Label: RENFLEXIS- infliximab injection, powder, lyophilized, for solution

- NDC Code(s): 0006-4305-01, 0006-4305-02
- Packager: Merck Sharp & Dohme LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated January 26, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use RENFLEXIS safely and effectively. See full prescribing information for RENFLEXIS.
RENFLEXIS (infliximab-abda) for injection, for intravenous use
Initial U.S. Approval: 2017
RENFLEXIS (infliximab-abda) is biosimilar* to REMICADE (infliximab). (1)WARNING: SERIOUS INFECTIONS and MALIGNANCY
See full prescribing information for complete boxed warning.
- •
- Increased risk of serious infections leading to hospitalization or death, including tuberculosis (TB), bacterial sepsis, invasive fungal infections (such as histoplasmosis) and infections due to other opportunistic pathogens.
- •
- Discontinue RENFLEXIS if a patient develops a serious infection.
- •
- Perform test for latent TB; if positive, start treatment for TB prior to starting RENFLEXIS. Monitor all patients for active TB during treatment, even if initial latent TB test is negative. (5.1)
- •
- Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with tumor necrosis factor (TNF) blockers, including infliximab products.
- •
- Postmarketing cases of fatal hepatosplenic T-cell lymphoma (HSTCL) have been reported in patients treated with TNF blockers including infliximab products. Almost all had received azathioprine or 6- mercaptopurine concomitantly with a TNF-blocker at or prior to diagnosis. The majority of cases were reported in patients with Crohn's disease or ulcerative colitis, most of whom were adolescent or young adult males. (5.2)
INDICATIONS AND USAGE
RENFLEXIS is a tumor necrosis factor (TNF) blocker indicated for:
Crohn's Disease:
- •
- reducing signs and symptoms and inducing and maintaining clinical remission in adult patients with moderately to severely active disease who have had an inadequate response to conventional therapy. (1.1)
- •
- reducing the number of draining enterocutaneous and rectovaginal fistulas and maintaining fistula closure in adult patients with fistulizing disease. (1.1)
Pediatric Crohn's Disease:
- •
- reducing signs and symptoms and inducing and maintaining clinical remission in pediatric patients with moderately to severely active disease who have had an inadequate response to conventional therapy. (1.2)
Ulcerative Colitis:
- •
- reducing signs and symptoms, inducing and maintaining clinical remission and mucosal healing, and eliminating corticosteroid use in adult patients with moderately to severely active disease who have had an inadequate response to conventional therapy. (1.3)
Pediatric Ulcerative Colitis:
- •
- reducing signs and symptoms and inducing and maintaining clinical remission in pediatric patients with moderately to severely active disease who have had an inadequate response to conventional therapy. (1.4)
Rheumatoid Arthritis in combination with methotrexate:
- •
- reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in patients with moderately to severely active disease. (1.5)
Ankylosing Spondylitis:
- •
- reducing signs and symptoms in patients with active disease. (1.6)
Psoriatic Arthritis:
- •
- reducing signs and symptoms of active arthritis, inhibiting the progression of structural damage, and improving physical function. (1.7)
Plaque Psoriasis:
- •
- treatment of adult patients with chronic severe (i.e., extensive and/or disabling) plaque psoriasis who are candidates for systemic therapy and when other systemic therapies are medically less appropriate. (1.8)
DOSAGE AND ADMINISTRATION
RENFLEXIS is administered by intravenous infusion over a period of not less than 2 hours. (2.11)
Crohn's Disease:
- •
- 5 mg/kg at 0, 2 and 6 weeks, then every 8 weeks. Some adult patients who initially respond to treatment may benefit from increasing the dose to 10 mg/kg if they later lose their response. (2.1)
Pediatric Crohn's Disease:
- •
- 5 mg/kg at 0, 2 and 6 weeks, then every 8 weeks. (2.2)
Ulcerative Colitis:
- •
- 5 mg/kg at 0, 2 and 6 weeks, then every 8 weeks. (2.3)
Pediatric Ulcerative Colitis:
- •
- 5 mg/kg at 0, 2 and 6 weeks, then every 8 weeks. (2.4)
Rheumatoid Arthritis:
- •
- In conjunction with methotrexate, 3 mg/kg at 0, 2 and 6 weeks, then every 8 weeks. Some patients may benefit from increasing the dose up to 10 mg/kg or treating as often as every 4 weeks. (2.5)
Ankylosing Spondylitis:
- •
- 5 mg/kg at 0, 2 and 6 weeks, then every 6 weeks. (2.6)
Psoriatic Arthritis and Plaque Psoriasis:
DOSAGE FORMS AND STRENGTHS
For injection: 100 mg of lyophilized infliximab-abda in a 20 mL vial for intravenous infusion. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- •
- Serious infections – do not give RENFLEXIS during an active infection. If an infection develops, monitor carefully and stop RENFLEXIS if infection becomes serious. (5.1)
- •
- Invasive fungal infections – for patients who develop a systemic illness on RENFLEXIS, consider empiric antifungal therapy for those who reside or travel to regions where mycoses are endemic (5.1)
- •
- Malignancies – the incidence of malignancies, including invasive cervical cancer and lymphoma, was greater in TNF blocker treated patients than in controls. Due to the risk of HSTCL carefully assess the risk/benefit especially if the patient has Crohn's disease or ulcerative colitis, is male, and is receiving azathioprine or 6-mercaptopurine treatment. (5.2)
- •
- Hepatitis B virus reactivation – test for HBV infection before starting RENFLEXIS. Monitor HBV carriers during and several months after therapy. If reactivation occurs, stop RENFLEXIS and begin anti-viral therapy. (5.3)
- •
- Hepatotoxicity – severe hepatic reactions, some fatal or necessitating liver transplantation. Stop RENFLEXIS in cases of jaundice and/or marked liver enzyme elevations. (5.4)
- •
- Heart failure – new onset or worsening symptoms may occur. (4, 5.5)
- •
- Cytopenias – advise patients to seek immediate medical attention if signs and symptoms develop, and consider stopping RENFLEXIS. (5.6)
- •
- Hypersensitivity – serious infusion reactions including anaphylaxis or serum sickness-like reactions may occur. (5.7)
- •
- Cardiovascular and Cerebrovascular Reactions – Cerebrovascular accidents, myocardial infarctions (some fatal), and arrhythmias have been reported during and within 24 hours of initiation of infliximab product infusion. Monitor patients during RENFLEXIS infusion and if serious reaction occurs, discontinue infusion. (5.8)
- •
- Demyelinating disease – exacerbation or new onset may occur. (5.9)
- •
- Lupus-like syndrome – stop RENFLEXIS if syndrome develops. (5.14)
- •
- Live vaccines or therapeutic infectious agents – should not be given with RENFLEXIS. Bring pediatric patients up to date with all vaccinations prior to initiating RENFLEXIS. At least a six month waiting period following birth is recommended before the administration of live vaccines to infants exposed in utero to infliximab products (5.15)
ADVERSE REACTIONS
Most common adverse reactions (>10%) – infections (e.g. upper respiratory, sinusitis, and pharyngitis), infusion-related reactions, headache, and abdominal pain. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., at 1-877-888-4231 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Use with anakinra or abatacept– increased risk of serious infections (7.1)
USE IN SPECIFIC POPULATIONS
Pediatric Use – Infliximab products have not been studied in children with Crohn's disease or ulcerative colitis <6 years of age. (8.4)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
- *
- Biosimilar means that the biological product is approved based on data demonstrating that it is highly similar to an FDA-approved biological product, known as a reference product, and that there are no clinically meaningful differences between the biosimilar product and the reference product. Biosimilarity of RENFLEXIS has been demonstrated for the condition(s) of use (e.g. indication(s), dosing regimen(s)), strength(s), dosage form(s), and route(s) of administration described in its Full Prescribing Information.
Revised: 1/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SERIOUS INFECTIONS and MALIGNANCY
1 INDICATIONS AND USAGE
1.1 Crohn's Disease
1.2 Pediatric Crohn's Disease
1.3 Ulcerative Colitis
1.4 Pediatric Ulcerative Colitis
1.5 Rheumatoid Arthritis
1.6 Ankylosing Spondylitis
1.7 Psoriatic Arthritis
1.8 Plaque Psoriasis
2 DOSAGE AND ADMINISTRATION
2.1 Crohn's Disease
2.2 Pediatric Crohn's Disease
2.3 Ulcerative Colitis
2.4 Pediatric Ulcerative Colitis
2.5 Rheumatoid Arthritis
2.6 Ankylosing Spondylitis
2.7 Psoriatic Arthritis
2.8 Plaque Psoriasis
2.9 Monitoring to Assess Safety
2.10 Administration Instructions Regarding Infusion Reactions
2.11 General Considerations and Instructions for Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
5.2 Malignancies
5.3 Hepatitis B Virus Reactivation
5.4 Hepatotoxicity
5.5 Patients with Heart Failure
5.6 Hematologic Reactions
5.7 Hypersensitivity
5.8 Cardiovascular and Cerebrovascular Reactions During and After Infusion
5.9 Neurologic Reactions
5.10 Use with Anakinra
5.11 Use with Abatacept
5.12 Concurrent Administration with other Biological Therapeutics
5.13 Switching between Biological Disease-Modifying Antirheumatic Drugs (DMARDs)
5.14 Autoimmunity
5.15 Live Vaccines/Therapeutic Infectious Agents
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Use with Anakinra or Abatacept
7.2 Use with Tocilizumab
7.3 Use with Other Biological Therapeutics
7.4 Methotrexate (MTX) and Other Concomitant Medications
7.5 Immunosuppressants
7.6 Cytochrome P450 Substrates
7.7 Live Vaccines/Therapeutic Infectious Agents
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Crohn's Disease
14.2 Pediatric Crohn's Disease
14.3 Ulcerative Colitis
14.4 Pediatric Ulcerative Colitis
14.5 Rheumatoid Arthritis
14.6 Ankylosing Spondylitis
14.7 Psoriatic Arthritis
14.8 Plaque Psoriasis
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SERIOUS INFECTIONS and MALIGNANCY
SERIOUS INFECTIONS
Patients treated with infliximab products are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
RENFLEXIS should be discontinued if a patient develops a serious infection or sepsis.
Reported infections include:
- •
- Active tuberculosis, including reactivation of latent tuberculosis. Patients with tuberculosis have frequently presented with disseminated or extrapulmonary disease. Patients should be tested for latent tuberculosis before RENFLEXIS use and during therapy.1,2 Treatment for latent infection should be initiated prior to RENFLEXIS use.
- •
- Invasive fungal infections, including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, and pneumocystosis. Patients with histoplasmosis or other invasive fungal infections may present with disseminated, rather than localized, disease. Antigen and antibody testing for histoplasmosis may be negative in some patients with active infection. Empiric anti-fungal therapy should be considered in patients at risk for invasive fungal infections who develop severe systemic illness.
- •
- Bacterial, viral and other infections due to opportunistic pathogens, including Legionella and Listeria.
The risks and benefits of treatment with RENFLEXIS should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with RENFLEXIS, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy.
MALIGNANCY
Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers, including infliximab products [see Warnings and Precautions (5.2)].
Postmarketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have been reported in patients treated with TNF blockers including infliximab products. These cases have had a very aggressive disease course and have been fatal. Almost all patients had received treatment with azathioprine or 6-mercaptopurine concomitantly with a TNF-blocker at or prior to diagnosis. The majority of reported cases have occurred in patients with Crohn's disease or ulcerative colitis and most were in adolescent and young adult males.
-
1 INDICATIONS AND USAGE
1.1 Crohn's Disease
RENFLEXIS is indicated for reducing signs and symptoms and inducing and maintaining clinical remission in adult patients with moderately to severely active Crohn's disease who have had an inadequate response to conventional therapy.
RENFLEXIS is indicated for reducing the number of draining enterocutaneous and rectovaginal fistulas and maintaining fistula closure in adult patients with fistulizing Crohn's disease.
1.2 Pediatric Crohn's Disease
RENFLEXIS is indicated for reducing signs and symptoms and inducing and maintaining clinical remission in pediatric patients 6 years of age and older with moderately to severely active Crohn's disease who have had an inadequate response to conventional therapy.
1.3 Ulcerative Colitis
RENFLEXIS is indicated for reducing signs and symptoms, inducing and maintaining clinical remission and mucosal healing, and eliminating corticosteroid use in adult patients with moderately to severely active ulcerative colitis who have had an inadequate response to conventional therapy.
1.4 Pediatric Ulcerative Colitis
RENFLEXIS is indicated for reducing signs and symptoms and inducing and maintaining clinical remission in pediatric patients 6 years of age and older with moderately to severely active ulcerative colitis who have had an inadequate response to conventional therapy.
1.5 Rheumatoid Arthritis
RENFLEXIS, in combination with methotrexate, is indicated for reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in patients with moderately to severely active rheumatoid arthritis.
1.6 Ankylosing Spondylitis
RENFLEXIS is indicated for reducing signs and symptoms in patients with active ankylosing spondylitis.
1.7 Psoriatic Arthritis
RENFLEXIS is indicated for reducing signs and symptoms of active arthritis, inhibiting the progression of structural damage, and improving physical function in patients with psoriatic arthritis.
1.8 Plaque Psoriasis
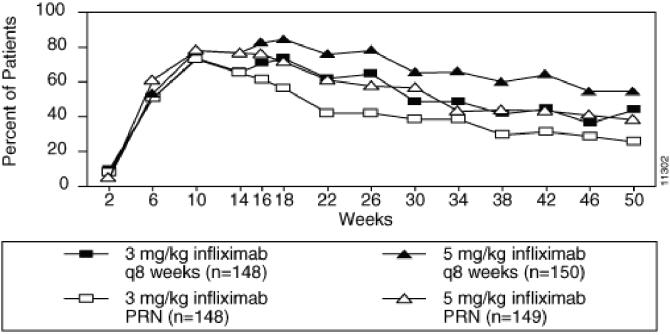
RENFLEXIS is indicated for the treatment of adult patients with chronic severe (i.e., extensive and/or disabling) plaque psoriasis who are candidates for systemic therapy and when other systemic therapies are medically less appropriate. RENFLEXIS should only be administered to patients who will be closely monitored and have regular follow-up visits with a physician [see Boxed Warnings, Warnings and Precautions (5)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Crohn's Disease
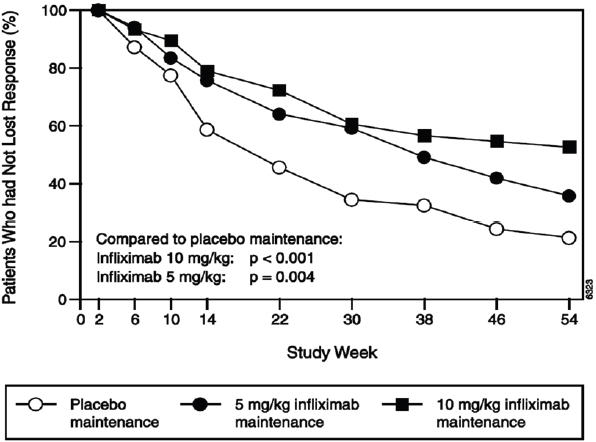
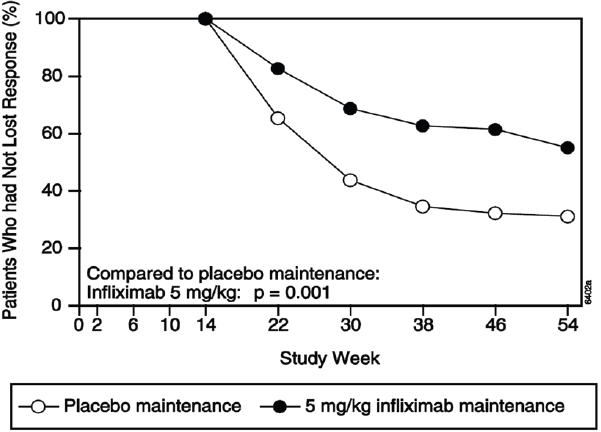
The recommended dose of RENFLEXIS is 5 mg/kg given as an intravenous induction regimen at 0, 2 and 6 weeks followed by a maintenance regimen of 5 mg/kg every 8 weeks thereafter for the treatment of adults with moderately to severely active Crohn's disease or fistulizing Crohn's disease. For adult patients who respond and then lose their response, consideration may be given to treatment with 10 mg/kg. Patients who do not respond by Week 14 are unlikely to respond with continued dosing and consideration should be given to discontinue RENFLEXIS in these patients.
