies (14.4)]. MULTAQ is contraindicated in patients in atrial fibrillation (AF) who will not or cannot be cardioverted into normal sinus rhythm [see Contraindications (4), Warnings and Precautions (5.2)].
-
1 INDICATIONS AND USAGE
MULTAQ® is indicated to reduce the risk of hospitalization for atrial fibrillation in patients in sinus rhythm with a history of paroxysmal or persistent atrial fibrillation (AF) [see Clinical Studies (14)].
-
2 DOSAGE AND ADMINISTRATION
The recommended dosage of MULTAQ is 400 mg twice daily in adults. MULTAQ should be taken as one tablet with the morning meal and one tablet with the evening meal.
Treatment with Class I or III antiarrhythmics (e.g., amiodarone, flecainide, propafenone, quinidine, disopyramide, dofetilide, sotalol) or drugs that are strong inhibitors of CYP3A (e.g., ketoconazole) must be stopped before starting MULTAQ [see Contraindications (4)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
MULTAQ is contraindicated in patients with:
- •
- Permanent atrial fibrillation (patients in whom normal sinus rhythm will not or cannot be restored) [see Boxed Warning, Warnings and Precautions (5.2)]
- •
- Symptomatic heart failure with recent decompensation requiring hospitalization or NYHA Class IV symptoms [see Boxed Warning, Warnings and Precautions (5.1)]
- •
- Second or third-degree atrioventricular (AV) block, or sick sinus syndrome (except when used in conjunction with a functioning pacemaker)
- •
- Bradycardia <50 bpm
- •
- Concomitant use of strong CYP3A inhibitors, such as ketoconazole, itraconazole, voriconazole, cyclosporine, telithromycin, clarithromycin, nefazodone, and ritonavir [see Drug Interactions (7.2)]
- •
- Concomitant use of drugs or herbal products that prolong the QT interval and might increase the risk of torsade de pointes, such as phenothiazine antipsychotics, tricyclic antidepressants, certain oral macrolide antibiotics, and Class I and III antiarrhythmics
- •
- Liver or lung toxicity related to the previous use of amiodarone
- •
- QTc Bazett interval ≥500 ms or PR interval >280 ms
- •
- Severe hepatic impairment
- •
- Pregnancy (Category X): MULTAQ may cause fetal harm when administered to a pregnant woman. MULTAQ is contraindicated in women who are or may become pregnant. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus [see Use in Specific Populations (8.1)].
- •
- Nursing mothers [see Use in Specific Populations (8.3)]
- •
- Hypersensitivity to the active substance or to any of the excipients
-
5 WARNINGS AND PRECAUTIONS
5.1 Cardiovascular Death in NYHA Class IV or Decompensated Heart Failure
MULTAQ is contraindicated in patients with NYHA Class IV heart failure or symptomatic heart failure with recent decompensation requiring hospitalization because it doubles the risk of death.
5.2 Cardiovascular Death and Heart Failure in Permanent AF
MULTAQ doubles the risk of cardiovascular death (largely arrhythmic) and heart failure events in patients with permanent AF. Patients treated with dronedarone should undergo monitoring of cardiac rhythm no less often than every 3 months. Cardiovert patients who are in atrial fibrillation (if clinically indicated) or discontinue MULTAQ. MULTAQ offers no benefit in subjects in permanent AF.
5.3 Increased Risk of Stroke in Permanent AF
In a placebo-controlled study in patients with permanent atrial fibrillation, dronedarone was associated with an increased risk of stroke, particularly in the first two weeks of therapy [see Clinical Studies (14.4)]. MULTAQ should only be initiated in patients in sinus rhythm who are receiving appropriate antithrombotic therapy [see Drug Interactions (7.3)].
5.4 New Onset or Worsening Heart Failure
New onset or worsening of heart failure has been reported during treatment with MULTAQ in the postmarketing setting. In a placebo-controlled study in patients with permanent AF increased rates of heart failure were observed in patients with normal left ventricular function and no history of symptomatic heart failure, as well as those with a history of heart failure or left ventricular dysfunction.
Advise patients to consult a physician if they develop signs or symptoms of heart failure, such as weight gain, dependent edema, or increasing shortness of breath. If heart failure develops or worsens and requires hospitalization, discontinue MULTAQ.
5.5 Liver Injury
- Hepatocellular liver injury, including acute liver failure requiring transplant, has been reported in patients treated with MULTAQ in the postmarketing setting. Advise patients treated with MULTAQ to report immediately symptoms suggesting hepatic injury (such as anorexia, nausea, vomiting, fever, malaise, fatigue, right upper quadrant pain, jaundice, dark urine, or itching). Consider obtaining periodic hepatic serum enzymes, especially during the first 6 months of treatment, but it is not known whether routine periodic monitoring of serum enzymes will prevent the development of severe liver injury. If hepatic injury is suspected, promptly discontinue MULTAQ and test serum enzymes, aspartate aminotransferase (AST), alanine aminotransferase (ALT) and alkaline phosphatase, as well as serum bilirubin, to establish whether there is liver injury. If liver injury is found, institute appropriate treatment and investigate the probable cause. Do not restart MULTAQ in patients without another explanation for the observed liver injury.
5.6 Pulmonary Toxicity
Cases of interstitial lung disease including pneumonitis and pulmonary fibrosis have been reported in patients treated with MULTAQ in the postmarketing setting [see Adverse Reactions (6.2)]. Onset of dyspnea or non-productive cough may be related to pulmonary toxicity and patients should be carefully evaluated clinically. If pulmonary toxicity is confirmed, MULTAQ should be discontinued.
5.7 Hypokalemia and Hypomagnesemia with Potassium-Depleting Diuretics
Hypokalemia or hypomagnesemia may occur with concomitant administration of potassium-depleting diuretics. Potassium levels should be within the normal range prior to administration of MULTAQ and maintained in the normal range during administration of MULTAQ.
5.8 QT Interval Prolongation
Dronedarone induces a moderate (average of about 10 ms but much greater effects have been observed) QTc (Bazett) prolongation [see Clinical Pharmacology (12.2), Clinical Studies (14.1)]. If the QTc Bazett interval is ≥500 ms, discontinue MULTAQ [see Contraindications (4)].
