TOLBUTAMIDE- tolbutamide tablet
Mylan Pharmaceuticals Inc.
----------
DESCRIPTION
Tolbutamide is an oral blood-glucose-lowering drug of the sulfonylurea class. Tolbutamide is a pure, white, crystalline compound which is practically insoluble in water. The chemical name is benzenesulfonamide, N-[(butylamino)-carbonyl]-4-methyl-. Its structure can be represented as follows:
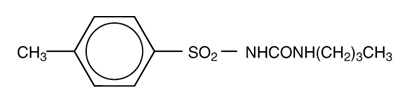
Tolbutamide is supplied as compressed tablets containing 500 mg of tolbutamide, USP.
Each tablet for oral administration contains 500 mg of tolbutamide and the following inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate and sodium starch glycolate.
CLINICAL PHARMACOLOGY
Actions
Tolbutamide appears to lower the blood glucose acutely by stimulating the release of insulin from the pancreas, an effect dependent upon functioning beta cells in the pancreatic islets. The mechanism by which tolbutamide lowers blood glucose during long-term administration has not been clearly established. With chronic administration in Type II diabetic patients, the blood-glucose-lowering effect persists despite a gradual decline in the insulin secretory response to the drug. Extrapancreatic effects may be involved in the mechanism of action of oral sulfonylurea hypoglycemic drugs.
Some patients who are initially responsive to oral hypoglycemic drugs, including tolbutamide, may become unresponsive or poorly responsive over time. Alternatively, tolbutamide may be effective in some patients who have become unresponsive to one or more of the other sulfonylurea drugs.
Pharmacokinetics
When administered orally, tolbutamide is readily absorbed from the gastrointestinal tract. Absorption is not impaired and glucose lowering and insulin releasing effects are not altered if the drug is taken with food. Detectable levels are present in the plasma within 20 minutes after oral ingestion of a 500 mg tolbutamide tablet, with peak levels occurring at 3 to 4 hours and only small amounts detectable at 24 hours. The half-life of tolbutamide is 4.5 to 6.5 hours. As tolbutamide has no p-amino group, it cannot be acetylated, which is one of the common modes of metabolic degradation for the antibacterial sulfonamides. However, the presence of the p-methyl group renders tolbutamide susceptible to oxidation, and this appears to be the principal manner of its metabolic degradation in man. The p-methyl group is oxidized to form a carboxyl group, converting tolbutamide into the totally inactive metabolite 1-butyl-3-p-carboxy-phenylsulfonylurea, which can be recovered in the urine within 24 hours in amounts accounting for up to 75% of the administered dose.
The major tolbutamide metabolite has been found to have no hypoglycemic or other action when administered orally and IV to both normal and diabetic subjects. This tolbutamide metabolite is highly soluble over the critical acid range of urinary pH values, and its solubility increases with increase in pH. Because of the marked solubility of the tolbutamide metabolite, crystalluria does not occur. A second metabolite, 1-butyl-3-(p-hydroxymethyl) phenyl sulfonylurea also occurs to a limited extent. It is an inactive metabolite.
The administration of 3 grams of tolbutamide to either nondiabetic or tolbutamide-responsive diabetic subjects will, in both instances, occasion a gradual lowering of blood glucose. Increasing the dose to 6 grams does not usually cause a response which is significantly different from that produced by the 3 gram dose. Following the administration of a 3 gram dose of tolbutamide solution, non-diabetic fasting adults exhibit a 30% or greater reduction in blood glucose within one hour, following which the blood glucose gradually returns to the fasting level over 6 to 12 hours. Following the administration of a 3 gram dose of tolbutamide solution, tolbutamide responsive diabetic patients show a gradually progressive blood glucose lowering effect, the maximal response being reached between 5 to 8 hours after ingestion of a single 3 gram dose. The blood glucose then rises gradually and by the 24th hour has usually returned to pretest levels. The magnitude of the reduction, when expressed in terms of percent of the pretest blood glucose, tends to be similar to the response seen in the nondiabetic subject.
INDICATIONS AND USAGE
Tolbutamide tablets are indicated as an adjunct to diet to lower the blood glucose in patients with non-insulin-dependent diabetes mellitus (type II) whose hyperglycemia cannot be controlled by diet alone.
In initiating treatment for non-insulin-dependent diabetes, diet should be emphasized as the primary form of treatment. Caloric restriction and weight loss are essential in the obese diabetic patient. Proper dietary management alone may be effective in controlling the blood glucose and symptoms of hyperglycemia. The importance of regular physical activity should also be stressed, and cardiovascular risk factors should be identified and corrective measures taken where possible.