2.2 Pediatric Crohn's Disease
The recommended dose of RENFLEXIS for pediatric patients 6 years and older with moderately to severely active Crohn's disease is 5 mg/kg given as an intravenous induction regimen at 0, 2 and 6 weeks followed by a maintenance regimen of 5 mg/kg every 8 weeks.
2.3 Ulcerative Colitis
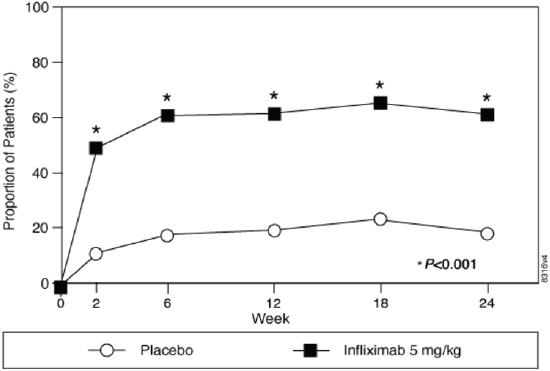
The recommended dose of RENFLEXIS is 5 mg/kg given as an intravenous induction regimen at 0, 2 and 6 weeks followed by a maintenance regimen of 5 mg/kg every 8 weeks thereafter for the treatment of adult patients with moderately to severely active ulcerative colitis.
2.4 Pediatric Ulcerative Colitis
The recommended dose of RENFLEXIS for pediatric patients 6 years and older with moderately to severely active ulcerative colitis is 5 mg/kg given as an intravenous induction regimen at 0, 2 and 6 weeks followed by a maintenance regimen of 5 mg/kg every 8 weeks.
2.5 Rheumatoid Arthritis
The recommended dose of RENFLEXIS is 3 mg/kg given as an intravenous induction regimen at 0, 2 and 6 weeks followed by a maintenance regimen of 3 mg/kg every 8 weeks thereafter for the treatment of moderately to severely active rheumatoid arthritis. RENFLEXIS should be given in combination with methotrexate. For patients who have an incomplete response, consideration may be given to adjusting the dose up to 10 mg/kg or treating as often as every 4 weeks bearing in mind that risk of serious infections is increased at higher doses [see Adverse Reactions (6.1)].
2.6 Ankylosing Spondylitis
The recommended dose of RENFLEXIS is 5 mg/kg given as an intravenous induction regimen at 0, 2 and 6 weeks followed by a maintenance regimen of 5 mg/kg every 6 weeks thereafter for the treatment of active ankylosing spondylitis.
2.7 Psoriatic Arthritis
The recommended dose of RENFLEXIS is 5 mg/kg given as an intravenous induction regimen at 0, 2 and 6 weeks followed by a maintenance regimen of 5 mg/kg every 8 weeks thereafter for the treatment of psoriatic arthritis. RENFLEXIS can be used with or without methotrexate.
2.8 Plaque Psoriasis
The recommended dose of RENFLEXIS is 5 mg/kg given as an intravenous induction regimen at 0, 2 and 6 weeks followed by a maintenance regimen of 5 mg/kg every 8 weeks thereafter for the treatment of chronic severe (i.e., extensive and/or disabling) plaque psoriasis.
2.9 Monitoring to Assess Safety
Prior to initiating RENFLEXIS and periodically during therapy, patients should be evaluated for active tuberculosis and tested for latent infection [see Warnings and Precautions (5.1)].
2.10 Administration Instructions Regarding Infusion Reactions
Adverse effects during administration of infliximab products have included flu-like symptoms, headache, dyspnea, hypotension, transient fever, chills, gastrointestinal symptoms, and skin rashes. Anaphylaxis might occur at any time during RENFLEXIS infusion. Approximately 20% of patients in all clinical trials of infliximab experienced an infusion reaction compared with 10% of placebo-treated patients [see Adverse Reactions (6.1)]. Prior to infusion with RENFLEXIS, premedication may be administered at the physician's discretion. Premedication could include antihistamines (anti-H1 +/- anti-H2), acetaminophen and/or corticosteroids.
During infusion, mild to moderate infusion reactions may improve following slowing or suspension of the infusion, and upon resolution of the reaction, reinitiation at a lower infusion rate and/or therapeutic administration of antihistamines, acetaminophen, and/or corticosteroids. For patients that do not tolerate the infusion following these interventions, RENFLEXIS should be discontinued.
During or following infusion, patients who have severe infusion-related hypersensitivity reactions should be discontinued from further RENFLEXIS treatment. The management of severe infusion reactions should be dictated by the signs and symptoms of the reaction. Appropriate personnel and medication should be available to treat anaphylaxis if it occurs.
2.11 General Considerations and Instructions for Preparation and Administration
RENFLEXIS is intended for use under the guidance and supervision of a physician. The reconstituted infusion solution should be prepared by a trained medical professional using aseptic technique by the following procedure:
- 1.
- Calculate the dose, total volume of reconstituted RENFLEXIS solution required and the number of RENFLEXIS vials needed. Each RENFLEXIS vial contains 100 mg of the infliximab-abda antibody.
- 2.
- Reconstitute each RENFLEXIS vial with 10 mL of Sterile Water for Injection, USP, using a syringe equipped with a 21-gauge or smaller needle as follows: Remove the flip-top from the vial and wipe the top with an alcohol swab. Insert the syringe needle into the vial through the center of the rubber stopper and direct the stream of Sterile Water for Injection, USP, to the glass wall of the vial. Gently swirl the solution by rotating the vial to dissolve the lyophilized powder. Avoid prolonged or vigorous agitation. DO NOT SHAKE. Foaming of the solution on reconstitution is not unusual. Allow the reconstituted solution to stand for 5 minutes. The reconstituted solution concentration is 10 mg/mL. The solution should be colorless to light yellow and opalescent, and the solution may develop a few translucent particles as infliximab-abda is a protein. Do not use if the lyophilized cake has not fully dissolved or if opaque particles, discoloration, or other foreign particles are present.
- 3.
- Dilute the total volume of the reconstituted RENFLEXIS solution dose to 250 mL with sterile 0.9% Sodium Chloride Injection, USP, by withdrawing a volume equal to the volume of reconstituted RENFLEXIS from the 0.9% Sodium Chloride Injection, USP, 250 mL bottle or bag. Do not dilute the reconstituted RENFLEXIS solution with any other diluent. Slowly add the total volume of reconstituted RENFLEXIS solution to the 250 mL infusion bottle or bag. Gently mix. The resulting infusion concentration should range between 0.4 mg/mL and 4 mg/mL.
- 4.
- The RENFLEXIS infusion should begin within 3 hours of reconstitution and dilution. The infusion must be administered over a period of not less than 2 hours and must use an infusion set with an in-line, sterile, non-pyrogenic, low-protein-binding filter (pore size of 1.2 μm or less). The vials do not contain antibacterial preservatives. Therefore, any unused portion of the infusion solution should not be stored for reuse.
- 5.
- No physical biochemical compatibility studies have been conducted to evaluate the coadministration of RENFLEXIS with other agents. RENFLEXIS should not be infused concomitantly in the same intravenous line with other agents.
- 6.
- Parenteral drug products should be inspected visually before and after reconstitution for particulate matter and discoloration prior to administration, whenever solution and container permit. If visibly opaque particles, discoloration or other foreign particulates are observed, the solution should not be used.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
RENFLEXIS at doses > 5 mg/kg should not be administered to patients with moderate to severe heart failure. In a randomized study evaluating infliximab in patients with moderate to severe heart failure (New York Heart Association [NYHA] Functional Class III/IV), infliximab treatment at 10 mg/kg was associated with an increased incidence of death and hospitalization due to worsening heart failure [see Warnings and Precautions (5.5) and Adverse Reactions (6.1)].
RENFLEXIS should not be re-administered to patients who have experienced a severe hypersensitivity reaction to infliximab products. Additionally, RENFLEXIS should not be administered to patients with known hypersensitivity to inactive components of the product or to any murine proteins.
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
Patients treated with infliximab products are at increased risk for developing serious infections involving various organ systems and sites that may lead to hospitalization or death.
Opportunistic infections due to bacterial, mycobacterial, invasive fungal, viral, or parasitic organisms including aspergillosis, blastomycosis, candidiasis, coccidioidomycosis, cryptococcosis, histoplasmosis, legionellosis, listeriosis, pneumocystosis, salmonellosis and tuberculosis have been reported with TNF-blockers. Patients have frequently presented with disseminated rather than localized disease.
Treatment with RENFLEXIS should not be initiated in patients with an active infection, including clinically important localized infections. Patients greater than 65 years of age, patients with co-morbid conditions and/or patients taking concomitant immunosuppressants such as corticosteroids or methotrexate may be at greater risk of infection. The risks and benefits of treatment should be considered prior to initiating therapy in patients:
- •
- with chronic or recurrent infection;
- •
- who have been exposed to tuberculosis;
- •
- with a history of an opportunistic infection;
- •
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses, such as histoplasmosis, coccidioidomycosis, or blastomycosis; or
- •
- with underlying conditions that may predispose them to infection.
Tuberculosis
Cases of reactivation of tuberculosis or new tuberculosis infections have been observed in patients receiving infliximab products, including patients who have previously received treatment for latent or active tuberculosis. Cases of active tuberculosis have also occurred in patients being treated with infliximab products during treatment for latent tuberculosis.
Patients should be evaluated for tuberculosis risk factors and tested for latent infection prior to initiating RENFLEXIS and periodically during therapy. Treatment of latent tuberculosis infection prior to therapy with TNF blocking agents has been shown to reduce the risk of tuberculosis reactivation during therapy. Induration of 5 mm or greater with tuberculin skin testing should be considered a positive test result when assessing if treatment for latent tuberculosis is needed prior to initiating RENFLEXIS, even for patients previously vaccinated with Bacille Calmette-Guérin (BCG).
Anti-tuberculosis therapy should also be considered prior to initiation of RENFLEXIS in patients with a past history of latent or active tuberculosis in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent tuberculosis but having risk factors for tuberculosis infection. Consultation with a physician with expertise in the treatment of tuberculosis is recommended to aid in the decision whether initiating anti-tuberculosis therapy is appropriate for an individual patient.
Tuberculosis should be strongly considered in patients who develop a new infection during RENFLEXIS treatment, especially in patients who have previously or recently traveled to countries with a high prevalence of tuberculosis, or who have had close contact with a person with active tuberculosis.
Monitoring
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with RENFLEXIS, including the development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy. Tests for latent tuberculosis infection may also be falsely negative while on therapy with RENFLEXIS.
RENFLEXIS should be discontinued if a patient develops a serious infection or sepsis. A patient who develops a new infection during treatment with RENFLEXIS should be closely monitored, undergo a prompt and complete diagnostic workup appropriate for an immunocompromised patient, and appropriate antimicrobial therapy should be initiated.
Invasive Fungal Infections
For patients who reside or travel in regions where mycoses are endemic, invasive fungal infection should be suspected if they develop a serious systemic illness. Appropriate empiric antifungal therapy should be considered while a diagnostic workup is being performed. Antigen and antibody testing for histoplasmosis may be negative in some patients with active infection. When feasible, the decision to administer empiric antifungal therapy in these patients should be made in consultation with a physician with expertise in the diagnosis and treatment of invasive fungal infections and should take into account both the risk for severe fungal infection and the risks of antifungal therapy.
5.2 Malignancies
Malignancies, some fatal, have been reported among children, adolescents and young adults who received treatment with TNF-blocking agents (initiation of therapy ≤ 18 years of age), including infliximab products. Approximately half of these cases were lymphomas, including Hodgkin's and non-Hodgkin's lymphoma. The other cases represented a variety of malignancies, including rare malignancies that are usually associated with immunosuppression and malignancies that are not usually observed in children and adolescents. The malignancies occurred after a median of 30 months (range 1 to 84 months) after the first dose of TNF blocker therapy. Most of the patients were receiving concomitant immunosuppressants. These cases were reported post-marketing and are derived from a variety of sources, including registries and spontaneous postmarketing reports.
Lymphomas
In the controlled portions of clinical trials of all the TNF-blocking agents, more cases of lymphoma have been observed among patients receiving a TNF blocker compared with control patients. In the controlled and open-label portions of infliximab clinical trials, 5 patients developed lymphomas among 5707 patients treated with infliximab (median duration of follow-up 1.0 years) vs. 0 lymphomas in 1600 control patients (median duration of follow-up 0.4 years). In rheumatoid arthritis patients, 2 lymphomas were observed for a rate of 0.08 cases per 100 patient-years of follow-up, which is approximately three-fold higher than expected in the general population. In the combined clinical trial population for rheumatoid arthritis, Crohn's disease, psoriatic arthritis, ankylosing spondylitis, ulcerative colitis, and plaque psoriasis, 5 lymphomas were observed for a rate of 0.10 cases per 100 patient-years of follow-up, which is approximately four-fold higher than expected in the general population. Patients with Crohn's disease, rheumatoid arthritis or plaque psoriasis, particularly patients with highly active disease and/or chronic exposure to immunosuppressant therapies, may be at a higher risk (up to several fold) than the general population for the development of lymphoma, even in the absence of TNF-blocking therapy. Cases of acute and chronic leukemia have been reported with postmarketing TNF-blocker use in rheumatoid arthritis and other indications. Even in the absence of TNF blocker therapy, patients with rheumatoid arthritis may be at a higher risk (approximately 2-fold) than the general population for the development of leukemia.
Hepatosplenic T-cell lymphoma (HSTCL)
Postmarketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have been reported in patients treated with TNF blockers including infliximab products. These cases have had a very aggressive disease course and have been fatal. Almost all patients had received treatment with the immunosuppressants azathioprine or 6-mercaptopurine concomitantly with a TNF-blocker at or prior to diagnosis. The majority of reported cases have occurred in patients with Crohn's disease or ulcerative colitis and most were in adolescent and young adult males. It is uncertain whether the occurrence of HSTCL is related to TNF-blockers or TNF-blockers in combination with these other immunosuppressants. When treating patients, consideration of whether to use RENFLEXIS alone or in combination with other immunosuppressants such as azathioprine or 6-mercaptopurine should take into account a possibility that there is a higher risk of HSTCL with combination therapy versus an observed increased risk of immunogenicity and hypersensitivity reactions with infliximab product monotherapy from the clinical trial data from studies with infliximab [see Warnings and Precautions (5.7) and Adverse Reactions (6.1)].
Skin Cancer
Melanoma and Merkel cell carcinoma have been reported in patients treated with TNF blocker therapy, including infliximab products [see Adverse Reactions (6.2)]. Periodic skin examination is recommended for all patients, particularly those with risk factors for skin cancer.
Cervical Cancer
A population-based retrospective cohort study using data from Swedish national health registries found a 2 to 3 fold increase in the incidence of invasive cervical cancer in women with rheumatoid arthritis treated with infliximab compared to biologics-naïve patients or the general population, particularly those over 60 years of age. A causal relationship between infliximab products and cervical cancer cannot be excluded. Periodic screening should continue in women treated with RENFLEXIS [see Adverse Reactions (6.2)].
Other Malignancies
In the controlled portions of clinical trials of some TNF-blocking agents including infliximab products, more malignancies (excluding lymphoma and nonmelanoma skin cancer [NMSC]) have been observed in patients receiving those TNF-blockers compared with control patients. During the controlled portions of infliximab trials in patients with moderately to severely active rheumatoid arthritis, Crohn's disease, psoriatic arthritis, ankylosing spondylitis, ulcerative colitis, and plaque psoriasis, 14 patients were diagnosed with malignancies (excluding lymphoma and NMSC) among 4019 infliximab-treated patients vs. 1 among 1597 control patients (at a rate of 0.52/100 patient-years among infliximab-treated patients vs. a rate of 0.11/100 patient-years among control patients), with median duration of follow-up 0.5 years for infliximab-treated patients and 0.4 years for control patients. Of these, the most common malignancies were breast, colorectal, and melanoma. The rate of malignancies among infliximab-treated patients was similar to that expected in the general population whereas the rate in control patients was lower than expected.