5.9 Renal Impairment and Failure
- Marked increase in serum creatinine, pre-renal azotemia and acute renal failure, often in the setting of heart failure [see Warnings and Precautions (5.4)] or hypovolemia, have been reported in patients taking MULTAQ. In most cases, these effects appear to be reversible upon drug discontinuation and with appropriate medical treatment. Monitor renal function periodically.
Small increases in creatinine levels (about 0.1 mg/dL) following dronedarone treatment initiation have been shown to be a result of inhibition of creatinine's tubular secretion. The elevation has a rapid onset, reaches a plateau after 7 days and is reversible after discontinuation.
5.10 Women of Childbearing Potential
Premenopausal women who have not undergone a hysterectomy or oophorectomy must use effective contraception while using MULTAQ. Dronedarone caused fetal harm in animal studies at doses equivalent to recommended human doses. Counsel women of childbearing potential regarding appropriate contraceptive choices [see Use in Specific Populations (8.1)].
-
6 ADVERSE REACTIONS
The following safety concerns are described elsewhere in the label:
- •
- New or worsening heart failure [see Warnings and Precautions (5.4)]
- •
- Liver Injury [see Warnings and Precautions (5.5)]
- •
- Pulmonary toxicity [see Warnings and Precautions (5.6)]
- •
- Hypokalemia and hypomagnesemia with potassium-depleting diuretics [see Warnings and Precautions (5.7)]
- •
- QT prolongation [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
The safety evaluation of dronedarone 400 mg twice daily in patients with AF or AFL is based on 5 placebo-controlled studies, ATHENA, EURIDIS, ADONIS, ERATO and DAFNE. In these studies, a total of 6285 patients were randomized and treated, 3282 patients with MULTAQ 400 mg twice daily, and 2875 with placebo. The mean exposure across studies was 12 months. In ATHENA, the maximum follow-up was 30 months.
In clinical trials, premature discontinuation because of adverse reactions occurred in 11.8% of the dronedarone-treated patients and in 7.7% of the placebo-treated group. The most common reasons for discontinuation of therapy with MULTAQ were gastrointestinal disorders (3.2% vs 1.8% in the placebo group) and QT prolongation (1.5% vs 0.5% in the placebo group).
The most frequent adverse reactions observed with MULTAQ 400 mg twice daily in the 5 studies were diarrhea, nausea, abdominal pain, vomiting, and asthenia.
Table 1 displays adverse reactions more common with dronedarone 400 mg twice daily than with placebo in AF or AFL patients, presented by system organ class and by decreasing order of frequency. Adverse laboratory and ECG effects are presented separately in Table 2.
Table 1: Adverse Drug Reactions that Occurred in at Least 1% of Patients and were More Frequent than Placebo Placebo Dronedarone 400 mg twice daily (N=2875) (N=3282) Gastrointestinal
Diarrhea
6%
9%
Nausea
3%
5%
Abdominal pain
3%
4%
Vomiting
1%
2%
Dyspeptic signs and symptoms
1%
2%
General
Asthenic conditions
5%
7%
Cardiac
Bradycardia
1%
3%
Skin and subcutaneous tissue
Including rashes (generalized, macular, maculo-papular, erythematous), pruritus, eczema, dermatitis, dermatitis allergic
3%
5%
Photosensitivity reaction and dysgeusia have also been reported at an incidence less than 1% in patients treated with MULTAQ.
The following laboratory data/ECG parameters were reported with MULTAQ 400 mg twice daily.
Table 2: Laboratory Data/ECG Parameters Not Necessarily Reported as Adverse Events Placebo MULTAQ
400 mg twice daily(N=2875)
(N=3282)
Early increases in creatinine ≥10%
21%
51%
(N=2237)
(N=2701)
QTc prolonged
19%
28%
Assessment of demographic factors such as gender or age on the incidence of treatment-emergent adverse events did not suggest an excess of adverse events in any particular subgroup.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of MULTAQ. Because these reactions are reported voluntarily from a population of an unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiac: New or worsening heart failure [see Warnings and Precautions (5.4)]
Atrial flutter with 1:1 atrioventricular conduction has been reported very rarely.
Respiratory: Interstitial lung disease including pneumonitis and pulmonary fibrosis [see Warnings and Precautions (5.6)]
-
7 DRUG INTERACTIONS
7.1 Pharmacodynamic Interactions
Drugs Prolonging the QT Interval (Inducing Torsade de Pointes)
Coadministration of drugs prolonging the QT interval (such as certain phenothiazines, tricyclic antidepressants, certain macrolide antibiotics, and Class I and III antiarrhythmics) is contraindicated because of the potential risk of torsade de pointes–type ventricular tachycardia [see Contraindications (4), Clinical Pharmacology (12.3)].
Digoxin
In the ANDROMEDA (patients with recently decompensated heart failure) and PALLAS (patients with permanent AF) trials baseline use of digoxin was associated with an increased risk of arrhythmic or sudden death in dronedarone-treated patients compared to placebo. In patients not taking digoxin, no difference in risk of sudden death was observed in the dronedarone versus placebo groups [see Clinical Studies (14.3)].
Digoxin can potentiate the electrophysiologic effects of dronedarone (such as decreased AV-node conduction). Dronedarone increases exposure to digoxin [see Drug Interactions (7.3), Clinical Pharmacology (12.3)].
Consider discontinuing digoxin. If digoxin treatment is continued, halve the dose of digoxin, monitor serum levels closely, and observe for toxicity.
Calcium Channel Blockers
Calcium channel blockers with depressant effects on the sinus and AV nodes could potentiate dronedarone's effects on conduction.
Give a low dose of calcium channel blockers initially and increase only after ECG verification of good tolerability [see Drug Interactions (7.3), Clinical Pharmacology (12.3)].
Beta-Blockers
In clinical trials, bradycardia was more frequently observed when dronedarone was given in combination with beta-blockers.
Give a low dose of beta-blockers initially, and increase only after ECG verification of good tolerability [see Drug Interactions (7.3), Clinical Pharmacology (12.3)].
7.2 Effects of Other Drugs on Dronedarone
Ketoconazole and Other Potent CYP3A Inhibitors
Concomitant use of ketoconazole as well as other potent CYP3A inhibitors such as itraconazole, voriconazole, ritonavir, clarithromycin, and nefazodone is contraindicated because exposure to dronedarone is significantly increased [see Contraindications (4), Clinical Pharmacology (12.3)].