If this treatment program fails to reduce symptoms and/or blood glucose, the use of an oral sulfonylurea or insulin should be considered. Use of tolbutamide tablets must be viewed by both the physician and patient as a treatment in addition to diet, and not as a substitute for diet or as a convenient mechanism for avoiding dietary restraint. Furthermore, loss of blood glucose control on diet alone may be transient, thus requiring only short-term administration of tolbutamide tablets.
During maintenance programs, tolbutamide tablets should be discontinued if satisfactory lowering of blood glucose is no longer achieved. Judgments should be based on regular clinical and laboratory evaluations.
In considering the use of tolbutamide tablets in asymptomatic patients, it should be recognized that controlling the blood glucose in non-insulin dependent diabetes has not been definitely established to be effective in preventing the long-term cardiovascular or neural complications of diabetes.
CONTRAINDICATIONS
Tolbutamide tablets are contraindicated in patients with:
- •
- Known hypersensitivity or allergy to the drug.
- •
- Diabetic ketoacidosis, with or without coma. This condition should be treated with insulin.
- •
- Type I diabetes, as sole therapy.
WARNINGS
SPECIAL WARNING ON INCREASED RISK OF CARDIOVASCULAR MORTALITY
The administration of oral hypoglycemic drugs has been reported to be associated with increased cardiovascular mortality as compared to treatment with diet alone or diet plus insulin. This warning is based on the study conducted by the University Group Diabetes Program (UGDP), a long-term prospective clinical trial designed to evaluate the effectiveness of glucose-lowering drugs in preventing or delaying vascular complications in patients with non-insulin-dependent diabetes. The study involved 823 patients who were randomly assigned to one of four treatment groups (Diabetes, 19 (supp.2):747-830, 1970).
UGDP reported that patients treated for 5 to 8 years with diet plus a fixed dose of tolbutamide (1.5 grams per day) had a rate of cardiovascular mortality approximately 2½ times that of patients treated with diet alone. A significant increase in total mortality was not observed, but the use of tolbutamide was discontinued based on the increase in cardiovascular mortality, thus limiting the opportunity for the study to show an increase in overall mortality. Despite controversy regarding the interpretation of these results, the findings of the UGDP study provide an adequate basis for this warning. The patient should be informed of the potential risks and advantages of tolbutamide and of alternative modes of therapy. Although only one drug in the sulfonylurea class (tolbutamide) was included in this study, it is prudent from a safety standpoint to consider that this warning may also apply to other oral hypoglycemic drugs in this class, in view of their close similarities in mode of action and chemical structure.
PRECAUTIONS
General
Hypoglycemia
All sulfonylurea drugs are capable of producing severe hypoglycemia. Proper patient selection, dosage, and instructions are important to avoid hypoglycemic episodes. Renal or hepatic insufficiency may cause elevated blood levels of tolbutamide and the latter may also diminish gluconeogenic capacity, both of which increase the risk of serious hypoglycemic reactions. Elderly, debilitated or malnourished patients, and those with adrenal or pituitary insufficiency are particularly susceptible to the hypoglycemic action of glucose-lowering drugs. Hypoglycemia may be difficult to recognize in the elderly, and in people who are taking beta-adrenergic blocking drugs. Hypoglycemia is more likely to occur when caloric intake is deficient, after severe or prolonged exercise, when alcohol is ingested, or when more than one glucose-lowering drug is used.
Loss of Control of Blood Glucose
When a patient stabilized on any diabetic regimen is exposed to stress such as fever, trauma, infection, or surgery, a loss of control may occur. At such times, it may be necessary to discontinue tolbutamide and administer insulin.
The effectiveness of any oral hypoglycemic drug, including tolbutamide, in lowering blood glucose to a desired level decreases in many patients over a period of time, which may be due to progression of the severity of the diabetes or to diminished responsiveness to the drug. This phenomenon is known as a secondary failure, to distinguish it from primary failure in which the drug is ineffective in an individual patient when first given. Adequate adjustment of dose and adherence to diet should be assessed before classifying a patient as a secondary failure.
Hemolytic Anemia
Treatment of patients with glucose 6-phosphate dehydrogenase (G6PD) deficiency with sulfonylurea agents can lead to hemolytic anemia. Because tolbutamide belongs to the class of sulfonylurea agents, caution should be used in patients with G6PD deficiency and a non-sulfonylurea alternative should be considered. In post-marketing reports, hemolytic anemia has also been reported in patients who did not have known G6PD deficiency.
Information for Patients
Patients should be informed of the potential risks and advantages of tolbutamide and of alternative modes of therapy. They should also be informed about the importance of adherence to dietary instructions, of a regular exercise program, and of regular testing of urine and/or blood glucose.
The risks of hypoglycemia, its symptoms and treatment, and conditions that predispose to its development should be explained to patients and responsible family members. Primary and secondary failure should also be explained.
Laboratory Tests
Blood and urine glucose should be monitored periodically. Measurement of glycosylated hemoglobin may be useful.