In a clinical trial exploring the use of infliximab in patients with moderate to severe chronic obstructive pulmonary disease (COPD), more malignancies, the majority of lung or head and neck origin, were reported in infliximab-treated patients compared with control patients. All patients had a history of heavy smoking [see Adverse Reactions (6.1)]. Prescribers should exercise caution when considering the use of RENFLEXIS in patients with moderate to severe COPD.
Psoriasis patients should be monitored for nonmelanoma skin cancers (NMSCs), particularly those patients who have had prior prolonged phototherapy treatment. In the maintenance portion of clinical trials for infliximab, NMSCs were more common in patients with previous phototherapy [see Adverse Reactions (6.1)].
The potential role of TNF-blocking therapy in the development of malignancies is not known [see Adverse Reactions (6.1)]. Rates in clinical trials for infliximab cannot be compared to rates in clinical trials of other TNF-blockers and may not predict rates observed in a broader patient population. Caution should be exercised in considering RENFLEXIS treatment in patients with a history of malignancy or in continuing treatment in patients who develop malignancy while receiving RENFLEXIS.
5.3 Hepatitis B Virus Reactivation
Use of TNF blockers, including infliximab products, has been associated with reactivation of hepatitis B virus (HBV) in patients who are chronic carriers of this virus. In some instances, HBV reactivation occurring in conjunction with TNF blocker therapy has been fatal. The majority of these reports have occurred in patients concomitantly receiving other medications that suppress the immune system, which may also contribute to HBV reactivation. Patients should be tested for HBV infection before initiating TNF blocker therapy, including RENFLEXIS. For patients who test positive for hepatitis B surface antigen, consultation with a physician with expertise in the treatment of hepatitis B is recommended. Adequate data are not available on the safety or efficacy of treating patients who are carriers of HBV with antiviral therapy in conjunction with TNF blocker therapy to prevent HBV reactivation. Patients who are carriers of HBV and require treatment with TNF blockers should be closely monitored for clinical and laboratory signs of active HBV infection throughout therapy and for several months following termination of therapy. In patients who develop HBV reactivation, TNF blockers should be stopped and antiviral therapy with appropriate supportive treatment should be initiated. The safety of resuming TNF blocker therapy after HBV reactivation is controlled is not known. Therefore, prescribers should exercise caution when considering resumption of TNF blocker therapy in this situation and monitor patients closely.
5.4 Hepatotoxicity
Severe hepatic reactions, including acute liver failure, jaundice, hepatitis and cholestasis, have been reported in postmarketing data in patients receiving infliximab products. Autoimmune hepatitis has been diagnosed in some of these cases. Severe hepatic reactions occurred between 2 weeks to more than 1 year after initiation of infliximab; elevations in hepatic aminotransferase levels were not noted prior to discovery of the liver injury in many of these cases. Some of these cases were fatal or necessitated liver transplantation. Patients with symptoms or signs of liver dysfunction should be evaluated for evidence of liver injury. If jaundice and/or marked liver enzyme elevations (e.g., ≥ 5 times the upper limit of normal) develop, RENFLEXIS should be discontinued, and a thorough investigation of the abnormality should be undertaken. In clinical trials, mild or moderate elevations of ALT and AST have been observed in patients receiving infliximab products without progression to severe hepatic injury [see Adverse Reactions (6.1)].
5.5 Patients with Heart Failure
Infliximab products have been associated with adverse outcomes in patients with heart failure, and should be used in patients with heart failure only after consideration of other treatment options. The results of a randomized study evaluating the use of infliximab in patients with heart failure (NYHA Functional Class III/IV) suggested higher mortality in patients who received 10 mg/kg infliximab, and higher rates of cardiovascular adverse events at doses of 5 mg/kg and 10 mg/kg. There have been post-marketing reports of worsening heart failure, with and without identifiable precipitating factors, in patients taking infliximab. There have also been rare post-marketing reports of new onset heart failure, including heart failure in patients without known pre-existing cardiovascular disease. Some of these patients have been under 50 years of age. If a decision is made to administer RENFLEXIS to patients with heart failure, they should be closely monitored during therapy, and RENFLEXIS should be discontinued if new or worsening symptoms of heart failure appear [see Contraindications (4) and Adverse Reactions (6.1)].
5.6 Hematologic Reactions
Cases of leukopenia, neutropenia, thrombocytopenia, and pancytopenia, some with a fatal outcome, have been reported in patients receiving infliximab products. The causal relationship to infliximab product therapy remains unclear. Although no high-risk group(s) has been identified, caution should be exercised in patients being treated with RENFLEXIS who have ongoing or a history of significant hematologic abnormalities. All patients should be advised to seek immediate medical attention if they develop signs and symptoms suggestive of blood dyscrasias or infection (e.g., persistent fever) while on RENFLEXIS. Discontinuation of RENFLEXIS therapy should be considered in patients who develop significant hematologic abnormalities.
5.7 Hypersensitivity
Infliximab products have been associated with hypersensitivity reactions that vary in their time of onset and required hospitalization in some cases. Most hypersensitivity reactions, which include anaphylaxis, urticaria, dyspnea, and/or hypotension, have occurred during or within 2 hours of infusion.
However, in some cases, serum sickness-like reactions have been observed in patients after initial therapy with infliximab products (i.e., as early as after the second dose), and when therapy with infliximab products was reinstituted following an extended period without treatment. Symptoms associated with these reactions include fever, rash, headache, sore throat, myalgias, polyarthralgias, hand and facial edema and/or dysphagia. These reactions were associated with a marked increase in antibodies to infliximab products, loss of detectable serum concentrations of infliximab products and possible loss of drug efficacy.
RENFLEXIS should be discontinued for severe hypersensitivity reactions. Medications for the treatment of hypersensitivity reactions (e.g., acetaminophen, antihistamines, corticosteroids and/or epinephrine) should be available for immediate use in the event of a reaction [see Adverse Reactions (6.1)].
In rheumatoid arthritis, Crohn's disease and psoriasis clinical trials, re-administration of infliximab after a period of no treatment resulted in a higher incidence of infusion reactions relative to regular maintenance treatment [see Adverse Reactions (6.1)]. In general, the benefit-risk of re-administration of RENFLEXIS after a period of no-treatment, especially as a re-induction regimen given at weeks 0, 2 and 6, should be carefully considered. In the case where RENFLEXIS maintenance therapy for psoriasis is interrupted, RENFLEXIS should be reinitiated as a single dose followed by maintenance therapy.
5.8 Cardiovascular and Cerebrovascular Reactions During and After Infusion
Serious cerebrovascular accidents, myocardial ischemia/infarction (some fatal), hypotension, hypertension, and arrhythmias have been reported during and within 24 hours of initiation of infliximab product infusion. Cases of transient visual loss have been reported during or within 2 hours of infusion of infliximab product. Monitor patients during infusion and if serious reaction occurs, discontinue infusion. Further management of reactions should be dictated by signs and symptoms [See Adverse Reactions (6)].
5.9 Neurologic Reactions
Agents that inhibit TNF have been associated with CNS manifestation of systemic vasculitis, seizure and new onset or exacerbation of clinical symptoms and/or radiographic evidence of central nervous system demyelinating disorders, including multiple sclerosis and optic neuritis, and peripheral demyelinating disorders, including Guillain-Barré syndrome. Prescribers should exercise caution in considering the use of RENFLEXIS in patients with these neurologic disorders and should consider discontinuation of RENFLEXIS if these disorders develop.
5.10 Use with Anakinra
Serious infections and neutropenia were seen in clinical studies with concurrent use of anakinra and another TNFα-blocking agent, etanercept, with no added clinical benefit compared to etanercept alone. Because of the nature of the adverse reactions seen with the combination of etanercept and anakinra therapy, similar toxicities may also result from the combination of anakinra and other TNFα-blocking agents. Therefore, the combination of RENFLEXIS and anakinra is not recommended.
5.11 Use with Abatacept
In clinical studies, concurrent administration of TNF-blocking agents and abatacept have been associated with an increased risk of infections including serious infections compared with TNF-blocking agents alone, without increased clinical benefit. Therefore, the combination of RENFLEXIS and abatacept is not recommended [see Drug Interactions (7.1)].
5.12 Concurrent Administration with other Biological Therapeutics
There is insufficient information regarding the concomitant use of infliximab products with other biological therapeutics used to treat the same conditions as RENFLEXIS. The concomitant use of RENFLEXIS with these biologics is not recommended because of the possibility of an increased risk of infection [see Drug Interactions (7.3)].
5.13 Switching between Biological Disease-Modifying Antirheumatic Drugs (DMARDs)
Care should be taken when switching from one biologic to another, since overlapping biological activity may further increase the risk of infection.
5.14 Autoimmunity
Treatment with infliximab products may result in the formation of autoantibodies and in the development of a lupus-like syndrome. If a patient develops symptoms suggestive of a lupus-like syndrome following treatment with RENFLEXIS, treatment should be discontinued [see Adverse Reactions (6.1)].
5.15 Live Vaccines/Therapeutic Infectious Agents
In patients receiving anti-TNF therapy, limited data are available on the response to vaccination with live vaccines or on the secondary transmission of infection by live vaccines. Use of live vaccines can result in clinical infections, including disseminated infections. The concurrent administration of live vaccines with RENFLEXIS is not recommended.
Fatal outcome due to disseminated BCG infection has been reported in an infant who received a BCG vaccine after in utero exposure to infliximab products. Infliximab products are known to cross the placenta and have been detected up to 6 months following birth. At least a six month waiting period following birth is recommended before the administration of any live vaccine to infants exposed in utero to infliximab products.
Other uses of therapeutic infectious agents such as live attenuated bacteria (e.g., BCG bladder instillation for the treatment of cancer) could result in clinical infections, including disseminated infections. It is recommended that therapeutic infectious agents not be given concurrently with RENFLEXIS.
It is recommended that all pediatric patients be brought up to date with all vaccinations prior to initiating RENFLEXIS therapy. The interval between vaccination and initiation of RENFLEXIS therapy should be in accordance with current vaccination guidelines.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Adults
The data described herein reflect exposure to infliximab in 4779 adult patients (1304 patients with rheumatoid arthritis, 1106 patients with Crohn's disease, 202 with ankylosing spondylitis, 293 with psoriatic arthritis, 484 with ulcerative colitis, 1373 with plaque psoriasis, and 17 patients with other conditions), including 2625 patients exposed beyond 30 weeks and 374 exposed beyond 1 year. [For information on adverse reactions in pediatric patients see Adverse Reactions (6.1).] One of the most-common reasons for discontinuation of treatment was infusion-related reactions (e.g., dyspnea, flushing, headache and rash).
Infusion-related Reactions
An infusion reaction was defined in clinical trials as any adverse event occurring during an infusion or within 1 hour after an infusion. In Phase 3 clinical studies, 18% of infliximab-treated patients experienced an infusion reaction compared to 5% of placebo-treated patients. Of infliximab-treated patients who had an infusion reaction during the induction period, 27% experienced an infusion reaction during the maintenance period. Of patients who did not have an infusion reaction during the induction period, 9% experienced an infusion reaction during the maintenance period.
Among all infliximab infusions, 3% were accompanied by nonspecific symptoms such as fever or chills, 1% were accompanied by cardiopulmonary reactions (primarily chest pain, hypotension, hypertension or dyspnea), and <1% were accompanied by pruritus, urticaria, or the combined symptoms of pruritus/urticaria and cardiopulmonary reactions. Serious infusion reactions occurred in <1% of patients and included anaphylaxis, convulsions, erythematous rash and hypotension. Approximately 3% of patients discontinued infliximab because of infusion reactions, and all patients recovered with treatment and/or discontinuation of the infusion. Infliximab infusions beyond the initial infusion were not associated with a higher incidence of reactions. The infusion reaction rates remained stable in psoriasis through 1 year in psoriasis Study I. In psoriasis Study II, the rates were variable over time and somewhat higher following the final infusion than after the initial infusion. Across the 3 psoriasis studies, the percent of total infusions resulting in infusion reactions (i.e., an adverse event occurring within 1 hour) was 7% in the 3 mg/kg group, 4% in the 5 mg/kg group, and 1% in the placebo group.
Patients who became positive for antibodies to infliximab were more likely (approximately two-to three-fold) to have an infusion reaction than were those who were negative. Use of concomitant immunosuppressant agents appeared to reduce the frequency of both antibodies to infliximab and infusion reactions [see Adverse Reactions (6.1) and Drug Interactions (7.4)].
Infusion Reactions following Re-administration
In a clinical trial of patients with moderate to severe psoriasis designed to assess the efficacy of long-term maintenance therapy versus re-treatment with an induction regimen of infliximab following disease flare, 4% (8/219) of patients in the re-treatment therapy arm experienced serious infusion reactions versus < 1% (1/222) in the maintenance therapy arm. Patients enrolled in this trial did not receive any concomitant immunosuppressant therapy. In this study, the majority of serious infusion reactions occurred during the second infusion at Week 2. Symptoms included, but were not limited to, dyspnea, urticaria, facial edema, and hypotension. In all cases, infliximab treatment was discontinued and/or other treatment instituted with complete resolution of signs and symptoms.
Delayed Reactions/Reactions Following Re-administration
In psoriasis studies, approximately 1% of infliximab-treated patients experienced a possible delayed hypersensitivity reaction, generally reported as serum sickness or a combination of arthralgia and/or myalgia with fever and/or rash. These reactions generally occurred within 2 weeks after repeat infusion.
Infections
In infliximab clinical studies, treated infections were reported in 36% of infliximab-treated patients (average of 51 weeks of follow-up) and in 25% of placebo-treated patients (average of 37 weeks of follow-up). The infections most frequently reported were respiratory tract infections (including sinusitis, pharyngitis, and bronchitis) and urinary tract infections. Among infliximab-treated patients, serious infections included pneumonia, cellulitis, abscess, skin ulceration, sepsis, and bacterial infection. In clinical trials, 7 opportunistic infections were reported; 2 cases each of coccidioidomycosis (1 case was fatal) and histoplasmosis (1 case was fatal), and 1 case each of pneumocystosis, nocardiosis and cytomegalovirus. Tuberculosis was reported in 14 patients, 4 of whom died due to miliary tuberculosis. Other cases of tuberculosis, including disseminated tuberculosis, also have been reported post-marketing. Most of these cases of tuberculosis occurred within the first 2 months after initiation of therapy with infliximab and may reflect recrudescence of latent disease [see Warnings and Precautions (5.1)]. In the 1year placebo-controlled studies RA I and RA II, 5.3% of patients receiving infliximab every 8 weeks with MTX developed serious infections as compared to 3.4% of placebo patients receiving MTX. Of 924 patients receiving infliximab, 1.7% developed pneumonia and 0.4% developed TB, when compared to 0.3% and 0.0% in the placebo arm respectively. In a shorter (22-week) placebo-controlled study of 1082 RA patients randomized to receive placebo, 3 mg/kg or 10 mg/kg infliximab infusions at 0, 2, and 6 weeks, followed by every 8 weeks with MTX, serious infections were more frequent in the 10 mg/kg infliximab group (5.3%) than the 3 mg/kg or placebo groups (1.7% in both). During the 54-week Crohn's II Study, 15% of patients with fistulizing Crohn's disease developed a new fistula-related abscess.
In infliximab clinical studies in patients with ulcerative colitis, infections treated with antimicrobials were reported in 27% of infliximab-treated patients (average of 41 weeks of follow-up) and in 18% of placebo-treated patients (average 32 weeks of follow-up). The types of infections, including serious infections, reported in patients with ulcerative colitis were similar to those reported in other clinical studies.
The onset of serious infections may be preceded by constitutional symptoms such as fever, chills, weight loss, and fatigue. The majority of serious infections, however, may also be preceded by signs or symptoms localized to the site of the infection.