Grapefruit Juice
Patients should avoid grapefruit juice beverages while taking MULTAQ because exposure to dronedarone is significantly increased [see Clinical Pharmacology (12.3)].
Rifampin and Other CYP3A Inducers
Avoid rifampin or other CYP3A inducers such as phenobarbital, carbamazepine, phenytoin, and St. John's wort because they decrease exposure to dronedarone significantly [see Clinical Pharmacology (12.3)].
Calcium Channel Blockers
Verapamil and diltiazem are moderate CYP3A inhibitors and increase dronedarone exposure. Give a low dose of calcium channel blockers initially and increase only after ECG verification of good tolerability [see Drug Interactions (7.3), Clinical Pharmacology (12.3)].
7.3 Effects of Dronedarone on Other Drugs
Simvastatin
Dronedarone increased simvastatin/simvastatin acid exposure. Avoid doses greater than 10 mg once daily of simvastatin [see Clinical Pharmacology (12.3)].
Other Statins
Because of multiple mechanisms of interaction with statins (CYPs and transporters), follow statin label recommendations for use with CYP3A and P-gp inhibitors such as dronedarone.
Calcium Channel Blockers
Dronedarone increased the exposure of calcium channel blockers (verapamil, diltiazem or nifedipine). Give a low dose of calcium channel blockers initially and increase only after ECG verification of good tolerability [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Sirolimus, Tacrolimus, and Other CYP3A Substrates with Narrow Therapeutic Range
Dronedarone can increase plasma concentrations of tacrolimus, sirolimus, and other CYP3A substrates with a narrow therapeutic range when given orally. Monitor plasma concentrations and adjust dosage appropriately.
Beta-Blockers and Other CYP2D6 Substrates
Dronedarone increased the exposure of propranolol and metoprolol. Give low doses of beta-blockers initially, and increase only after ECG verification of good tolerability. Other CYP2D6 substrates, including other beta-blockers, tricyclic antidepressants, and selective serotonin reuptake inhibitors (SSRIs) may have increased exposure upon coadministration with dronedarone [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
P-glycoprotein Substrates
- Digoxin
Dronedarone increased digoxin exposure by inhibiting the P-gp transporter. Consider discontinuing digoxin. If digoxin treatment is continued, halve the dose of digoxin, monitor serum levels closely, and observe for toxicity [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Dabigatran
Dronedarone increases dabigatran plasma exposures by inhibiting the P-gp transporter [see Clinical Pharmacology (12.3)]. In patients with moderate renal impairment (CrCL 30–50 mL/min), reduce the dose of dabigatran to 75 mg twice daily when concomitantly administered with dronedarone. In patients with severe renal impairment (CrCL 15–30 mL/min), concomitant use of dronedarone with dabigatran should be avoided.
Warfarin
When coadministered with dronedarone exposure to S-warfarin was slightly higher than when warfarin was administered alone. There were no clinically significant increases in INR [see Clinical Pharmacology (12.3)].
More patients experienced clinically significant INR elevations (≥5) usually within 1 week after starting dronedarone versus placebo in patients taking oral anticoagulants in ATHENA. However, no excess risk of bleeding was observed in the dronedarone group.
Postmarketing cases of increased INR with or without bleeding events have been reported in warfarin-treated patients initiated on dronedarone. Monitor INR after initiating dronedarone in patients taking warfarin.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category X [see Contraindications (4)]
MULTAQ may cause fetal harm when administered to a pregnant woman. In animal studies, dronedarone was teratogenic in rats at the maximum recommended human dose (MRHD), and in rabbits at half the MRHD. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
When pregnant rats received dronedarone at oral doses greater than or equal to the MRHD (on a mg/m2 basis), fetuses had increased rates of external, visceral and skeletal malformations (cranioschisis, cleft palate, incomplete evagination of pineal body, brachygnathia, partially fused carotid arteries, truncus arteriosus, abnormal lobation of the liver, partially duplicated inferior vena cava, brachydactyly, ectrodactyly, syndactyly, and anterior and/or posterior club feet). When pregnant rabbits received dronedarone, at a dose approximately half the MRHD (on a mg/m2 basis), fetuses had an increased rate of skeletal abnormalities (anomalous ribcage and vertebrae, pelvic asymmetry) at doses ≥20 mg/kg (the lowest dose tested and approximately half the MRHD on a mg/m2 basis).
Actual animal doses: rat (≥80 mg/kg/day); rabbit (≥20 mg/kg)
8.3 Nursing Mothers
It is not known whether MULTAQ is excreted in human milk. Dronedarone and its metabolites are excreted in rat milk. During a prenatal and postnatal study in rats, maternal dronedarone administration was associated with minor reduced body-weight gain in the offspring. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from MULTAQ, discontinue nursing or discontinue the drug [see Contraindications (4)].
8.4 Pediatric Use
Safety and efficacy in children below the age of 18 years have not been established.
8.5 Geriatric Use
More than 4500 patients with AF or AFL aged 65 years or above were included in the MULTAQ clinical program (of whom more than 2000 patients were 75 years or older). Efficacy and safety were similar in elderly and younger patients.
8.6 Renal Impairment
Patients with renal impairment were included in clinical studies. Because renal excretion of dronedarone is minimal [see Clinical Pharmacology (12.3)], no dosing alteration is needed.
8.7 Hepatic Impairment
Dronedarone is extensively metabolized by the liver. There is little clinical experience with moderate hepatic impairment and none with severe impairment. No dosage adjustment is recommended for moderate hepatic impairment [see Contraindications (4), Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
In the event of overdosage, monitor the patient's cardiac rhythm and blood pressure. Treatment should be supportive and based on symptoms.
It is not known whether dronedarone or its metabolites can be removed by dialysis (hemodialysis, peritoneal dialysis or hemofiltration).
There is no specific antidote available.
-
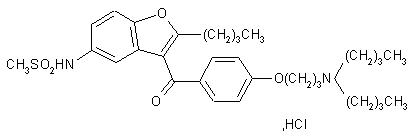
11 DESCRIPTION
Dronedarone HCl is a benzofuran derivative with the following chemical name:
N-{2-butyl-3-[4-(3-dibutylaminopropoxy)benzoyl]benzofuran-5-yl} methanesulfonamide, hydrochloride.