A metabolite of tolbutamide in urine may give a false positive reaction for albumin if measured by the acidification-after-boiling test, which causes the metabolite to precipitate. There is no interference with the sulfosalicylic acid test.
Drug Interactions
The hypoglycemia action of sulfonylurea may be potentiated by certain drugs including non-steroidal anti-inflammatory agents and other drugs that are highly protein bound, salicylates, sulfonamides, chloramphenicol, probenecid, coumarins, monoamine oxidase inhibitors, and beta-adrenergic blocking agents. When such drugs are administered to a patient receiving tolbutamide, the patient should be observed closely for hypoglycemia. When such drugs are withdrawn from a patient receiving tolbutamide, the patient should be observed closely for loss of control.
Certain drugs tend to produce hyperglycemia and may lead to loss of control. These drugs include the thiazides and other diuretics, corticosteroids, phenothiazines, thyroid products, estrogens, oral contraceptives, phenytoin, nicotinic acid, sympathomimetics, calcium channel blocking drugs, and isoniazid. When such drugs are administered to a patient receiving tolbutamide, the patient should be closely observed for loss of control. When such drugs are withdrawn from a patient receiving tolbutamide, the patient should be observed closely for hypoglycemia.
A potential interaction between oral miconazole and oral hypoglycemic agents leading to severe hypoglycemia has been reported. Whether this interaction also occurs with the intravenous, topical or vaginal preparations of miconazole is not known.
Carcinogenicity and Mutagenicity
Bioassay for carcinogenicity was performed in both sexes of rats and mice following ingestion of tolbutamide for 78 weeks. No evidence of carcinogenicity was found.
Tolbutamide has also been demonstrated to be nonmutagenic in the Ames salmonella/mammalian microsome mutagenicity test.
Pregnancy
Teratogenic Effects: Pregnancy Category C
Tolbutamide has been shown to be teratogenic in rats when given in doses 25 to 100 times the human dose. In some studies, pregnant rats given high doses of tolbutamide have shown ocular and bony abnormalities and increased mortality in offspring. Repeat studies in other species (rabbits) have not demonstrated a teratogenic effect. There are no adequate and well controlled studies in pregnant women. Tolbutamide is not recommended for the treatment of pregnant diabetic patients.
Serious consideration should also be given to the possible hazards of the use of tolbutamide in women of childbearing age and in those who might become pregnant while using the drug.
Because recent information suggests that abnormal blood glucose levels during pregnancy are associated with a higher incidence of congenital abnormalities, many experts recommend that insulin be used during pregnancy to maintain blood glucose levels as close to normal as possible.
Nonteratogenic Effects
Prolonged severe hypoglycemia (4 to 10 days) has been reported in neonates born to mothers who were receiving a sulfonylurea drug at the time of delivery. This has been reported more frequently with the use of agents with prolonged half-lives. If tolbutamide is used during pregnancy, it should be discontinued at least 2 weeks before the expected delivery date.
Nursing Mothers
Although it is not known whether tolbutamide is excreted in human milk, some sulfonylurea drugs are known to be excreted in human milk. Because the potential for hypoglycemia in nursing infants may exist, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. If the drug is discontinued and if diet alone is inadequate for controlling blood glucose, insulin therapy should be considered.
ADVERSE REACTIONS
Gastrointestinal Reactions
Cholestatic jaundice may occur rarely; tolbutamide should be discontinued if this occurs. Gastrointestinal disturbances, e.g., nausea, epigastric fullness, and heartburn, are the most common reactions and occur in 1.4% of patients treated during clinical trial. They tend to be dose related and may disappear when dosage is reduced.
Dermatologic Reactions
Allergic skin reactions, e.g., pruritus, erythema, urticaria, and morbilliform or maculopapular eruptions, occur in 1.1% of patients treated during clinical trials. These may be transient and may disappear despite continued use of tolbutamide; if skin reactions persist, the drug should be discontinued.
Porphyria cutanea tarda and photosensitivity reactions have been reported with sulfonylureas.
Hematologic Reactions
Leukopenia, agranulocytosis, thrombocytopenia, hemolytic anemia, aplastic anemia, and pancytopenia have been reported with sulfonylureas.
Metabolic Reactions
Hepatic porphyria and disulfiram-like reactions have been reported with sulfonylureas.
OVERDOSAGE
Overdosage of sulfonylureas including tolbutamide can produce hypoglycemia. Mild hypoglycemic symptoms without loss of consciousness or neurologic findings should be treated aggressively with oral glucose and adjustments in drug dosage and/or meal patterns. Close monitoring should continue until the physician is assured that the patient is out of danger. Severe hypoglycemic reactions with coma, seizure, or other neurological impairment occur infrequently, but constitute medical emergencies requiring immediate hospitalization. If hypoglycemic coma is diagnosed or suspected, the patient should be given a rapid intravenous injection of concentrated (50%) dextrose injection. This should be followed by a continuous infusion of a more dilute (10%) dextrose injection at a rate that will maintain the blood glucose at a level above 100 mg/dL. Patients should be closely monitored for a minimum of 24 to 48 hours since hypoglycemia may recur after apparent clinical recovery.