Autoantibodies/Lupus-like Syndrome
Approximately half of infliximab-treated patients in clinical trials who were antinuclear antibody (ANA) negative at baseline developed a positive ANA during the trial compared with approximately one-fifth of placebo-treated patients. Anti-dsDNA antibodies were newly detected in approximately one-fifth of infliximab-treated patients compared with 0% of placebo-treated patients. Reports of lupus and lupus-like syndromes, however, remain uncommon.
Malignancies
In controlled trials, more infliximab-treated patients developed malignancies than placebo-treated patients [see Warnings and Precautions (5.2)].
In a randomized controlled clinical trial exploring the use of infliximab in patients with moderate to severe COPD who were either current smokers or ex-smokers, 157 patients were treated with infliximab at doses similar to those used in rheumatoid arthritis and Crohn's disease. Of these infliximab-treated patients, 9 developed a malignancy, including 1 lymphoma, for a rate of 7.67 cases per 100 patient-years of follow-up (median duration of follow-up 0.8 years; 95% CI 3.51 -14.56). There was 1 reported malignancy among 77 control patients for a rate of 1.63 cases per 100 patient-years of follow-up (median duration of follow-up 0.8 years; 95% CI 0.04 -9.10). The majority of the malignancies developed in the lung or head and neck.
Patients with Heart Failure
In a randomized study evaluating infliximab in moderate to severe heart failure (NYHA Class III/IV; left ventricular ejection fraction ≤ 35%), 150 patients were randomized to receive treatment with 3 infusions of infliximab 10 mg/kg, 5 mg/kg, or placebo, at 0, 2, and 6 weeks. Higher incidences of mortality and hospitalization due to worsening heart failure were observed in patients receiving the 10 mg/kg infliximab dose. At 1 year, 8 patients in the 10 mg/kg infliximab group had died compared with 4 deaths each in the 5 mg/kg infliximab and the placebo groups. There were trends toward increased dyspnea, hypotension, angina, and dizziness in both the 10 mg/kg and 5 mg/kg infliximab treatment groups, versus placebo. Infliximab has not been studied in patients with mild heart failure (NYHA Class I/II) [see Contraindications (4) and Warnings and Precautions (5.5)].
Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other infliximab products may be misleading.
Treatment with infliximab products can be associated with the development of antibodies to infliximab products. An enzyme immunoassay (EIA) method was originally used to measure anti-infliximab antibodies in clinical studies of infliximab. The EIA method is subject to interference by serum infliximab, possibly resulting in an underestimation of the rate of patient antibody formation. A separate, drug-tolerant electrochemiluminescence immunoassay (ECLIA) method for detecting antibodies to infliximab was subsequently developed and validated. This method is 60-fold more sensitive than the original EIA. With the ECLIA method, all clinical samples can be classified as either positive or negative for antibodies to infliximab without the need for the inconclusive category.
The incidence of antibodies to infliximab was based on the original EIA method in all clinical studies of infliximab except for the Phase 3 study in pediatric patients with ulcerative colitis where the incidence of antibodies to infliximab was detected using both the EIA and ECLIA methods [see Adverse Reactions, Pediatric Ulcerative Colitis (6.1)].
The incidence of antibodies to infliximab in patients given a 3-dose induction regimen followed by maintenance dosing was approximately 10% as assessed through 1 to 2 years of treatment with infliximab. A higher incidence of antibodies to infliximab was observed in Crohn's disease patients receiving infliximab after drug-free intervals >16 weeks. In a study of psoriatic arthritis in which 191 patients received 5 mg/kg with or without MTX, antibodies to infliximab occurred in 15% of patients. The majority of antibody-positive patients had low titers. Patients who were antibody-positive were more likely to have higher rates of clearance, reduced efficacy and to experience an infusion reaction [see Adverse Reactions (6.1)] than were patients who were antibody negative. Antibody development was lower among rheumatoid arthritis and Crohn's disease patients receiving immunosuppressant therapies such as 6-MP/AZA or MTX.
In the psoriasis Study II, which included both the 5 mg/kg and 3 mg/kg doses, antibodies were observed in 36% of patients treated with 5 mg/kg every 8 weeks for 1 year, and in 51% of patients treated with 3 mg/kg every 8 weeks for 1 year. In the psoriasis Study III, which also included both the 5 mg/kg and 3 mg/kg doses, antibodies were observed in 20% of patients treated with 5 mg/kg induction (weeks 0, 2 and 6), and in 27% of patients treated with 3 mg/kg induction. Despite the increase in antibody formation, the infusion reaction rates in Studies I and II in patients treated with 5 mg/kg induction followed by every 8 week maintenance for 1 year and in Study III in patients treated with 5 mg/kg induction (14.1%-23.0%) and serious infusion reaction rates (<1%) were similar to those observed in other study populations. The clinical significance of apparent increased immunogenicity on efficacy and infusion reactions in psoriasis patients as compared to patients with other diseases treated with infliximab products over the long term is not known.
Hepatotoxicity
Severe liver injury, including acute liver failure and autoimmune hepatitis, has been reported in patients receiving infliximab products [see Warnings and Precautions (5.4)]. Reactivation of hepatitis B virus has occurred in patients receiving TNF-blocking agents, including infliximab products, who are chronic carriers of this virus [see Warnings and Precautions (5.3)].
In clinical trials in rheumatoid arthritis, Crohn's disease, ulcerative colitis, ankylosing spondylitis, plaque psoriasis, and psoriatic arthritis, elevations of aminotransferases were observed (ALT more common than AST) in a greater proportion of patients receiving infliximab than in controls (Table 1), both when infliximab was given as monotherapy and when it was used in combination with other immunosuppressive agents. In general, patients who developed ALT and AST elevations were asymptomatic, and the abnormalities decreased or resolved with either continuation or discontinuation of infliximab, or modification of concomitant medications.
Table 1 Proportion of patients with elevated ALT in clinical trials Proportion of patients with elevated ALT > 1 to < 3 × ULN ≥ 3 × ULN ≥ 5 × ULN Placebo Infliximab Placebo Infliximab Placebo Infliximab - *
- Placebo patients received methotrexate while infliximab patients received both infliximab and methotrexate. Median follow-up was 58 weeks.
- †
- Placebo patients in the 2 Phase 3 trials in Crohn's disease received an initial dose of 5 mg/kg infliximab at study start and were on placebo in the maintenance phase. Patients who were randomized to the placebo maintenance group and then later crossed over to infliximab are included in the infliximab group in ALT analysis. Median follow-up was 54 weeks.
- ‡
- Median follow-up was 30 weeks. Specifically, the median duration of follow-up was 30 weeks for placebo and 31 weeks for infliximab.
- §
- Median follow-up was 24 weeks for the placebo group and 102 weeks for the infliximab group.
- ¶
- Median follow-up was 39 weeks for the infliximab group and 18 weeks for the placebo group.
- #
- ALT values are obtained in 2 Phase 3 psoriasis studies with median follow-up of 50 weeks for infliximab and 16 weeks for placebo.
Rheumatoid arthritis*
24%
34%
3%
4%
< 1%
< 1%
Crohn's disease†
34%
39%
4%
5%
0%
2%
Ulcerative colitis‡
12%
17%
1%
2%
< 1%
< 1%
Ankylosing spondylitis§
15%
51%
0%
10%
0%
4%
Psoriatic arthritis¶
16%
50%
0%
7%
0%
2%
Plaque psoriasis#
24%
49%
<1%
8%
0%
3%
Adverse Reactions in Psoriasis Studies
During the placebo-controlled portion across the 3 clinical trials up to Week 16, the proportion of patients who experienced at least 1 serious adverse reaction (SAE; defined as resulting in death, life threatening, requires hospitalization, or persistent or significant disability/incapacity) was 0.5% in the 3 mg/kg infliximab group, 1.9% in the placebo group, and 1.6% in the 5 mg/kg infliximab group.
Among patients in the 2 Phase 3 studies, 12.4% of patients receiving infliximab 5 mg/kg every 8 weeks through 1 year of maintenance treatment experienced at least 1 SAE in Study I. In Study II, 4.1% and 4.7% of patients receiving infliximab 3 mg/kg and 5 mg/kg every 8 weeks, respectively, through 1 year of maintenance treatment experienced at least 1 SAE. One death due to bacterial sepsis occurred 25 days after the second infusion of 5 mg/kg infliximab. Serious infections included sepsis, and abscesses. In Study I, 2.7% of patients receiving infliximab 5 mg/kg every 8 weeks through 1 year of maintenance treatment experienced at least 1 serious infection. In Study II, 1.0% and 1.3% of patients receiving infliximab 3 mg/kg and 5 mg/kg, respectively, through 1 year of treatment experienced at least 1 serious infection. The most common serious infection (requiring hospitalization) was abscess (skin, throat, and peri-rectal) reported by 5 (0.7%) patients in the 5 mg/kg infliximab group. Two active cases of tuberculosis were reported: 6 weeks and 34 weeks after starting infliximab.
In the placebo-controlled portion of the psoriasis studies, 7 of 1123 patients who received infliximab at any dose were diagnosed with at least one NMSC compared to 0 of 334 patients who received placebo.
In the psoriasis studies, 1% (15/1373) of patients experienced serum sickness or a combination of arthralgia and/or myalgia with fever, and/or rash, usually early in the treatment course. Of these patients, 6 required hospitalization due to fever, severe myalgia, arthralgia, swollen joints, and immobility.
Other Adverse Reactions
Safety data are available from 4779 adult patients treated with infliximab, including 1304 with rheumatoid arthritis, 1106 with Crohn's disease, 484 with ulcerative colitis, 202 with ankylosing spondylitis, 293 with psoriatic arthritis, 1373 with plaque psoriasis and 17 with other conditions. [For information on other adverse reactions in pediatric patients, see Adverse Reactions (6.1)]. Adverse reactions reported in ≥ 5% of all patients with rheumatoid arthritis receiving 4 or more infusions are in Table 2. The types and frequencies of adverse reactions observed were similar in rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, plaque psoriasis and Crohn's disease patients treated with infliximab except for abdominal pain, which occurred in 26% of patients with Crohn's disease. In the Crohn's disease studies, there were insufficient numbers and duration of follow-up for patients who never received infliximab to provide meaningful comparisons.
Table 2 Adverse reactions occurring in 5% or more of patients receiving 4 or more infusions for rheumatoid arthritis Placebo Infliximab (n = 350) (n = 1129) Average weeks of follow-up
59
66
Gastrointestinal
Nausea
20%
21%
Abdominal pain
8%
12%
Diarrhea
12%
12%
Dyspepsia
7%
10%
Respiratory
Upper respiratory tract infection
25%
32%
Sinusitis
8%
14%
Pharyngitis
8%
12%
Coughing
8%
12%
Bronchitis
9%
10%
Skin and appendages disorders
Rash
5%
10%
Pruritus
2%
7%
Body as a whole-general disorders
Fatigue
7%
9%
Pain
7%
8%
Resistance mechanism disorders
Fever
4%
7%
Moniliasis
3%
5%
Central and peripheral nervous system disorders
Headache
14%
18%
Musculoskeletal system disorders
Arthralgia
7%
8%
Urinary system disorders
Urinary tract infection
6%
8%
Cardiovascular disorders, general
Hypertension
5%
7%
The most common serious adverse reactions observed in clinical trials of infliximab were infections [see Adverse Reactions (6.1)]. Other serious, medically relevant adverse reactions ≥ 0.2% or clinically significant adverse reactions by body system were as follows:
Body as a whole: allergic reaction, edema
Blood: pancytopenia
Cardiovascular: hypotension
Gastrointestinal: constipation, intestinal obstruction
Central and Peripheral Nervous: dizziness
Heart Rate and Rhythm: bradycardia
Liver and Biliary: hepatitis
Metabolic and Nutritional: dehydration
Platelet, Bleeding and Clotting: thrombocytopenia
Neoplasms: lymphoma
Red Blood Cell: anemia, hemolytic anemia
Resistance Mechanism: cellulitis, sepsis, serum sickness, sarcoidosis
Respiratory: lower respiratory tract infection (including pneumonia), pleurisy, pulmonary edema
Skin and Appendages: increased sweating
Vascular (Extracardiac): thrombophlebitis
White Cell and Reticuloendothelial: leukopenia, lymphadenopathy
Adverse Reactions in Pediatric Patients
Pediatric Crohn's Disease
There were some differences in the adverse reactions observed in the pediatric patients receiving infliximab compared to those observed in adults with Crohn's disease. These differences are discussed in the following paragraphs.
The following adverse reactions were reported more commonly in 103 randomized pediatric Crohn's disease patients administered 5 mg/kg infliximab through 54 weeks than in 385 adult Crohn's disease patients receiving a similar treatment regimen: anemia (11%), leukopenia (9%), flushing (9%), viral infection (8%), neutropenia (7%), bone fracture (7%), bacterial infection (6%), and respiratory tract allergic reaction (6%).
Infections were reported in 56% of randomized pediatric patients in Study Peds Crohn's and in 50% of adult patients in Study Crohn's I. In Study Peds Crohn's, infections were reported more frequently for patients who received every 8-week as opposed to every 12-week infusions (74% and 38%, respectively), while serious infections were reported for 3 patients in the every 8-week and 4 patients in the every 12-week maintenance treatment group. The most commonly reported infections were upper respiratory tract infection and pharyngitis, and the most commonly reported serious infection was abscess. Pneumonia was reported for 3 patients, (2 in the every 8week and 1 in the every 12-week maintenance treatment groups). Herpes zoster was reported for 2 patients in the every 8-week maintenance treatment group.
In Study Peds Crohn's, 18% of randomized patients experienced 1 or more infusion reactions, with no notable difference between treatment groups. Of the 112 patients in Study Peds Crohn's, there were no serious infusion reactions, and 2 patients had non-serious anaphylactoid reactions.
In Study Peds Crohn's, in which all patients received stable doses of 6-MP, AZA, or MTX, excluding inconclusive samples, 3 of 24 patients had antibodies to infliximab. Although 105 patients were tested for antibodies to infliximab, 81 patients were classified as inconclusive because they could not be ruled as negative due to assay interference by the presence of infliximab in the sample.
Elevations of ALT up to 3 times the upper limit of normal (ULN) were seen in 18% of pediatric patients in Crohn's disease clinical trials; 4% had ALT elevations ≥ 3 × ULN, and 1% had elevations ≥ 5 × ULN. (Median follow-up was 53 weeks.)
Pediatric Ulcerative Colitis
Overall, the adverse reactions reported in the pediatric ulcerative colitis trial and adult ulcerative colitis (Study UC I and Study UC II) studies were generally consistent. In a pediatric UC trial, the most common adverse reactions were upper respiratory tract infection, pharyngitis, abdominal pain, fever, and headache.
Infections were reported in 31 (52%) of 60 treated patients in the pediatric UC trial and 22 (37%) required oral or parenteral antimicrobial treatment. The proportion of patients with infections in the pediatric UC trial was similar to that in the pediatric Crohn's disease study (Study Peds Crohn's) but higher than the proportion in the adults' ulcerative colitis studies (Study UC I and Study UC II). The overall incidence of infections in the pediatric UC trial was 13/22 (59%) in the every 8 week maintenance treatment group. Upper respiratory tract infection (7/60 [12%]) and pharyngitis (5/60 [8%]) were the most frequently reported respiratory system infections. Serious infections were reported in 12% (7/60) of all treated patients.
In the pediatric UC trial, 58 patients were evaluated for antibodies to infliximab using the EIA as well as the drug-tolerant ECLIA. With the EIA, 4 of 58 (7%) patients had antibodies to infliximab. With the ECLIA, 30 of 58 (52%) patients had antibodies to infliximab [see Adverse Reactions, Immunogenicity (6.1)]. The higher incidence of antibodies to infliximab by the ECLIA method was due to the 60-fold higher sensitivity compared to the EIA method. While EIA-positive patients generally had undetectable trough infliximab concentrations, ECLIA-positive patients could have detectable trough concentrations of infliximab because the ECLIA assay is more sensitive and drug-tolerant.