Dronedarone HCl is a white fine powder that is practically insoluble in water and freely soluble in methylene chloride and methanol.
Its empirical formula is C31H44N2O5 S, HCl with a relative molecular mass of 593.2. Its structural formula is:
MULTAQ is provided as tablets for oral administration.
Each tablet of MULTAQ contains 400 mg of dronedarone (expressed as base).
The inactive ingredients are:
- Core of the tablets: hypromellose, starch, crospovidone, poloxamer 407, lactose monohydrate, colloidal silicon dioxide, magnesium stearate.
- Coating/polishing of the tablets: hypromellose, polyethylene glycol 6000, titanium dioxide, carnauba wax.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism of action of dronedarone is unknown. Dronedarone has antiarrhythmic properties belonging to all four Vaughan-Williams classes, but the contribution of each of these activities to the clinical effect is unknown.
12.2 Pharmacodynamics
Electrophysiological Effects
Dronedarone exhibits properties of all four Vaughn-Williams antiarrhythmic classes, although it is unclear which of these are important in producing dronedarone's clinical effects. The effect of dronedarone on 12-lead ECG parameters (heart rate, PR, and QTc) was investigated in healthy subjects following repeated oral doses up to 1600 mg once daily or 800 mg twice daily for 14 days and 1600 mg twice daily for 10 days. In the dronedarone 400 mg twice-daily group, there was no apparent effect on heart rate; a moderate heart rate lowering effect (about 4 bpm) was noted at 800 mg twice daily. There was a clear dose-dependent effect on PR-interval with an increase of +5 ms at 400 mg twice daily and up to +50 ms at 1600 mg twice daily. There was a moderate dose related effect on the QTc-interval with an increase of +10 ms at 400 mg twice daily and up to +25 ms with 1600 mg twice daily.
DAFNE Study
DAFNE was a dose-response study in patients with recurrent AF, evaluating the effect of dronedarone in comparison with placebo in maintaining sinus rhythm. The doses of dronedarone in this study were 400, 600, and 800 mg twice a day. In this small study, doses above 400 mg were not more effective and were less well tolerated.
12.3 Pharmacokinetics
Dronedarone is extensively metabolized and has low systemic bioavailability; its bioavailability is increased by meals. Its elimination half-life is 13 to 19 hours.
Absorption
Because of presystemic first pass metabolism the absolute bioavailability of dronedarone without food is low, about 4%. It increases to approximately 15% when dronedarone is administered with a high fat meal. After oral administration in fed conditions, peak plasma concentrations of dronedarone and the main circulating active metabolite (N-debutyl metabolite) are reached within 3 to 6 hours. After repeated administration of 400 mg twice daily, steady state is reached within 4 to 8 days of treatment and the mean accumulation ratio for dronedarone ranges from 2.6 to 4.5. The steady-state Cmax and exposure of the main N-debutyl metabolite is similar to that of the parent compound. The pharmacokinetics of dronedarone and its N-debutyl metabolite both deviate moderately from dose proportionality: a 2-fold increase in dose results in an approximate 2.5 to 3.0-fold increase with respect to Cmax and AUC.
Distribution
The in vitro plasma protein binding of dronedarone and its N-debutyl metabolite is >98% and not saturable. Both compounds bind mainly to albumin. After intravenous (IV) administration the volume of distribution at steady state is about 1400 L.
Metabolism
Dronedarone is extensively metabolized, mainly by CYP3A. The initial metabolic pathway includes N-debutylation to form the active N-debutyl metabolite, oxidative deamination to form the inactive propanoic acid metabolite, and direct oxidation. The metabolites undergo further metabolism to yield over 30 uncharacterized metabolites. The N-debutyl metabolite exhibits pharmacodynamic activity but is 1/10 to 1/3 as potent as dronedarone. Monoamine oxidases contribute partially to the metabolism of the active metabolite of dronedarone.
Excretion/Elimination
In a mass balance study with orally administered dronedarone (14C-labeled) approximately 6% of the labeled dose was excreted in urine, mainly as metabolites (no unchanged compound excreted in urine), and 84% was excreted in feces, mainly as metabolites. Dronedarone and its N-debutyl active metabolite accounted for less than 15% of the resultant radioactivity in the plasma.
After IV administration the plasma clearance of dronedarone ranges from 130 to 150 L/h. The elimination half-life of dronedarone ranges from 13 to 19 hours.
Special Populations
- Race
Pharmacokinetic differences related to race were not formally assessed. However, based on a cross study comparison, following single dose administration (400 mg), Asian males (Japanese) have about a 2-fold higher exposure than Caucasian males. The pharmacokinetics of dronedarone in other races has not been assessed.
- Elderly
Of the total number of subjects in clinical studies of dronedarone, 73% were 65 years of age and over and 34% were 75 and over. In patients aged 65 years old and above, dronedarone exposures are 23% higher than in patients less than 65 years old [see Use in Specific Populations (8.5)].
- Hepatic impairment
In subjects with moderate hepatic impairment, the mean dronedarone exposure increased by 1.3-fold relative to subjects with normal hepatic function and the mean exposure of the N-debutyl metabolite decreased by about 50%. Pharmacokinetic data were significantly more variable in subjects with moderate hepatic impairment.
The effect of severe hepatic impairment on the pharmacokinetics of dronedarone was not assessed [see Contraindications (4)].
- Renal impairment
Consistent with the low renal excretion of dronedarone, no pharmacokinetic difference was observed in subjects with mild or moderate renal impairment compared to subjects with normal renal function [see Use in Specific Populations (8.6)]. No pharmacokinetic difference was observed in patients with mild to severe renal impairment in comparison with patients with normal renal function.
Drug Interactions
Dronedarone is metabolized primarily by CYP3A and is a moderate inhibitor of CYP3A and CYP2D6. Dronedarone has no significant potential to inhibit CYP1A2, CYP2C9, CYP2C19, CYP2C8 and CYP2B6. It has the potential to inhibit P-glycoprotein (P-gp) transport. Dronedarone inhibits in vivo the tubular secretion of creatinine a substrate of the organic cation transporter (OCT2) [see Warnings and Precautions (5.9)].