DOSAGE AND ADMINISTRATION
There is no fixed dosage regimen for the management of diabetes mellitus with tolbutamide tablets or any other hypoglycemic agent. In addition to the usual monitoring of urinary glucose, the patient's blood glucose must also be monitored periodically to determine the minimum effective dose for the patient; to detect primary failure, i.e., inadequate lowering of blood glucose at the maximum recommended dose of medication; and to detect secondary failure, i.e., loss of an adequate blood glucose lowering response after an initial period of effectiveness. Glycosylated hemoglobin levels may also be of value in monitoring the patient's response to therapy.
Short-term administration of tolbutamide tablets may be sufficient during periods of transient loss of control in patients usually controlled well on diet.
Usual Starting Dose
The usual starting dose is 1 to 2 grams daily. This may be increased or decreased, depending on individual patient response. Failure to follow an appropriate dosage regimen may precipitate hypoglycemia. Patients who do not adhere to their prescribed dietary regimens are more prone to exhibit unsatisfactory response to drug therapy.
Transfer from Other Hypoglycemic Therapy
Patients Receiving Other Antidiabetic Therapy
Transfer of patients from other oral antidiabetes regimens to tolbutamide tablets should be done conservatively. When transferring patients from oral hypoglycemic agents other than chlorpropamide to tolbutamide, no transition period and no initial or priming doses are necessary. When transferring patients from chlorpropamide, however, particular care should be exercised during the first 2 weeks because of the prolonged retention of chlorpropamide, in the body and the possibility that subsequent overlapping drug effects might provoke hypoglycemia.
Patients Receiving Insulin
Patients requiring 20 units or less of insulin daily may be placed directly on tolbutamide tablets and insulin abruptly discontinued. Patients whose insulin requirement is between 20 and 40 units daily may be started on therapy with tolbutamide tablets with a concurrent 30% to 50% reduction in insulin dose, with further daily reduction of the insulin when response to tolbutamide tablets is observed. In patients requiring more than 40 units of insulin daily, therapy with tolbutamide tablets may be initiated in conjunction with a 20% reduction in insulin dose the first day, with further careful reduction of insulin as response is observed. Occasionally, conversion to tolbutamide tablets in the hospital may be advisable in candidates who require more than 40 units of insulin daily. During this conversion period when both insulin and tolbutamide tablets are being used hypoglycemia may rarely occur. During insulin withdrawal, patients should test their urine for glucose and acetone at least 3 times daily and report results to their physician. The appearance of persistent acetonuria with glycosuria indicates that the patient is type I diabetic who requires insulin therapy.
Usual Maintenance Dose
The maintenance dose is in the range of 0.25 to 3 grams daily. Maintenance doses above 2 grams are seldom required.
Dosage Interval
The total daily dose may be taken either in the morning or in divided doses through the day. While either schedule is usually effective, the divided dose system is preferred by some clinicians from the standpoint of digestive tolerance.
In elderly patients, debilitated or malnourished patients, and patients with impaired renal or hepatic function, the initial and maintenance dosing should be conservative to avoid hypoglycemic reactions (see PRECAUTIONS).
HOW SUPPLIED
Tolbutamide Tablets, USP are available containing 500 mg of tolbutamide, USP. The tablets are white to off-white round, scored tablets debossed with M to the left of the score and 13 to the right of the score on one side of the tablet and blank on the other side. They are available as follows:
NDC 0378-0215-01
bottles of 100 tablets
NDC 0378-0215-05
bottles of 500 tablets
Store at 20° to 25°C (68° to 77°F). [See USP for Controlled Room Temperature.]
Protect from light.
Dispense in a tight, light-resistant container as defined in the USP using a child-resistant closure.
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505
REVISED FEBRUARY 2009
TOLB:R14
PRINCIPAL DISPLAY PANEL - 500 mg
NDC 0378-0215-01
TOLBUTamide
Tablets, USP
500 mg
Rx only 100 Tablets
Each tablet contains
Tolbutamide, USP 500 mg
Dispense in a tight, light-resistant
container as defined in the USP
using a child-resistant closure.
Keep container tightly closed.
Keep this and all medication
out of the reach of children.
Store at 20° to 25°C (68° to 77°F).
[See USP Controlled Room
Temperature.]
Protect from light.
Usual Dosage: See accompanying
prescribing information.
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505 U.S.A.
Mylan.com
RM0215A12
| TOLBUTAMIDE
tolbutamide tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Mylan Pharmaceuticals Inc. (059295980) |