Elevations of ALT up to 3 times the upper limit of normal (ULN) were seen in 17% (10/60) of pediatric patients in the pediatric UC trial; 7% (4/60) had ALT elevations ≥ 3 × ULN, and 2% (1/60) had elevations ≥ 5 × ULN (median follow-up was 49 weeks).
Overall, 8 of 60 (13%) treated patients experienced one or more infusion reactions, including 4 of 22 (18%) patients in the every 8-week treatment maintenance group. No serious infusion reactions were reported.
In the pediatric UC trial, 45 patients were in the 12 to 17 year age group and 15 in the 6 to 11 year age group. The numbers of patients in each subgroup are too small to make any definitive conclusions about the effect of age on safety events. There were higher proportions of patients with serious adverse events (40% vs. 18%) and discontinuation due to adverse events (40% vs. 16%) in the younger age group than in the older age group. While the proportion of patients with infections was also higher in the younger age group (60% vs. 49%), for serious infections, the proportions were similar in the two age groups (13% in the 6 to 11 year age group vs. 11% in the 12 to 17 year age group). Overall proportions of adverse reactions, including infusion reactions, were similar between the 6 to 11 and 12 to 17 year age groups (13%).
6.2 Postmarketing Experience
Adverse reactions have been identified during post approval use of infliximab products in adult and pediatric patients. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following adverse reactions, some with fatal outcome, have been reported during post-approval use of infliximab products: neutropenia [see Warnings and Precautions (5.6)], agranulocytosis (including infants exposed in utero to infliximab products), interstitial lung disease (including pulmonary fibrosis/interstitial pneumonitis and rapidly progressive disease), idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, pericardial effusion, systemic and cutaneous vasculitis, erythema multiforme, Stevens-Johnson Syndrome, toxic epidermal necrolysis, peripheral demyelinating disorders (such as Guillain-Barré syndrome, chronic inflammatory demyelinating polyneuropathy, and multifocal motor neuropathy), new onset and worsening psoriasis (all subtypes including pustular, primarily palmoplantar), transverse myelitis, and neuropathies (additional neurologic reactions have also been observed) [see Warnings and Precautions (5.9)], acute liver failure, jaundice, hepatitis, and cholestasis [see Warnings and Precautions (5.4)], serious infections [see Warnings and Precautions (5.1)], malignancies, including leukemia, melanoma, Merkel cell carcinoma, and cervical cancer [see Warnings and Precautions (5.2)] and vaccine breakthrough infection including bovine tuberculosis (disseminated BCG infection) following vaccination in an infant exposed in utero to infliximab products [see Warnings and Precautions (5.15)].
Infusion-related Reactions
In post-marketing experience, cases of anaphylactic reactions, including anaphylactic shock, laryngeal/pharyngeal edema and severe bronchospasm, and seizure have been associated with administration of infliximab products.
Cases of transient visual loss have been reported in association with infliximab products during or within 2 hours of infusion. Cerebrovascular accidents, myocardial ischemia/infarction (some fatal), and arrhythmia occurring within 24 hours of initiation of infusion have also been reported [see Warnings and Precautions (5.8)].
Adverse Reactions in Pediatric Patients
The following serious adverse reactions have been reported in the post-marketing experience in children: infections (some fatal) including opportunistic infections and tuberculosis, infusion reactions, and hypersensitivity reactions.
Serious adverse reactions in the post-marketing experience with infliximab products in the pediatric population have also included malignancies, including hepatosplenic T-cell lymphomas [see Boxed WARNINGS and Warnings and Precautions (5.2)], transient hepatic enzyme abnormalities, lupus-like syndromes, and the development of autoantibodies.
-
7 DRUG INTERACTIONS
7.1 Use with Anakinra or Abatacept
An increased risk of serious infections was seen in clinical studies of other TNFα-blocking agents used in combination with anakinra or abatacept, with no added clinical benefit. Because of the nature of the adverse reactions seen with these combinations with TNF-blocker therapy, similar toxicities may also result from the combination of anakinra or abatacept with other TNFα-blocking agents. Therefore, the combination of RENFLEXIS and anakinra or abatacept is not recommended [see Warnings and Precautions (5.10 and 5.11)].
7.2 Use with Tocilizumab
The use of tocilizumab in combination with biological DMARDs such as TNF antagonists, including RENFLEXIS, should be avoided because of the possibility of increased immunosuppression and increased risk of infection.
7.3 Use with Other Biological Therapeutics
The combination of RENFLEXIS with other biological therapeutics used to treat the same conditions as RENFLEXIS is not recommended [see Warnings and Precautions (5.12)].
7.4 Methotrexate (MTX) and Other Concomitant Medications
Specific drug interaction studies, including interactions with MTX, have not been conducted. The majority of patients in rheumatoid arthritis or Crohn's disease clinical studies received one or more concomitant medications. In rheumatoid arthritis, concomitant medications besides MTX were nonsteroidal anti-inflammatory agents (NSAIDs), folic acid, corticosteroids and/or narcotics. Concomitant Crohn's disease medications were antibiotics, antivirals, corticosteroids, 6-MP/AZA and aminosalicylates. In psoriatic arthritis clinical trials, concomitant medications included MTX in approximately half of the patients as well as NSAIDs, folic acid and corticosteroids. Concomitant MTX use may decrease the incidence of anti-drug antibody production and increase infliximab product concentrations.
7.5 Immunosuppressants
Patients with Crohn's disease who received immunosuppressants tended to experience fewer infusion reactions compared to patients on no immunosuppressants [see Adverse Reactions (6.1)]. Serum infliximab concentrations appeared to be unaffected by baseline use of medications for the treatment of Crohn's disease including corticosteroids, antibiotics (metronidazole or ciprofloxacin) and aminosalicylates.
7.6 Cytochrome P450 Substrates
The formation of CYP450 enzymes may be suppressed by increased levels of cytokines (e.g., TNFα, IL-1, IL-6, IL-10, IFN) during chronic inflammation. Therefore, it is expected that for a molecule that antagonizes cytokine activity, such as infliximab products, the formation of CYP450 enzymes could be normalized. Upon initiation or discontinuation of RENFLEXIS in patients being treated with CYP450 substrates with a narrow therapeutic index, monitoring of the effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) is recommended and the individual dose of the drug product may be adjusted as needed.
7.7 Live Vaccines/Therapeutic Infectious Agents
It is recommended that live vaccines not be given concurrently with RENFLEXIS. It is also recommended that live vaccines not be given to infants after in utero exposure to infliximab products for at least 6 months following birth [see Warnings and Precautions (5.15)].
It is recommended that therapeutic infectious agents not be given concurrently with RENFLEXIS [see Warnings and Precautions (5.15)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from published literature on the use of infliximab products during pregnancy have not reported a clear association with infliximab products and adverse pregnancy outcomes. Infliximab products cross the placenta and infants exposed in utero should not be administered live vaccines for at least 6 months after birth [see Clinical Considerations]. In a development study conducted in mice using an analogous antibody, no evidence of maternal toxicity, embryotoxicity or teratogenicity was observed [see Data].
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Fetal/Neonatal adverse reactions
Infliximab products cross the placenta, and have been detected in the serum of infants up to 6 months following birth. Consequently, these infants may be at increased risk of infection, including disseminated infection which can become fatal. At least a six month waiting period following birth is recommended before the administration of live vaccines (e.g., BCG vaccine or other live vaccines, such as the rotavirus vaccine) to these infants [see Warnings and Precautions (5.15)]. Cases of agranulocytosis in infants exposed in utero have also been reported [see Adverse Reactions (6.2)].
Data
Animal Data
Because infliximab products do not cross-react with TNFα in species other than humans and chimpanzees, animal reproduction studies have not been conducted with infliximab products. An embryofetal development study was conducted in pregnant mice using an analogous antibody that selectively inhibits the functional activity of mouse TNFα. This antibody, administered during the period of organogenesis on gestation day 6 and 12 at IV doses up to 40 mg/kg produced no evidence of maternal toxicity, embryotoxicity, or teratogenicity. Doses of 10 to 15 mg/kg in pharmacodynamic animal models with the anti-TNF analogous antibody produced maximal pharmacologic effectiveness.
8.2 Lactation
Risk Summary
Available information is insufficient to inform the amount of infliximab products present in human milk, and the effects on the breastfed infant. There are no data on the effects of infliximab products on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for an infliximab product and any potential adverse effects on the breastfed infant from infliximab products or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of infliximab products have been established in pediatric patients 6 to 17 years of age for induction and maintenance treatment of Crohn's disease or ulcerative colitis. However, infliximab products have not been studied in children with Crohn's disease or ulcerative colitis <6 years of age.
Pediatric Crohn's Disease
RENFLEXIS is indicated for reducing signs and symptoms and inducing and maintaining clinical remission in pediatric patients with moderately to severely active Crohn's disease who have had an inadequate response to conventional therapy [see Boxed Warnings, Warnings and Precautions (5), Indications and Usage (1.2), Dosage and Administration (2.2), Clinical Studies (14.2) and Adverse Reactions (6.1)].
Infliximab has been studied only in combination with conventional immunosuppressive therapy in pediatric Crohn's disease. The longer term (greater than 1 year) safety and effectiveness of infliximab products in pediatric Crohn's disease patients have not been established in clinical trials.
Pediatric Ulcerative Colitis
The safety and effectiveness of infliximab products for reducing signs and symptoms and inducing and maintaining clinical remission in pediatric patients aged 6 years and older with moderately to severely active ulcerative colitis who have had an inadequate response to conventional therapy are supported by evidence from adequate and well-controlled studies of infliximab in adults. Additional safety and pharmacokinetic data were collected in 60 pediatric patients aged 6 years and older [see Clinical Pharmacology (12.3), Dosage and Administration (2.4), Adverse Reactions (6.1), and Clinical Studies (14.4)]. The effectiveness of infliximabin inducing and maintaining mucosal healing could not be established. Although 41 patients had a Mayo endoscopy subscore of 0 or 1 at the Week 8 endoscopy, the induction phase was open-label and lacked a control group. Only 9 patients had an optional endoscopy at Week 54.
In the pediatric UC trial, approximately half of the patients were on concomitant immunomodulators (AZA, 6-MP, MTX) at study start. Due to the risk of HSTCL, a careful risk-benefit assessment should be made when RENFLEXIS is used in combination with other immunosuppressants.
The longer term (greater than 1 year) safety and effectiveness of infliximab products in pediatric ulcerative colitis patients have not been established in clinical trials.
Juvenile Rheumatoid Arthritis (JRA)
The safety and efficacy of infliximab in patients with juvenile rheumatoid arthritis (JRA) were evaluated in a multicenter, randomized, placebo-controlled, double-blind study for 14 weeks, followed by a double-blind, all-active treatment extension, for a maximum of 44 weeks. Patients with active JRA between the ages of 4 and 17 years who had been treated with MTX for at least 3 months were enrolled. Concurrent use of folic acid, oral corticosteroids (≤0.2 mg/kg/day of prednisone or equivalent), NSAIDs, and/or disease modifying antirheumatic drugs (DMARDs) was permitted.
Doses of 3 mg/kg infliximab or placebo were administered intravenously at Weeks 0, 2 and 6. Patients randomized to placebo crossed-over to receive 6 mg/kg infliximab at Weeks 14, 16, and 20, and then every 8 weeks through Week 44. Patients who completed the study continued to receive open-label treatment with infliximab for up to 2 years in a companion extension study.
The study failed to establish the efficacy of infliximab in the treatment of JRA. Key observations in the study included a high placebo response rate and a higher rate of immunogenicity than what has been observed in adults. Additionally, a higher rate of clearance of infliximab was observed than had been observed in adults [see Clinical Pharmacology (12.3)].
A total of 60 patients with JRA were treated with doses of 3 mg/kg and 57 patients were treated with doses of 6 mg/kg. The proportion of patients with infusion reactions who received 3 mg/kg infliximab was 35% (21/60) over 52 weeks compared with 18% (10/57) in patients who received 6 mg/kg over 38 weeks. The most common infusion reactions reported were vomiting, fever, headache, and hypotension. In the 3 mg/kg infliximab group, 4 patients had a serious infusion reaction and 3 patients reported a possible anaphylactic reaction (2 of which were among the serious infusion reactions). In the 6 mg/kg infliximab group, 2 patients had a serious infusion reaction, 1 of whom had a possible anaphylactic reaction. Two of the 6 patients who experienced serious infusion reactions received infliximab by rapid infusion (duration of less than 2 hours). Antibodies to infliximab developed in 38% (20/53) of patients who received 3 mg/kg infliximab compared with 12% (6/49) of patients who received 6 mg/kg.
A total of 68% (41/60) of patients who received 3 mg/kg infliximab in combination with MTX experienced an infection over 52 weeks compared with 65% (37/57) of patients who received 6 mg/kg infliximab in combination with MTX over 38 weeks. The most commonly reported infections were upper respiratory tract infection and pharyngitis, and the most commonly reported serious infection was pneumonia. Other notable infections included primary varicella infection in 1 patient and herpes zoster in 1 patient.
8.5 Geriatric Use
In rheumatoid arthritis and plaque psoriasis clinical trials, no overall differences were observed in effectiveness or safety in 181 patients with rheumatoid arthritis and 75 patients with plaque psoriasis, aged 65 or older who received infliximab, compared to younger patients -although the incidence of serious adverse reactions in patients aged 65 or older was higher in both infliximab and control groups compared to younger patients. In Crohn's disease, ulcerative colitis, ankylosing spondylitis and psoriatic arthritis studies, there were insufficient numbers of patients aged 65 and over to determine whether they respond differently from patients aged 18 to 65. There is a greater incidence of infections in the elderly population in general. The incidence of serious infections in infliximab-treated patients 65 years and older was greater than in those under 65 years of age; therefore caution should be used in treating the elderly [see Adverse Reactions (6.1)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
Infliximab-abda, the active ingredient in RENFLEXIS, is a chimeric IgG1κ monoclonal antibody (composed of human constant and murine variable regions) specific for human tumor necrosis factor-alpha (TNFα). It has a molecular weight of approximately 149.1 kilodaltons. Infliximab-abda is produced in a recombinant cell line and is purified by a series of steps that includes measures to inactivate and remove viruses.
RENFLEXIS (infliximab-abda) for Injection is supplied as a sterile, white, lyophilized powder for intravenous infusion. Following reconstitution with 10 mL of Sterile Water for Injection, USP, the resulting pH is approximately 6. Each mL contains 10 mg infliximab-abda, dibasic sodium phosphate heptahydrate (0.12 mg), monobasic sodium phosphate monohydrate (0.63 mg), polysorbate 80 (0.05 mg), and sucrose (50 mg). No preservatives are present.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Infliximab products neutralize the biological activity of TNFα by binding with high affinity to the soluble and transmembrane forms of TNFα and inhibit binding of TNFα with its receptors. Infliximab products do not neutralize TNFβ (lymphotoxin-α), a related cytokine that utilizes the same receptors as TNFα. Biological activities attributed to TNFα include: induction of pro-inflammatory cytokines such as interleukins (IL) 1 and 6, enhancement of leukocyte migration by increasing endothelial layer permeability and expression of adhesion molecules by endothelial cells and leukocytes, activation of neutrophil and eosinophil functional activity, induction of acute phase reactants and other liver proteins, as well as tissue degrading enzymes produced by synoviocytes and/or chondrocytes. Cells expressing transmembrane TNFα bound by infliximab products can be lysed in vitro or in vivo. Infliximab products inhibit the functional activity of TNFα in a wide variety of in vitro bioassays utilizing human fibroblasts, endothelial cells, neutrophils, B and T-lymphocytes and epithelial cells. The relationship of these biological response markers to the mechanism(s) by which infliximab products exert their clinical effects is unknown. Anti-TNFα antibodies reduce disease activity in the cotton-top tamarin colitis model, and decrease synovitis and joint erosions in a murine model of collagen-induced arthritis. Infliximab products prevent disease in transgenic mice that develop polyarthritis as a result of constitutive expression of human TNFα, and when administered after disease onset, allow eroded joints to heal.