In vitro dronedarone and the metabolites SR35021 and SR90154 show no significant potential to inhibit the organic anion transporters OAT1 and OAT3 or the organic cation transporter OCT1. However, in vitro data indicate that SR90154 is likely to inhibit the organic anion transporting polypeptides (OATP1B1, OATP1B3) in vivo.
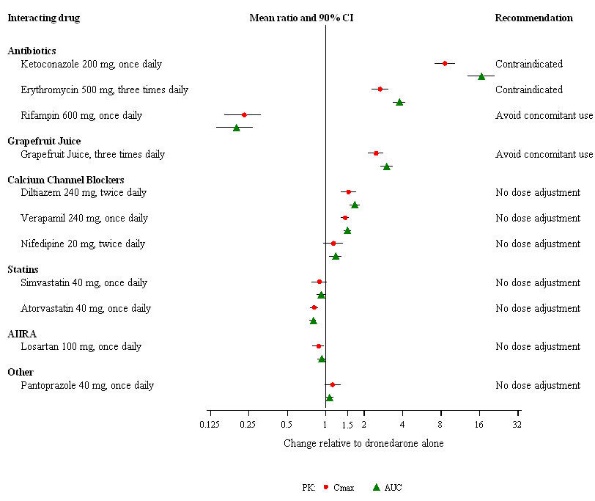
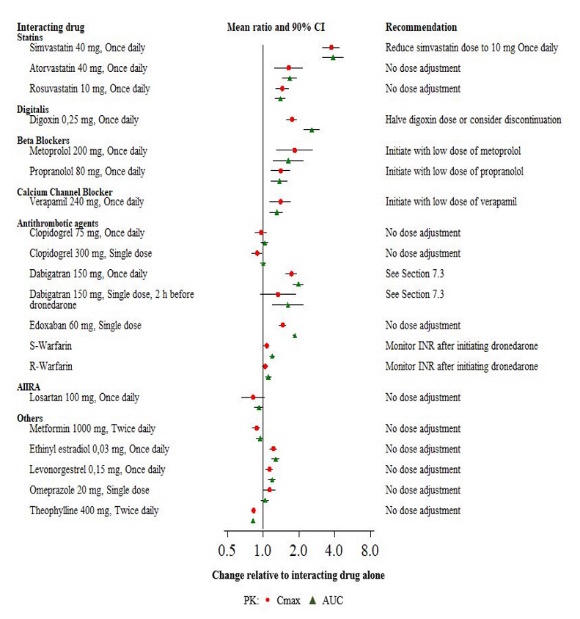
Pharmacokinetic measures indicating the magnitude of these interactions are presented in Figure 1 (impact of coadministered drugs on dronedarone) and Figure 2 (impact of dronedarone on coadministered drugs).
Figure 1: The Impact of Coadministered Drugs on the Pharmacokinetics of Dronedarone and Recommendations for Dronedarone Coadministration or Dose Adjustment
Figure 2: The Impact of Dronedarone on Coadministered Drugs and Recommendations for Dose Adjustment of Coadministered Drug
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In studies in which dronedarone was administered to rats and mice for up to 2 years at doses of up to 70 mg/kg/day and 300 mg/kg/day, respectively, there was an increased incidence of histiocytic sarcomas in dronedarone-treated male mice (300 mg/kg/day or 5× the maximum recommended human dose based on AUC comparisons), mammary adenocarcinomas in dronedarone-treated female mice (300 mg/kg/day or 8× MRHD based on AUC comparisons) and hemangiomas in dronedarone-treated male rats (70 mg/kg/day or 5× MRHD based on AUC comparisons).
Dronedarone did not demonstrate genotoxic potential in the in vivo mouse micronucleus test, the Ames bacterial mutation assay, the unscheduled DNA synthesis assay, or an in vitro chromosomal aberration assay in human lymphocytes. S-9 processed dronedarone, however, was positive in a V79 transfected Chinese hamster V79 assay.
In fertility studies conducted with female rats, dronedarone given prior to breeding and implantation caused an increase in irregular estrus cycles and cessation of cycling at doses ≥10 mg/kg (equivalent to 0.12 × the MRHD on a mg/m2 basis).
Corpora lutea, implantations and live fetuses were decreased at 100 mg/kg (equivalent to 1.2 × the MRHD on a mg/m2 basis). There were no reported effects on mating behavior or fertility of male rats at doses of up to 100 mg/kg/day.
13.3 Developmental Toxicity
Dronedarone was teratogenic in rats given oral doses ≥80 mg/kg/day (a dose equivalent to the MRHD on a mg/m2 basis), with fetuses showing external, visceral and skeletal malformations (cranioschisis, cleft palate, incomplete evagination of pineal body, brachygnathia, partially fused carotid arteries, truncus arteriosus, abnormal lobation of the liver, partially duplicated inferior vena cava, brachydactyly, ectrodactyly, syndactyly, and anterior and/or posterior club feet). In rabbits, dronedarone caused an increase in skeletal abnormalities (anomalous ribcage and vertebrae, pelvic asymmetry) at doses ≥20 mg/kg (the lowest dose tested and approximately half the MRHD on a mg/m2 basis).
-
14 CLINICAL STUDIES
14.1 ATHENA
ATHENA was a multicenter, multinational, double blind, and randomized placebo-controlled study of dronedarone in 4628 patients with a recent history of AF/AFL who were in sinus rhythm or who were to be converted to sinus rhythm. The objective of the study was to determine whether dronedarone could delay death from any cause or hospitalization for cardiovascular reasons.
Initially patients were to be ≥70 years old, or <70 years old with at least one risk factor (including hypertension, diabetes, prior cerebrovascular accident, left atrial diameter ≥50 mm or LVEF <0.40). The inclusion criteria were later changed such that patients were to be ≥75 years old, or ≥70 years old with at least one risk factor. Patients had to have both AF/AFL and sinus rhythm documented within the previous 6 months. Patients could have been in AF/AFL or in sinus rhythm at the time of randomization, but patients not in sinus rhythm were expected to be either electrically or chemically converted to normal sinus rhythm after anticoagulation.
Subjects were randomized and treated for up to 30 months (median follow-up: 22 months) with either MULTAQ 400 mg twice daily (2301 patients) or placebo (2327 patients), in addition to conventional therapy for cardiovascular diseases that included beta-blockers (71%), ACE inhibitors or angiotensin II receptor blockers (ARBs) (69%), digoxin (14%), calcium antagonists (14%), statins (39%), oral anticoagulants (60%), aspirin (44%), other chronic antiplatelet therapy (6%) and diuretics (54%).
The primary endpoint of the study was the time to first hospitalization for cardiovascular reasons or death from any cause. Time to death from any cause, time to first hospitalization for cardiovascular reasons, and time to cardiovascular death and time to all causes of death were also explored.
Patients ranged in age from 23 to 97 years; 42% were 75 years old or older. Forty-seven percent (47%) of patients were female and a majority was Caucasian (89%). Seventy-one percent (71%) of those enrolled had no history of heart failure. The median ejection fraction was 60%. Twenty-nine percent (29%) of patients had heart failure, mostly NYHA class II (17%). The majority had hypertension (86%) and structural heart disease (60%).
Results are shown in Table 3. MULTAQ reduced the combined endpoint of cardiovascular hospitalization or death from any cause by 24.2% when compared to placebo. This difference was entirely attributable to its effect on cardiovascular hospitalization, principally hospitalization related to AF.
Other endpoints, death from any cause and first hospitalization for cardiovascular reasons, are shown in Table 3. Secondary endpoints count all first events of a particular type, whether or not they were preceded by a different type of event.
Table 3: Incidence of Endpoint Events Placebo MULTAQ
400 mg BID(N=2327) (N=2301) HR 95% CI p-Value Primary endpoint
- Cardiovascular hospitalization or death from any cause
913 (39.2%)
727 (31.6%)
0.76
[0.68–0.83]
<0.0001
Components of the endpoint (as first event)
- •
- Cardiovascular hospitalization
856 (36.8%)
669 (29.1%)
- •
- Death from any cause
57 (2.4%)
58 (2.5%)
- Secondary endpoints (any time in study)
- •
- Death from any cause
135 (5.8%)
115 (5.0%)
0.86
[0.67–1.11]
0.24
- •
- Cardiovascular hospitalization
856 (36.8%)
669 (29.1%)
0.74
[0.67–0.82]
<0.0001
Components of the cardiovascular hospitalization endpoint (as first event)
- •
- AF and other supraventricular rhythm disorders
456 (19.6%)
292 (12.7%)
0.61
[0.53–0.71]
<0.0001
- •
- Other
400 (17.2%)
377 (16.4%)
0.89
[0.77–1.03]
0.11
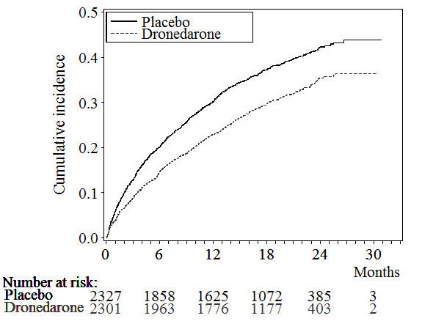
The Kaplan-Meier cumulative incidence curves showing the time to first event are displayed in Figure 3. The event curves separated early and continued to diverge over the 30-month follow-up period.
Figure 3: Kaplan-Meier Cumulative Incidence Curves from Randomization to First Cardiovascular Hospitalization or Death from Any Cause
Reasons for hospitalization included major bleeding (1% in both groups), syncope (1% in both groups), and ventricular arrhythmia (<1% in both groups).
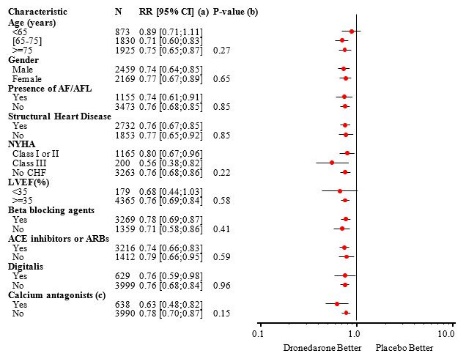
The reduction in cardiovascular hospitalization or death from any cause was generally consistent in all subgroups based on baseline characteristics or medications (ACE inhibitors or ARBs; beta-blockers, digoxin, statins, calcium channel blockers, diuretics) (see Figure 4).
- Figure 4: Relative Risk (MULTAQ vs Placebo) Estimates with 95% Confidence Intervals According to Selected Baseline Characteristics: First Cardiovascular Hospitalization or Death from Any Cause.
(a) Determined from Cox regression model
(b) P-value of interaction between baseline characteristics and treatment based on Cox regression model
(c) Calcium antagonists with heart rate lowering effects restricted to diltiazem, verapamil and bepridil14.2 EURIDIS and ADONIS
In EURIDIS and ADONIS, a total of 1237 patients in sinus rhythm with a prior episode of AF or AFL were randomized in an outpatient setting and treated with either MULTAQ 400 mg twice daily (n=828) or placebo (n=409) on top of conventional therapies (including oral anticoagulants, beta-blockers, ACE inhibitors or ARBs, chronic antiplatelet agents, diuretics, statins, digoxin, and calcium channel blockers). Patients had at least one ECG-documented AF/AFL episode during the 3 months prior to study entry but were in sinus rhythm for at least one hour. Patients ranged in age from 20 to 88 years, with the majority being Caucasian (97%), male (70%) patients. The most common comorbidities were hypertension (56.8%) and structural heart disease (41.5%), including coronary heart disease (21.8%). Patients were followed for 12 months.
In the pooled data from EURIDIS and ADONIS as well as in the individual trials, dronedarone delayed the time to first recurrence of AF/AFL (primary endpoint), lowering the risk of first AF/AFL recurrence during the 12-month study period by about 25%, with an absolute difference in recurrence rate of about 11% at 12 months.
14.3 ANDROMEDA
Patients recently hospitalized with symptomatic heart failure and severe left ventricular systolic dysfunction (wall motion index ≤1.2) were randomized to either MULTAQ 400 mg twice daily or matching placebo, with a primary composite end point of all-cause mortality or hospitalization for heart failure. Patients enrolled in ANDROMEDA were predominantly NYHA Class II (40%) and III (57%), and only 25% had AF at randomization. After enrollment of 627 patients and a median follow-up of 63 days, the trial was terminated because of excess mortality in the dronedarone group. Twenty-five (25) patients in the dronedarone group died versus 12 patients in the placebo group (hazard ratio 2.13; 95% CI: 1.07 to 4.25). The main reason for death was worsening heart failure. Baseline digoxin therapy was reported in 6/16 dronedarone patients versus 1/16 placebo patients who died of arrhythmia. In patients without baseline use of digoxin, no excess risk of arrhythmic death was observed in the dronedarone versus placebo groups.