12.2 Pharmacodynamics
Elevated concentrations of TNFα have been found in involved tissues and fluids of patients with rheumatoid arthritis, Crohn's disease, ulcerative colitis, ankylosing spondylitis, psoriatic arthritis and plaque psoriasis. In rheumatoid arthritis, treatment with infliximab products reduced infiltration of inflammatory cells into inflamed areas of the joint as well as expression of molecules mediating cellular adhesion [E-selectin, intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1)], chemoattraction [IL-8 and monocyte chemotactic protein (MCP-1)] and tissue degradation [matrix metalloproteinase (MMP) 1 and 3]. In Crohn's disease, treatment with infliximab products reduced infiltration of inflammatory cells and TNFα production in inflamed areas of the intestine, and reduced the proportion of mononuclear cells from the lamina propria able to express TNFα and interferon. After treatment with infliximab products, patients with rheumatoid arthritis or Crohn's disease exhibited decreased levels of serum IL-6 and C-reactive protein (CRP) compared to baseline. Peripheral blood lymphocytes from infliximab product-treated patients showed no significant decrease in number or in proliferative responses to in vitro mitogenic stimulation when compared to cells from untreated patients. In psoriatic arthritis, treatment with infliximab products resulted in a reduction in the number of T-cells and blood vessels in the synovium and psoriatic skin lesions as well as a reduction of macrophages in the synovium. In plaque psoriasis, treatment with infliximab products may reduce the epidermal thickness and infiltration of inflammatory cells. The relationship between these pharmacodynamic activities and the mechanism(s) by which infliximab products exert their clinical effects is unknown.
12.3 Pharmacokinetics
In adults, single intravenous (IV) infusions of 3 mg/kg to 20 mg/kg of infliximab showed a linear relationship between the dose administered and the maximum serum concentration. The volume of distribution at steady state was independent of dose and indicated that infliximab was distributed primarily within the vascular compartment. Pharmacokinetic results for single doses of 3 mg/kg to 10 mg/kg in rheumatoid arthritis, 5 mg/kg in Crohn's disease, and 3 mg/kg to 5 mg/kg in plaque psoriasis indicate that the median terminal half-life of infliximab is 7.7 to 9.5 days.
Following an initial dose of infliximab, repeated infusions at 2 and 6 weeks resulted in predictable concentration-time profiles following each treatment. No systemic accumulation of infliximab occurred upon continued repeated treatment with 3 mg/kg or 10 mg/kg at 4-or 8week intervals. Development of antibodies to infliximab increased infliximab clearance. At 8 weeks after a maintenance dose of 3 to 10 mg/kg of infliximab, median infliximab serum concentrations ranged from approximately 0.5 to 6 mcg/mL; however, infliximab concentrations were not detectable (<0.1 mcg/mL) in patients who became positive for antibodies to infliximab. No major differences in clearance or volume of distribution were observed in patient subgroups defined by age, weight, or gender. It is not known if there are differences in clearance or volume of distribution in patients with marked impairment of hepatic or renal function.
Infliximab pharmacokinetic characteristics (including peak and trough concentrations and terminal half-life) were similar in pediatric (aged 6 to 17 years) and adult patients with Crohn's disease or ulcerative colitis following the administration of 5 mg/kg infliximab.
Population pharmacokinetic analysis showed that in children with juvenile rheumatoid arthritis (JRA) with a body weight of up to 35 kg receiving 6 mg/kg infliximab and children with JRA with body weight greater than 35 kg up to adult body weight receiving 3 mg/kg infliximab, the steady state area under the concentration curve (AUCss) was similar to that observed in adults receiving 3 mg/kg of infliximab.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The significance of the results of nonclinical studies for human risk is unknown. A repeat dose toxicity study was conducted with mice given cV1q anti-mouse TNFα to evaluate tumorigenicity. CV1q is an analogous antibody that inhibits the function of TNFα in mice. Animals were assigned to 1 of 3 dose groups: control, 10 mg/kg or 40 mg/kg cV1q given weekly for 6 months. The weekly doses of 10 mg/kg and 40 mg/kg are 2 and 8 times, respectively, the human dose of 5 mg/kg for Crohn's disease. Results indicated that cV1q did not cause tumorigenicity in mice. No clastogenic or mutagenic effects of infliximab were observed in the in vivo mouse micronucleus test or the Salmonella-Escherichia coli (Ames) assay, respectively. Chromosomal aberrations were not observed in an assay performed using human lymphocytes. It is not known whether infliximab products can impair fertility in humans. No impairment of fertility was observed in a fertility and general reproduction toxicity study with the analogous mouse antibody used in the 6-month chronic toxicity study.
-
14 CLINICAL STUDIES
14.1 Crohn's Disease
Active Crohn's Disease
The safety and efficacy of single and multiple doses of infliximab were assessed in 2 randomized, double-blind, placebo-controlled clinical studies in 653 patients with moderate to severely active Crohn's disease [Crohn's Disease Activity Index (CDAI) ≥ 220 and ≤ 400] with an inadequate response to prior conventional therapies. Concomitant stable doses of aminosalicylates, corticosteroids and/or immunomodulatory agents were permitted and 92% of patients continued to receive at least one of these medications.
In the single-dose trial of 108 patients, 16% (4/25) of placebo patients achieved a clinical response (decrease in CDAI ≥ 70 points) at Week 4 vs. 81% (22/27) of patients receiving 5 mg/kg infliximab (p < 0.001, two-sided, Fisher's Exact test). Additionally, 4% (1/25) of placebo patients and 48% (13/27) of patients receiving 5 mg/kg infliximab achieved clinical remission (CDAI < 150) at Week 4.
In a multidose trial (ACCENT I [Study Crohn's I]), 545 patients received 5 mg/kg at Week 0 and were then randomized to one of three treatment groups; the placebo maintenance group received placebo at Weeks 2 and 6, and then every 8 weeks; the 5 mg/kg maintenance group received 5 mg/kg at Weeks 2 and 6, and then every 8 weeks; and the 10 mg/kg maintenance group received 5 mg/kg at Weeks 2 and 6, and then 10 mg/kg every 8 weeks. Patients in response at Week 2 were randomized and analyzed separately from those not in response at Week 2. Corticosteroid taper was permitted after Week 6.
At Week 2, 57% (311/545) of patients were in clinical response. At Week 30, a significantly greater proportion of these patients in the 5 mg/kg and 10 mg/kg maintenance groups achieved clinical remission compared to patients in the placebo maintenance group (Table 3). Additionally, a significantly greater proportion of patients in the 5 mg/kg and 10 mg/kg infliximab maintenance groups were in clinical remission and were able to discontinue corticosteroid use compared to patients in the placebo maintenance group at Week 54 (Table 3).
Table 3 Clinical remission and steroid withdrawal Single 5-mg/kg Dose* Three-Dose Induction† Placebo Maintenance Infliximab Maintenance q 8 wks 5 mg/kg 10 mg/kg Week 30
25/102
41/104
48/105
Clinical remission
25%
39%
46%
P-value‡
0.022
0.001
Week 54
6/54
14/56
18/53
Patients in remission able to discontinue corticosteroid use§
11%
25%
34%
P-value‡
0.059
0.005
Patients in the infliximab maintenance groups (5 mg/kg and 10 mg/kg) had a longer time to loss of response than patients in the placebo maintenance group (Figure 1). At Weeks 30 and 54, significant improvement from baseline was seen among the 5 mg/kg and 10 mg/kg infliximab-treated groups compared to the placebo group in the disease-specific inflammatory bowel disease questionnaire (IBDQ), particularly the bowel and systemic components, and in the physical component summary score of the general health-related quality of life questionnaire SF-36.
Figure 1 Kaplan-Meier estimate of the proportion of patients who had not lost response through Week 54
In a subset of 78 patients who had mucosal ulceration at baseline and who participated in an endoscopic substudy, 13 of 43 patients in the infliximab maintenance group had endoscopic evidence of mucosal healing compared to 1 of 28 patients in the placebo group at Week 10. Of the infliximab-treated patients showing mucosal healing at Week 10, 9 of 12 patients also showed mucosal healing at Week 54.
Patients who achieved a response and subsequently lost response were eligible to receive infliximab on an episodic basis at a dose that was 5 mg/kg higher than the dose to which they were randomized. The majority of such patients responded to the higher dose. Among patients who were not in response at Week 2, 59% (92/157) of infliximab maintenance patients responded by Week 14 compared to 51% (39/77) of placebo maintenance patients. Among patients who did not respond by Week 14, additional therapy did not result in significantly more responses [see Dosage and Administration (2)].
Fistulizing Crohn's Disease
The safety and efficacy of infliximab were assessed in 2 randomized, double-blind, placebo-controlled studies in patients with fistulizing Crohn's disease with fistula(s) that were of at least 3 months duration. Concurrent use of stable doses of corticosteroids, 5-aminosalicylates, antibiotics, MTX, 6-mercaptopurine (6-MP) and/or azathioprine (AZA) was permitted.
In the first trial, 94 patients received 3 doses of either placebo or infliximab at Weeks 0, 2 and 6. Fistula response (≥ 50% reduction in number of enterocutaneous fistulas draining upon gentle compression on at least 2 consecutive visits without an increase in medication or surgery for Crohn's disease) was seen in 68% (21/31) of patients in the 5 mg/kg infliximab group (P = 0.002) and 56% (18/32) of patients in the 10 mg/kg infliximab group (P = 0.021) vs. 26% (8/31) of patients in the placebo arm. The median time to onset of response and median duration of response in infliximab-treated patients was 2 and 12 weeks, respectively. Closure of all fistulas was achieved in 52% of infliximab-treated patients compared with 13% of placebo-treated patients (P < 0.001).
In the second trial (ACCENT II [Study Crohn's II]), patients who were enrolled had to have at least 1 draining enterocutaneous (perianal, abdominal) fistula. All patients received 5 mg/kg infliximab at Weeks 0, 2 and 6. Patients were randomized to placebo or 5 mg/kg infliximab maintenance at Week 14. Patients received maintenance doses at Week 14 and then every 8 weeks through Week 46. Patients who were in fistula response (fistula response was defined the same as in the first trial) at both Weeks 10 and 14 were randomized separately from those not in response. The primary endpoint was time from randomization to loss of response among those patients who were in fistula response.
Among the randomized patients (273 of the 296 initially enrolled), 87% had perianal fistulas and 14% had abdominal fistulas. Eight percent also had rectovaginal fistulas. Greater than 90% of the patients had received previous immunosuppressive and antibiotic therapy.
At Week 14, 65% (177/273) of patients were in fistula response. Patients randomized to infliximab maintenance had a longer time to loss of fistula response compared to the placebo maintenance group (Figure 2). At Week 54, 38% (33/87) of infliximab-treated patients had no draining fistulas compared with 22% (20/90) of placebo-treated patients (P=0.02). Compared to placebo maintenance, patients on infliximab maintenance had a trend toward fewer hospitalizations.
Figure 2 Life table estimates of the proportion of patients who had not lost fistula response through Week 54
Patients who achieved a fistula response and subsequently lost response were eligible to receive infliximab maintenance therapy at a dose that was 5 mg/kg higher than the dose to which they were randomized. Of the placebo maintenance patients, 66% (25/38) responded to 5 mg/kg infliximab, and 57% (12/21) of infliximab maintenance patients responded to 10 mg/kg.
Patients who had not achieved a response by Week 14 were unlikely to respond to additional doses of infliximab.
Similar proportions of patients in either group developed new fistulas (17% overall) and similar numbers developed abscesses (15% overall).
14.2 Pediatric Crohn's Disease
The safety and efficacy of infliximab were assessed in a randomized, open-label study (Study Peds Crohn's) in 112 pediatric patients aged 6 to 17 years old with moderately to severely active Crohn's disease and an inadequate response to conventional therapies. The median age was 13 years and the median Pediatric Crohn's Disease Activity Index (PCDAI) was 40 (on a scale of 0 to 100). All patients were required to be on a stable dose of 6-MP, AZA, or MTX; 35% were also receiving corticosteroids at baseline.
All patients received induction dosing of 5 mg/kg infliximab at Weeks 0, 2, and 6. At Week 10, 103 patients were randomized to a maintenance regimen of 5 mg/kg infliximab given either every 8 weeks or every 12 weeks.
At Week 10, 88% of patients were in clinical response (defined as a decrease from baseline in the PCDAI score of ≥ 15 points and total PCDAI score of ≤ 30 points), and 59% were in clinical remission (defined as PCDAI score of ≤ 10 points).
The proportion of pediatric patients achieving clinical response at Week 10 compared favorably with the proportion of adults achieving a clinical response in Study Crohn's I. The study definition of clinical response in Study Peds Crohn's was based on the PCDAI score, whereas the CDAI score was used in the adult Study Crohn's I.
At both Week 30 and Week 54, the proportion of patients in clinical response was greater in the every 8-week treatment group than in the every 12-week treatment group (73% vs. 47% at Week 30, and 64% vs. 33% at Week 54). At both Week 30 and Week 54, the proportion of patients in clinical remission was also greater in the every 8-week treatment group than in the every 12week treatment group (60% vs. 35% at Week 30, and 56% vs. 24% at Week 54), (Table 4).
For patients in Study Peds Crohn's receiving corticosteroids at baseline, the proportion of patients able to discontinue corticosteroids while in remission at Week 30 was 46% for the every 8-week maintenance group and 33% for the every 12-week maintenance group. At Week 54, the proportion of patients able to discontinue corticosteroids while in remission was 46% for the every 8-week maintenance group and 17% for the every 12-week maintenance group.
14.3 Ulcerative Colitis
The safety and efficacy of infliximab were assessed in 2 randomized, double-blind, placebo-controlled clinical studies in 728 patients with moderately to severely active ulcerative colitis (UC) (Mayo score5 6 to 12 [of possible range 0 to 12], Endoscopy subscore ≥ 2) with an inadequate response to conventional oral therapies (Studies UC I and UC II). Concomitant treatment with stable doses of aminosalicylates, corticosteroids and/or immunomodulatory agents was permitted. Corticosteroid taper was permitted after Week 8. Patients were randomized at week 0 to receive either placebo, 5 mg/kg infliximab or 10 mg/kg infliximab at Weeks 0, 2, 6, and every 8 weeks thereafter through Week 46 in Study UC I, and at Weeks 0, 2, 6, and every 8 weeks thereafter through Week 22 in Study UC II. In Study UC II, patients were allowed to continue blinded therapy to Week 46 at the investigator's discretion.
Patients in Study UC I had failed to respond or were intolerant to oral corticosteroids, 6-MP, or AZA. Patients in Study UC II had failed to respond or were intolerant to the above treatments and/or aminosalicylates. Similar proportions of patients in Studies UC I and UC II were receiving corticosteroids (61% and 51%, respectively), 6-MP/AZA (49% and 43%) and aminosalicylates (70% and 75%) at baseline. More patients in Study UC II than UC I were taking solely aminosalicylates for UC (26% vs. 11%, respectively). Clinical response was defined as a decrease from baseline in the Mayo score by ≥30% and ≥3 points, accompanied by a decrease in the rectal bleeding subscore of ≥1 or a rectal bleeding subscore of 0 or 1.
Clinical Response, Clinical Remission, and Mucosal Healing
In both Study UC I and Study UC II, greater percentages of patients in both infliximab groups achieved clinical response, clinical remission and mucosal healing than in the placebo group. Each of these effects was maintained through the end of each trial (Week 54 in Study UC I, and Week 30 in Study UC II). In addition, a greater proportion of patients in infliximab groups demonstrated sustained response and sustained remission than in the placebo groups (Table 5).
Of patients on corticosteroids at baseline, greater proportions of patients in the infliximab treatment groups were in clinical remission and able to discontinue corticosteroids at Week 30 compared with the patients in the placebo treatment groups (22% in infliximab treatment groups vs. 10% in placebo group in Study UC I; 23% in infliximab treatment groups vs. 3% in placebo group in Study UC II). In Study UC I, this effect was maintained through Week 54 (21% in infliximab treatment groups vs. 9% in placebo group). The infliximab-associated response was generally similar in the 5 mg/kg and 10 mg/kg dose groups.