There were also excess hospitalizations for cardiovascular reasons in the dronedarone group (71 vs 51 for placebo) [see Boxed Warning, Contraindications (4)].
14.4 PALLAS
Patients with permanent AF (AF documented in 2 weeks prior to randomization and at least 6 months prior to randomization in whom cardioversion had failed or was not planned) and additional risk factors for thromboembolism (coronary artery disease, prior stroke or TIA, symptomatic heart failure, LVEF <40%, peripheral arterial occlusive disease, or age >75 with hypertension and diabetes) were randomized to dronedarone 400 mg twice daily or placebo.
After enrollment of 3236 patients (placebo=1617 and dronedarone=1619) and a median follow up of 3.7 months for placebo and 3.9 for dronedarone, the study was terminated because of a significant increase in:
- •
- Mortality: 25 dronedarone versus 13 placebo (HR, 1.94; CI: 0.99 to 3.79). The majority of deaths in the dronedarone group were classified as arrhythmic/sudden deaths (HR, 3.26; CI: 1.06 to 10.0). Baseline digoxin therapy was reported in 11/13 dronedarone patients who died of arrhythmia. None of the arrhythmic deaths on placebo (4) reported use of digoxin. In patients without baseline use of digoxin, no excess risk of arrhythmic death was observed in the dronedarone versus placebo groups.
- •
- Stroke: 23 dronedarone versus 10 placebo (HR, 2.32; CI: 1.11 to 4.88). The increased risk of stroke observed with dronedarone was observed in the first two weeks of therapy (10 dronedarone vs 1 placebo), most of the subjects treated with dronedarone did not have an INR of 2.0 to 3.0 [see Warnings and Precautions (5.3)].
- •
- Hospitalizations for heart failure in the dronedarone group: 43 dronedarone versus 24 placebo (HR, 1.81; CI: 1.10 to 2.99).
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Administration Instructions
MULTAQ should be administered with a meal. Warn patients not to take MULTAQ with grapefruit juice.
If a dose is missed, patients should take the next dose at the regularly scheduled time and should not double the dose.
Advise patients to consult a physician before stopping treatment with MULTAQ.
New Onset or Worsening Heart Failure
Advise patients to consult a physician if they develop signs or symptoms of heart failure such as acute weight gain, dependent edema, or increasing shortness of breath.
Advise patients to inform their physician of any history of heart failure, rhythm disturbance other than atrial fibrillation or flutter or predisposing conditions such as uncorrected hypokalemia.
-
MEDICATION GUIDE
Medication Guide
MULTAQ (MUL-tak)
(dronedarone) TabletsRead this Medication Guide before you start taking MULTAQ and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment.
What is the most important information I should know about MULTAQ?
MULTAQ can cause serious side effects.
Do not take MULTAQ if you:
- 1.
- have symptoms of heart failure that recently worsened and you were hospitalized, or if you have severe heart failure.
MULTAQ doubles your risk of dying if you have these conditions. Heart failure means your heart does not pump blood through your body as well as it should.
Call your doctor right away if you have any signs or symptoms of heart failure during treatment with MULTAQ:
- •
- shortness of breath or wheezing at rest
- •
- wheezing, chest tightness or coughing up frothy sputum at rest, nighttime or after minor exercise
- •
- trouble sleeping or waking up at night because of breathing problems
- •
- using more pillows to prop yourself up at night so you can breathe more easily
- •
- gaining more than 5 pounds quickly
- •
- increasing swelling of feet or legs
- 1.
- have a type of atrial fibrillation (irregular heart rhythm) called permanent atrial fibrillation (AF).
- You and your doctor may decide not to try to change your heart rhythm back to a normal heart rhythm or your heart rhythm cannot be changed back to a normal rhythm. If you have permanent AF and take MULTAQ, you have a higher risk of death, stroke, and needing to be treated in a hospital for your heart failure.
- Your doctor will monitor your heart rhythm regularly to make sure your heartbeat keeps a normal rhythm.
Call your doctor right away if you notice that your pulse is irregular during treatment with MULTAQ. This is a sign that you are in atrial fibrillation.
MULTAQ may cause liver problems, including life-threatening liver failure. Your doctor may order blood tests to check your liver before you start taking MULTAQ and during treatment. In some cases, MULTAQ treatment may need to be stopped.
Call your doctor right away if you develop any of these signs and symptoms of liver problems during treatment with MULTAQ:
- •
- loss of appetite, nausea, vomiting
- •
- fever, feeling unwell, unusual tiredness
- •
- itching
- •
- yellowing of the skin or the whites of the eyes (jaundice)
- •
- unusual darkening of the urine
- •
- right upper stomach area pain or discomfort
What is MULTAQ?
MULTAQ is a prescription medicine used to lower the chance that you will need to go into the hospital for atrial fibrillation. It is meant for people who have had certain types of atrial fibrillation (paroxysmal or persistent AF) in the past, but are now in normal rhythm.
It is not known if MULTAQ is safe and effective in children younger than age 18 years old.
Who should not take MULTAQ?
See "What is the most important information I should know about taking MULTAQ?"
Do not take MULTAQ if:
- •
- you have a certain type of heart problem called heart block, and you do not have an implanted pacemaker
- •
- you have a slow heart rate, less than 50 beats each minute
- •
- you have severe liver problems or had liver or lung problems after using amiodarone (a medicine for abnormal heart rhythm)
- •
-
you take certain medicines that can change the amount of MULTAQ that gets into your body. Do not use these medicines with MULTAQ:
- •
- Nefazodone for depression
- •
- Norvir® (ritonavir) for HIV infection
- •
- Nizoral® (ketoconazole), and Sporanox® (itraconazole), and Vfend® (voriconazole) for fungal infections
- •
- Telithromycin and Biaxin® (clarithromycin) for bacterial infections
- •
- Cyclosporine for organ transplant
- •
-
You take certain medicines that can lead to a dangerous abnormal heart rhythm:Ask your doctor if you are not sure if your medicine is one that is listed above.
- •
- Some medicines for mental illness called phenothiazines
- •
- Some medicines for depression called tricyclic antidepressants
- •
- Some medicines for abnormal heart rhythm or fast heartbeat
- •
- Some medicines for bacterial infection
- •
-
You are pregnant or plan to become pregnant. It is not known if MULTAQ will harm your unborn baby. Talk to your doctor if you are pregnant or plan to become pregnant.
- •
- Women who may become pregnant should use effective birth control (contraception) while taking MULTAQ. Talk to your doctor about the best birth control methods for you.
- •
- You are breastfeeding or plan to breastfeed. It is not known if MULTAQ passes into your breast milk. You and your doctor should decide if you will take MULTAQ or breastfeed. You should not do both.
- •
- You are allergic to dronedarone or any of the other ingredients in MULTAQ. See the end of this Medication Guide for a complete list of ingredients in MULTAQ.
What should I tell my doctor before taking MULTAQ?
Before taking MULTAQ, tell your doctor if you:
- •
- have any other heart problems
- •
- have any other medical conditions
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins and herbal supplements. MULTAQ and certain other medicines can react with each other, causing serious side effects.
Especially tell your doctor and pharmacist if you take:
- •
- medicine for high blood pressure, chest pain, or other heart conditions
- •
- statin medicine to lower blood cholesterol
- •
- medicine for TB (tuberculosis)
- •
- medicine for seizures
- •
- digoxin (Lanoxin)
- •
- warfarin (Coumadin, Jantoven), a blood thinner medicine
- •
- medicine for organ transplant
- •
- herbal supplement called St. John's wort
Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist when you get a new medicine.
How should I take MULTAQ?
- •
- Take MULTAQ exactly as your doctor tells you.
- •
- Take MULTAQ two times a day with food, once with your morning meal and once with your evening meal.
- •
- Do not stop taking MULTAQ without first talking to your doctor, even if you are feeling well for a long time.
- •
- If you miss a dose, wait and take your next dose at your regular time. Do not take 2 doses at the same time. Do not try to make up for a missed dose.
- •
- If you take too much MULTAQ, call your doctor or go to the nearest hospital emergency room right away.
What should I avoid while taking MULTAQ?
Do not drink grapefruit juice while you are being treated with MULTAQ. Grapefruit juice can increase the amount of MULTAQ in your blood and increase the likelihood that you will have a side effect of MULTAQ.
What are the possible side effects of MULTAQ?
MULTAQ may cause serious side effects, including:
- •
- See "What is the most important information I should know about MULTAQ?"
- •
- Slowed heartbeat (bradycardia)
- •
- Inflammation of the lungs, including scarring and thickening. Call your doctor if you develop shortness of breath or a dry cough during treatment with MULTAQ.
- •
- Low potassium and magnesium levels in your blood. This can happen if you take certain water pills (diuretics) during treatment with MULTAQ. Your doctor may check you for this problem before and during treatment.
- •
- Changes in kidney function blood tests after starting MULTAQ. Your doctor may check you for this during treatment.
The most common side effects of MULTAQ include:
- •
- diarrhea
- •
- nausea
- •
- vomiting
- •
- stomach area (abdominal) pain
- •
- indigestion
- •
- feeling tired and weak
- •
- skin problems such as redness, rash, and itching
Tell your doctor about any side effect that bothers you or that does not go away. These are not all the possible side effects of MULTAQ. For more information ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store MULTAQ?
Store MULTAQ at room temperature (59°F–86°F or 15°C–30°C).
Keep MULTAQ and all medicines out of the reach of children.
General information about MULTAQ
Medicines are sometimes used for purposes other than those listed in a Medication Guide. Do not use MULTAQ for a condition for which it was not prescribed. Do not give MULTAQ to other people, even if they have the same symptoms or condition. It may harm them.
This Medication Guide summarizes the most important information about MULTAQ. If you would like more information:
- •
- Talk with your doctor
- •
- Ask your doctor or pharmacist for information about MULTAQ that was written for health-care professionals
- •
- For the latest information and Medication Guide, visit www.sanofi-aventis.us or call sanofi-aventis Medical Information Services at 1-800-633-1610 option 1. The Medication Guide may have changed since this copy was printed.
What are the ingredients in MULTAQ?
Active ingredient: dronedarone
Inactive ingredients: hypromellose, starch, crospovidone, poloxamer 407, lactose monohydrate, colloidal silicon dioxide, magnesium stearate, polyethylene glycol 6000, titanium dioxide, carnauba wax
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: November 2020
Manufactured by:
sanofi-aventis U.S. LLC
Bridgewater, NJ 08807
A SANOFI COMPANY©sanofi-aventis 2020
All rights reserved.Distributed By:
Cardinal Health
Dublin, OH 43017
L7605 Rev. A
MULTAQ is a trademark of sanofi-aventis.
The brands listed are the registered trademarks of their respective owners and are not trademarks of sanofi-aventis U.S. LLC. - Package/Label Display Panel
-
INGREDIENTS AND APPEARANCE
MULTAQ
dronedarone tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55154-8104(NDC:0024-4142) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Dronedarone (UNII: JQZ1L091Y2) (Dronedarone - UNII:JQZ1L091Y2) Dronedarone 400 mg Inactive Ingredients Ingredient Name Strength HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) starch, corn (UNII: O8232NY3SJ) CROSPOVIDONE (120 .MU.M) (UNII: 68401960MK) poloxamer 407 (UNII: TUF2IVW3M2) lactose monohydrate (UNII: EWQ57Q8I5X) silicon dioxide (UNII: ETJ7Z6XBU4) magnesium stearate (UNII: 70097M6I30) polyethylene glycol 6000 (UNII: 30IQX730WE) titanium dioxide (UNII: 15FIX9V2JP) carnauba wax (UNII: R12CBM0EIZ) Product Characteristics Color WHITE Score no score Shape OVAL Size 17mm Flavor Imprint Code 4142 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55154-8104-2 1260 in 1 BOTTLE; Type 0: Not a Combination Product 07/01/2009 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022425 07/01/2009 Labeler - Cardinal Health 107, LLC (118546603)