Table 5 Response, remission and mucosal healing in ulcerative colitis studies Study UC I Study UC II Placebo 5 mg/kg
infliximab10 mg/kg
infliximabPlacebo 5 mg/kg
infliximab10 mg/kg
infliximab- *
- Defined as a decrease from baseline in the Mayo score by ≥30% and ≥3 points, accompanied by a decrease in the rectal bleeding subscore of ≥1 or a rectal bleeding subscore of 0 or 1. (The Mayo score consists of the sum of four subscores: stool frequency, rectal bleeding, physician's global assessment and endoscopy findings.)
- †
- Patients who had a prohibited change in medication, had an ostomy or colectomy, or discontinued study infusions due to lack of efficacy are considered to not be in clinical response, clinical remission or mucosal healing from the time of the event onward.
- ‡
- P < 0.001,
- §
- P < 0.01
- ¶
- Defined as a Mayo score ≤2 points, no individual subscore >1.
- #
- Defined as a 0 or 1 on the endoscopy subscore of the Mayo score.
Patients randomized
121
121
122
123
121
120
Week 8
37%
69%‡
62%‡
29%
65%‡
69%‡
Week 30
30%
52%‡
51%§
26%
47%‡
60%‡
Week 54
20%
45%‡
44%‡
NA
NA
NA
Sustained Response†
(Clinical response at both Week 8 and 30)
23%
49%‡
46%‡
15%
41%‡
53%‡
(Clinical response at Week 8, 30 and 54)
14%
39%‡
37%‡
NA
NA
NA
Week 8
15%
39%‡
32%§
6%
34%‡
28%‡
Week 30
16%
34%§
37%‡
11%
26%§
36%‡
Week 54
17%
35%§
34%§
NA
NA
NA
Sustained Remission†
(Clinical response at both Week 8 and 30)
8%
23%§
26%‡
2%
15%‡
23%‡
(Clinical response at Week 8, 30 and 54)
7%
20%§
20%§
NA
NA
NA
Week 8
34%
62%‡
59%‡
31%
60%‡
62%‡
Week 30
25%
50%‡
49%‡
30%
46%§
57%‡
Week 54
18%
45%‡
47%‡
NA
NA
NA
The improvement with infliximab was consistent across all Mayo subscores through Week 54 (Study UC I shown in Table 6; Study UC II through Week 30 was similar).
Table 6 Proportion of patients in Study UC I with Mayo subscores indicating inactive or mild disease through Week 54 Study UC I Infliximab Placebo 5 mg/kg 10 mg/kg (n = 121) (n = 121) (n = 122) Stool frequency
Baseline
17%
17%
10%
Week 8
35%
60%
58%
Week 30
35%
51%
53%
Week 54
31%
52%
51%
Rectal bleeding
Baseline
54%
40%
48%
Week 8
74%
86%
80%
Week 30
65%
74%
71%
Week 54
62%
69%
67%
Physician's Global Assessment
Baseline
4%
6%
3%
Week 8
44%
74%
64%
Week 30
36%
57%
55%
Week 54
26%
53%
53%
Endoscopy findings
Baseline
0%
0%
0%
Week 8
34%
62%
59%
Week 30
26%
51%
52%
Week 54
21%
50%
51%
14.4 Pediatric Ulcerative Colitis
The safety and effectiveness of infliximab products for reducing signs and symptoms and inducing and maintaining clinical remission in pediatric patients aged 6 years and older with moderately to severely active ulcerative colitis who have had an inadequate response to conventional therapy are supported by evidence from adequate and well-controlled studies of infliximab in adults. Additional safety and pharmacokinetic data were collected in an open-label pediatric UC trial in 60 pediatric patients aged 6 through 17 years (median age 14.5 years) with moderately to severely active ulcerative colitis (Mayo score of 6 to 12; Endoscopic subscore ≥ 2) and an inadequate response to conventional therapies. At baseline, the median Mayo score was 8, 53% of patients were receiving immunomodulator therapy (6-MP/AZA/MTX), and 62% of patients were receiving corticosteroids (median dose 0.5 mg/kg/day in prednisone equivalents). Discontinuation of immunomodulators and corticosteroid taper were permitted after Week 0.
All patients received induction dosing of 5 mg/kg infliximab at Weeks 0, 2, and 6. Patients who did not respond to infliximab at Week 8 received no further infliximab and returned for safety follow-up. At Week 8, 45 patients were randomized to a maintenance regimen of 5 mg/kg infliximab given either every 8 weeks through Week 46 or every 12 weeks through Week 42. Patients were allowed to change to a higher dose and/or more frequent administration schedule if they experienced loss of response.
Clinical response at Week 8 was defined as a decrease from baseline in the Mayo score by ≥ 30% and ≥ 3 points, including a decrease in the rectal bleeding subscore by ≥ 1 points or achievement of a rectal bleeding subscore of 0 or 1.
Clinical remission at Week 8 was measured by the Mayo score, defined as a Mayo score of ≤ 2 points with no individual subscore >1. Clinical remission was also assessed at Week 8 and Week 54 using the Pediatric Ulcerative Colitis Activity Index (PUCAI)6 score and was defined by a PUCAI score of <10 points.
Endoscopies were performed at baseline and at Week 8. A Mayo endoscopy subscore of 0 indicated normal or inactive disease and a subscore of 1 indicated mild disease (erythema, decreased vascular pattern, or mild friability).
Of the 60 patients treated, 44 were in clinical response at Week 8. Of 32 patients taking concomitant immunomodulators at baseline, 23 achieved clinical response at Week 8, compared to 21 of 28 of those not taking concomitant immunomodulators at baseline. At Week 8, 24 of 60 patients were in clinical remission as measured by the Mayo score and 17 of 51 patients were in remission as measured by the PUCAI score.
At Week 54, 8 of 21 patients in the every 8-week maintenance group and 4 of 22 patients in the every 12-week maintenance group achieved remission as measured by the PUCAI score.
During maintenance phase, 23 of 45 randomized patients (9 in the every 8-week group and 14 in the every 12-week group) required an increase in their dose and/or increase in frequency of infliximab administration due to loss of response. Nine of the 23 patients who required a change in dose had achieved remission at Week 54. Seven of those patients received the 10 mg/kg every 8-week dosing.
14.5 Rheumatoid Arthritis
The safety and efficacy of infliximab were assessed in 2 multicenter, randomized, doubleblind, pivotal trials: ATTRACT (Study RA I) and ASPIRE (Study RA II). Concurrent use of stable doses of folic acid, oral corticosteroids (≤ 10 mg/day) and/or non-steroidal anti-inflammatory drugs (NSAIDs) was permitted.
Study RA I was a placebo-controlled study of 428 patients with active rheumatoid arthritis despite treatment with MTX. Patients enrolled had a median age of 54 years, median disease duration of 8.4 years, median swollen and tender joint count of 20 and 31 respectively, and were on a median dose of 15 mg/wk of MTX. Patients received either placebo + MTX or one of 4 doses/schedules of infliximab + MTX: 3 mg/kg or 10 mg/kg of infliximab by IV infusion at Weeks 0, 2 and 6 followed by additional infusions every 4 or 8 weeks in combination with MTX.
Study RA II was a placebo-controlled study of 3 active treatment arms in 1004 MTX naive patients of 3 or fewer years' duration active rheumatoid arthritis. Patients enrolled had a median age of 51 years with a median disease duration of 0.6 years, median swollen and tender joint count of 19 and 31, respectively, and >80% of patients had baseline joint erosions. At randomization, all patients received MTX (optimized to 20 mg/wk by Week 8) and either placebo, 3 mg/kg or 6 mg/kg infliximab at Weeks 0, 2, and 6 and every 8 weeks thereafter.
Data on use of infliximab products without concurrent MTX are limited [see Adverse Reactions (6.1)].
Clinical Response
In Study RA I, all doses/schedules of infliximab + MTX resulted in improvement in signs and symptoms as measured by the American College of Rheumatology response criteria (ACR 20) with a higher percentage of patients achieving an ACR 20, 50 and 70 compared to placebo + MTX (Table 7). This improvement was observed at Week 2 and maintained through Week 102. Greater effects on each component of the ACR 20 were observed in all patients treated with infliximab + MTX compared to placebo + MTX (Table 8). More patients treated with infliximab reached a major clinical response than placebo-treated patients (Table 7).
In Study RA II, after 54 weeks of treatment, both doses of infliximab + MTX resulted in statistically significantly greater response in signs and symptoms compared to MTX alone as measured by the proportion of patients achieving ACR 20, 50 and 70 responses (Table 7). More patients treated with infliximab reached a major clinical response than placebo-treated patients (Table 7).
Table 7 ACR response (percent of patients) Study RA I Study RA II Infliximab + MTX Infliximab + MTX 3 mg/kg 10 mg/kg 3 mg/kg 6 mg/kg Response Placebo + MTX q8 wks q4 wks q8 wks q4 wks Placebo + MTX q8 wks q8 wks (n = 88) (n = 86) (n = 86) (n = 87) (n = 81) (n = 274) (n = 351) (n = 355) ACR20
Week 30
20%
50%*
50%*
52%*
58%*
N/A
N/A
N/A
Week 54
17%
42%*
48%*
59%*
59%*
54%
62%†
66%*
ACR50
Week 30
5%
27%*
29%*
31%*
26%*
N/A
N/A
N/A
Week 54
9%
21%†
34%*
40%*
38%*
32%
46%*
50%*
ACR70
Week 30
0%
8%‡
11%‡
18%*
11%*
N/A
N/A
N/A
Week 54
2%
11%†
18%*
26%*
19%*
21%
33%‡
37%*
Major clinical response§
0%
7%
8%‡
15%*
6%†
8%
12%
17%*
Table 8 Components of ACR 20 at baseline and 54 weeks (Study RA I) Placebo + MTX Infliximab + MTX* (n = 88) (n = 340) Parameter (medians) Baseline Week54 Baseline Week54 No. of Tender Joints
24
16
32
8
No. of Swollen Joints
19
13
20
7
Pain†
6.7
6.1
6.8
3.3
Physician's Global Assessment†
6.5
5.2
6.2
2.1
Patient's Global Assessment†
6.2
6.2
6.3
3.2
Disability Index (HAQ-DI)‡
1.8
1.5
1.8
1.3
CRP (mg/dL)
3.0
2.3
2.4
0.6
Radiographic Response
Structural damage in both hands and feet was assessed radiographically at Week 54 by the change from baseline in the van der Heijde-modified Sharp (vdH-S) score, a composite score of structural damage that measures the number and size of joint erosions and the degree of joint space narrowing in hands/wrists and feet.3
In Study RA I, approximately 80% of patients had paired X-ray data at 54 weeks and approximately 70% at 102 weeks. The inhibition of progression of structural damage was observed at 54 weeks (Table 9) and maintained through 102 weeks.
In Study RA II, >90% of patients had at least 2 evaluable X-rays. Inhibition of progression of structural damage was observed at Weeks 30 and 54 (Table 9) in the infliximab + MTX groups compared to MTX alone. Patients treated with infliximab + MTX demonstrated less progression of structural damage compared to MTX alone, whether baseline acute-phase reactants (ESR and CRP) were normal or elevated: patients with elevated baseline acute-phase reactants treated with MTX alone demonstrated a mean progression in vdH-S score of 4.2 units compared to patients treated with infliximab + MTX who demonstrated 0.5 units of progression; patients with normal baseline acute phase reactants treated with MTX alone demonstrated a mean progression in vdH-S score of 1.8 units compared to infliximab + MTX who demonstrated 0.2 units of progression. Of patients receiving infliximab + MTX, 59% had no progression (vdH-S score ≤ 0 unit) of structural damage compared to 45% of patients receiving MTX alone. In a subset of patients who began the study without erosions, infliximab + MTX maintained an erosion-free state at 1 year in a greater proportion of patients than MTX alone, 79% (77/98) vs. 58% (23/40), respectively (P<0.01). Fewer patients in the infliximab + MTX groups (47%) developed erosions in uninvolved joints compared to MTX alone (59%).
Table 9 Radiographic change from baseline to Week 54 Study RA I Study RA II Infliximab + MTX Infliximab + MTX Placebo + MTX
(n = 64)3 mg/kg
q8 wks
(n = 71)10 mg/kg
q8 wks
(n = 77)Placebo + MTX
(n = 282)3 mg/kg
q8 wks
(n = 359)6 mg/kg
q8 wks
(n = 363)- *
- P < 0.001 for each outcome against placebo.
Total score
Baseline
Mean
79
78
65
11.3
11.6
11.2
Median
55
57
56
5.1
5.2
5.3
Change from baseline
Mean
6.9
1.3*
0.2*
3.7
0.4*
0.5*
Median
4.0
0.5
0.5
0.4
0.0
0.0
Erosion Score
Baseline
Mean
44
44
33
8.3
8.8
8.3
Median
25
29
22
3.0
3.8
3.8
Change from baseline
Mean
4.1
0.2*
0.2*
3.0
0.3*
0.1*
Median
2.0
0.0
0.5
0.3
0.0
0.0
JSN Score
Baseline
Mean
36
34
31
3.0
2.9
2.9
Median
26
29
24
1.0
1.0
1.0
Change from baseline
Mean
2.9
1.1*
0.0*
0.6
0.1*
0.2
Median
1.5
0.0
0.0
0.0
0.0
0.0
Physical Function Response
Physical function and disability were assessed using the Health Assessment Questionnaire (HAQ-DI) and the general health-related quality of life questionnaire SF-36.
In Study RA I, all doses/schedules of infliximab + MTX showed significantly greater improvement from baseline in HAQ-DI and SF-36 physical component summary score averaged over time through Week 54 compared to placebo + MTX, and no worsening in the SF-36 mental component summary score. The median (interquartile range) improvement from baseline to Week 54 in HAQ-DI was 0.1 (-0.1, 0.5) for the placebo + MTX group and 0.4 (0.1, 0.9) for infliximab + MTX (p<0.001). Both HAQ-DI and SF-36 effects were maintained through Week 102. Approximately 80% of patients in all doses/schedules of infliximab + MTX remained in the trial through 102 weeks.
In Study RA II, both infliximab treatment groups showed greater improvement in HAQ-DI from baseline averaged over time through Week 54 compared to MTX alone; 0.7 for infliximab + MTX vs. 0.6 for MTX alone (P≤0.001). No worsening in the SF-36 mental component summary score was observed.
14.6 Ankylosing Spondylitis
The safety and efficacy of infliximab were assessed in a randomized, multicenter, double-blind, placebo-controlled study in 279 patients with active ankylosing spondylitis. Patients were between 18 and 74 years of age, and had ankylosing spondylitis as defined by the modified New York criteria for Ankylosing Spondylitis.4 Patients were to have had active disease as evidenced by both a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score >4 (possible range 0-10) and spinal pain >4 (on a Visual Analog Scale [VAS] of 0-10). Patients with complete ankylosis of the spine were excluded from study participation, and the use of Disease Modifying Anti-Rheumatic Drugs (DMARDs) and systemic corticosteroids were prohibited. Doses of infliximab 5 mg/kg or placebo were administered intravenously at Weeks 0, 2, 6, 12 and 18.
At 24 weeks, improvement in the signs and symptoms of ankylosing spondylitis, as measured by the proportion of patients achieving a 20% improvement in ASAS response criteria (ASAS 20), was seen in 60% of patients in the infliximab-treated group vs. 18% of patients in the placebo group (p<0.001). Improvement was observed at Week 2 and maintained through Week 24 (Figure 3 and Table 10).
Figure 3 Proportion of patients achieving ASAS 20 response
At 24 weeks, the proportions of patients achieving a 50% and a 70% improvement in the signs and symptoms of ankylosing spondylitis, as measured by ASAS response criteria (ASAS 50 and ASAS 70, respectively), were 44% and 28%, respectively, for patients receiving infliximab, compared to 9% and 4%, respectively, for patients receiving placebo (P < 0.001, infliximab vs. placebo). A low level of disease activity (defined as a value < 20 [on a scale of 0-100 mm] in each of the 4 ASAS response parameters) was achieved in 22% of infliximab-treated patients vs. 1% in placebo-treated patients (P < 0.001).
Table 10 Components of ankylosing spondylitis disease activity Placebo
(n = 78)Infliximab 5 mg/kg
(n = 201)Baseline 24 Weeks Baseline 24 Weeks P-value - *
- Measured on a VAS with 0 = "none" and 10 = "severe"
- †
- Bath Ankylosing Spondylitis Functional Index (BASFI), average of 10 questions
- ‡
- Inflammation, average of last 2 questions on the 6-question BASDAI
- §
- CRP normal range 0-1.0 mg/dL
- ¶
- Spinal mobility normal values: modified Schober's test: > 4 cm; chest expansion: > 6 cm; tragus to wall: < 15 cm; lateral spinal flexion: > 10 cm
ASAS 20 response Criteria (Mean)
Patient Global Assessment*
6.6
6.0
6.8
3.8
< 0.001
Spinal pain*
7.3
6.5
7.6
4.0
< 0.001
BASFI†
5.8
5.6
5.7
3.6
< 0.001
Inflammation‡
6.9
5.8
6.9
3.4
< 0.001
Acute Phase Reactants
Median CRP§ (mg/dL)
1.7
1.5
1.5
0.4
< 0.001
Spinal Mobility (cm, Mean)
Modified Schober's test¶
4.0
5.0
4.3
4.4
0.75
Chest expansion¶
3.6
3.7
3.3
3.9
0.04
Tragus to wall¶
17.3
17.4
16.9
15.7
0.02
Lateral spinal flexion¶
10.6
11.0
11.4
12.9
0.03
The median improvement from baseline in the general health-related quality-of-life questionnaire SF-36 physical component summary score at Week 24 was 10.2 for the infliximab group vs. 0.8 for the placebo group (P < 0.001). There was no change in the SF-36 mental component summary score in either the infliximab group or the placebo group.
Results of this study were similar to those seen in a multicenter double-blind, placebo-controlled study of 70 patients with ankylosing spondylitis.
14.7 Psoriatic Arthritis
Safety and efficacy of infliximab were assessed in a multicenter, double-blind, placebo-controlled study in 200 adult patients with active psoriatic arthritis despite DMARD or NSAID therapy (≥ 5 swollen joints and ≥ 5 tender joints) with 1 or more of the following subtypes: arthritis involving DIP joints (n = 49), arthritis mutilans (n = 3), asymmetric peripheral arthritis (n = 40), polyarticular arthritis (n = 100), and spondylitis with peripheral arthritis (n = 8). Patients also had plaque psoriasis with a qualifying target lesion ≥ 2 cm in diameter. Forty-six percent of patients continued on stable doses of methotrexate (≤ 25 mg/week). During the 24-week double-blind phase, patients received either 5 mg/kg infliximab or placebo at Weeks 0, 2, 6, 14, and 22 (100 patients in each group). At Week 16, placebo patients with < 10% improvement from baseline in both swollen and tender joint counts were switched to infliximab induction (early escape). At Week 24, all placebo-treated patients crossed over to infliximab induction. Dosing continued for all patients through Week 46.
Clinical Response
Treatment with infliximab resulted in improvement in signs and symptoms, as assessed by the ACR criteria, with 58% of infliximab-treated patients achieving ACR 20 at Week 14, compared with 11% of placebo-treated patients (P < 0.001). The response was similar regardless of concomitant use of methotrexate. Improvement was observed as early as Week 2. At 6 months, the ACR 20/50/70 responses were achieved by 54%, 41%, and 27%, respectively, of patients receiving infliximab compared to 16%, 4%, and 2%, respectively, of patients receiving placebo. Similar responses were seen in patients with each of the subtypes of psoriatic arthritis, although few patients were enrolled with the arthritis mutilans and spondylitis with peripheral arthritis subtypes.
Compared to placebo, treatment with infliximab resulted in improvements in the components of the ACR response criteria, as well as in dactylitis and enthesopathy (Table 11). The clinical response was maintained through Week 54. Similar ACR responses were observed in an earlier randomized, placebo-controlled study of 104 psoriatic arthritis patients, and the responses were maintained through 98 weeks in an open-label extension phase.
Table 11 Components of ACR 20 and percentage of patients with 1 or more joints with dactylitis and percentage of patients with enthesopathy at baseline and Week 24 Placebo Infliximab 5 mg/kg* Patient randomized (n = 100) (n = 100) Baseline Week 24 Baseline Week 24 - *
- P<0.001 for percent change from baseline in all components of ACR 20 at Week 24, P<0.05 for % of patients with dactylitis, and P=0.004 for % of patients with enthesopathy at Week 24
- †
- Scale 0-68
- ‡
- Scale 0-66
- §
- Visual Analog Scale (0=best, 10=worst)
- ¶
- Health Assessment Questionnaire, measurement of 8 categories: dressing and grooming, arising, eating, walking, hygiene, reach, grip, and activities (0=best, 3=worst)
- #
- Normal range 0-0.6 mg/dL
Parameter (medians)
No. of Tender Joints†
24
20
20
6
No. of Swollen Joints‡
12
9
12
3
Pain§
6.4
5.6
5.9
2.6
Physician's Global Assessment§
6.0
4.5
5.6
1.5
Patient's Global Assessment§
6.1
5.0
5.9
2.5
Disability Index (HAQ-DI)¶
1.1
1.1
1.1
0.5
CRP (mg/dL)#
1.2
0.9
1.0
0.4
% Patients with 1 or more digits with dactylitis
41
33
40
15
% Patients with enthesopathy
35
36
42
22
Improvement in Psoriasis Area and Severity Index (PASI) in psoriatic arthritis patients with baseline body surface area (BSA) ≥ 3% (n = 87 placebo, n = 83 infliximab) was achieved at Week 14, regardless of concomitant methotrexate use, with 64% of infliximab-treated patients achieving at least 75% improvement from baseline vs. 2% of placebo-treated patients; improvement was observed in some patients as early as Week 2. At 6 months, the PASI 75 and PASI 90 responses were achieved by 60% and 39%, respectively, of patients receiving infliximab compared to 1% and 0%, respectively, of patients receiving placebo. The PASI response was generally maintained through Week 54. [See also Clinical Studies (14.8)].
Radiographic Response
Structural damage in both hands and feet was assessed radiographically by the change from baseline in the van der Heijde-Sharp (vdH-S) score, modified by the addition of hand DIP joints. The total modified vdH-S score is a composite score of structural damage that measures the number and size of joint erosions and the degree of joint space narrowing (JSN) in the hands and feet. At Week 24, infliximab-treated patients had less radiographic progression than placebo-treated patients (mean change of -0.70 vs. 0.82, P < 0.001). infliximab-treated patients also had less progression in their erosion scores (-0.56 vs 0.51) and JSN scores (-0.14 vs 0.31).
The patients in the infliximab group demonstrated continued inhibition of structural damage at Week 54. Most patients showed little or no change in the vdH-S score during this 12-month study (median change of 0 in both patients who initially received infliximab or placebo). More patients in the placebo group (12%) had readily apparent radiographic progression compared with the infliximab group (3%).
Physical Function
Physical function status was assessed using the HAQ Disability Index (HAQ-DI) and the SF-36 Health Survey. infliximab-treated patients demonstrated significant improvement in physical function as assessed by HAQ-DI (median percent improvement in HAQ-DI score from baseline to Week 14 and 24 of 43% for infliximab-treated patients vs 0% for placebo-treated patients).
During the placebo-controlled portion of the trial (24 weeks), 54% of infliximab-treated patients achieved a clinically meaningful improvement in HAQ-DI (≥0.3 unit decrease) compared to 22% of placebo-treated patients. infliximab-treated patients also demonstrated greater improvement in the SF-36 physical and mental component summary scores than placebo-treated patients. The responses were maintained for up to 2 years in an open-label extension study.
14.8 Plaque Psoriasis
The safety and efficacy of infliximab were assessed in 3 randomized, double-blind, placebo-controlled studies in patients 18 years of age and older with chronic, stable plaque psoriasis involving ≥10% BSA, a minimum PASI score of 12, and who were candidates for systemic therapy or phototherapy. Patients with guttate, pustular, or erythrodermic psoriasis were excluded from these studies. No concomitant anti-psoriatic therapies were allowed during the study, with the exception of low-potency topical corticosteroids on the face and groin after Week 10 of study initiation.
Study I (EXPRESS) evaluated 378 patients who received placebo or infliximab at a dose of 5 mg/kg at Weeks 0, 2, and 6 (induction therapy), followed by maintenance therapy every 8 weeks. At Week 24, the placebo group crossed over to infliximab induction therapy (5 mg/kg), followed by maintenance therapy every 8 weeks. Patients originally randomized to infliximab continued to receive infliximab 5 mg/kg every 8 weeks through Week 46. Across all treatment groups, the median baseline PASI score was 21 and the baseline Static Physician Global Assessment (sPGA) score ranged from moderate (52% of patients) to marked (36%) to severe (2%). In addition, 75% of patients had a BSA > 20%. Seventy-one percent of patients previously received systemic therapy, and 82% received phototherapy.
Study II (EXPRESS II) evaluated 835 patients who received placebo or infliximab at doses of 3 mg/kg or 5 mg/kg at Weeks 0, 2, and 6 (induction therapy). At Week 14, within each infliximab dose group, patients were randomized to either scheduled (every 8 weeks) or as needed (PRN) maintenance treatment through Week 46. At Week 16, the placebo group crossed over to infliximab induction therapy (5 mg/kg), followed by maintenance therapy every 8 weeks. Across all treatment groups, the median baseline PASI score was 18, and 63% of patients had a BSA > 20%. Fifty-five percent of patients previously received systemic therapy, and 64% received a phototherapy.
Study III (SPIRIT) evaluated 249 patients who had previously received either psoralen plus ultraviolet A treatment (PUVA) or other systemic therapy for their psoriasis. These patients were randomized to receive either placebo or infliximab at doses of 3 mg/kg or 5 mg/kg at Weeks 0, 2, and 6. At Week 26, patients with a sPGA score of moderate or worse (greater than or equal to 3 on a scale of 0 to 5) received an additional dose of the randomized treatment. Across all treatment groups, the median baseline PASI score was 19, and the baseline sPGA score ranged from moderate (62% of patients) to marked (22%) to severe (3%). In addition, 75% of patients had a BSA >20%. Of the enrolled patients, 114 (46%) received the Week 26 additional dose.
In Studies I, II and III, the primary endpoint was the proportion of patients who achieved a reduction in score of at least 75% from baseline at Week 10 by the PASI (PASI 75). In Study I and Study III, another evaluated outcome included the proportion of patients who achieved a score of "cleared" or "minimal" by the sPGA. The sPGA is a 6-category scale ranging from "5 = severe" to "0 = cleared" indicating the physician's overall assessment of the psoriasis severity focusing on induration, erythema, and scaling. Treatment success, defined as "cleared" or "minimal," consisted of none or minimal elevation in plaque, up to faint red coloration in erythema, and none or minimal fine scale over < 5% of the plaque.
Study II also evaluated the proportion of patients who achieved a score of "clear" or "excellent" by the relative Physician's Global Assessment (rPGA). The rPGA is a 6-category scale ranging from "6 = worse" to "1 = clear" that was assessed relative to baseline. Overall lesions were graded with consideration to the percent of body involvement as well as overall induration, scaling, and erythema. Treatment success, defined as "clear" or "excellent," consisted of some residual pinkness or pigmentation to marked improvement (nearly normal skin texture; some erythema may be present). The results of these studies are presented in Table 12.
Table 12 Psoriasis studies I, II, and III, Week 10 percentage of patients who achieved PASI 75 and percentage who achieved treatment "success" with Physician's Global Assessment Placebo Infliximab 3 mg/kg 5 mg/kg Psoriasis Study I -patients randomized*
77
-
301
PASI 75
2 (3%)
-
242 (80%)†
sPGA
3 (4%)
-
242 (80%)†
Psoriasis Study II -patients randomized*
208
313
314
PASI 75
4 (2%)
220 (70%)†
237 (75%)†
rPGA
2 (1%)
217 (69%)†
234 (75%)†
Psoriasis Study III -patients randomized‡
51
99
99
PASI 75
3 (6%)
71 (72%)†
87 (88%)†
sPGA
5 (10%)
71 (72%)†
89 (90%)†
In Study I, in the subgroup of patients with more extensive psoriasis who had previously received phototherapy, 85% of patients on 5 mg/kg infliximab achieved a PASI 75 at Week 10 compared with 4% of patients on placebo.
In Study II, in the subgroup of patients with more extensive psoriasis who had previously received phototherapy, 72% and 77% of patients on 3 mg/kg and 5 mg/kg infliximab achieved a PASI 75 at Week 10 respectively compared with 1% on placebo. In Study II, among patients with more extensive psoriasis who had failed or were intolerant to phototherapy, 70% and 78% of patients on 3 mg/kg and 5 mg/kg infliximab achieved a PASI 75 at Week 10 respectively, compared with 2% on placebo.
Maintenance of response was studied in a subset of 292 and 297 infliximab-treated patients in the 3 mg/kg and 5 mg/kg groups; respectively, in Study II. Stratified by PASI response at Week 10 and investigational site, patients in the active treatment groups were re-randomized to either a scheduled or as needed maintenance (PRN) therapy, beginning on Week 14.
The groups that received a maintenance dose every 8 weeks appear to have a greater percentage of patients maintaining a PASI 75 through week 50 as compared to patients who received the as-needed or PRN doses, and the best response was maintained with the 5 mg/kg every 8-week dose. These results are shown in Figure 4. At Week 46, when infliximab serum concentrations were at trough level, in the every 8-week dose group, 54% of patients in the 5 mg/kg group compared to 36% in the 3 mg/kg group achieved PASI 75. The lower percentage of PASI 75 responders in the 3 mg/kg every 8-week dose group compared to the 5 mg/kg group was associated with a lower percentage of patients with detectable trough serum infliximab levels.
This may be related in part to higher antibody rates [see Adverse Reactions (6.1)]. In addition, in a subset of patients who had achieved a response at Week 10, maintenance of response appears to be greater in patients who received infliximab every 8 weeks at the 5 mg/kg dose. Regardless of whether the maintenance doses are PRN or every 8 weeks, there is a decline in response in a subpopulation of patients in each group over time. The results of Study I through Week 50 in the 5 mg/kg every 8 weeks maintenance dose group were similar to the results from Study II.
Figure 4 Proportion of patients achieving ≥75% improvement in PASI from baseline through Week 50; patients randomized at Week 14
Efficacy and safety of infliximab treatment beyond 50 weeks have not been evaluated in patients with plaque psoriasis.
-
15 REFERENCES
- 1.
- American Thoracic Society, Centers for Disease Control and Prevention. Targeted tuberculin testing and treatment of latent tuberculosis infection. Am J Respir Crit Care Med 2000;161:S221-S247.
- 2.
- See latest Centers for Disease Control guidelines and recommendations for tuberculosis testing in immunocompromised patients.
- 3.
- van der Heijde DM, van Leeuwen MA, van Riel PL, et al. Biannual radiographic assessments of hands and feet in a three-year prospective follow-up of patients with early rheumatoid arthritis. Arthritis Rheum. 1992;35(1):26-34.
- 4.
- van der Linden S, Valkenburg HA, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria. Arthritis Rheum. 1984;27(4):361-368.
- 5.
- Schroeder KW, Tremaine WJ, Ilstrup DM. Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study. N Engl J Med. 1987;317(26):1625-1629.
- 6.
- Turner D, Otley AR, Mack D, et al. Development, validation, and evaluation of a pediatric ulcerative colitis activity index: A prospective multicenter study. Gastroenterology. 2007;133:423–432.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Each RENFLEXIS (infliximab-abda) for Injection 100 mg vial is individually packaged in a carton.
NDC 0006-4305-02
100 mg vial
Each single dose vial contains 100 mg of lyophilized infliximab-abda for final reconstitution volume of 10 mL.
Storage and Stability
Store unopened RENFLEXIS vials in a refrigerator at 2ºC to 8ºC (36ºF to 46ºF).
If needed, unopened RENFLEXIS vials may be stored at room temperatures up to a maximum of 30ºC (86°F) for a single period of up to 6 months but not exceeding the original expiration date. Once removed from the refrigerator, RENFLEXIS cannot be returned to the refrigerator.
[For storage conditions of the reconstituted product, see Dosage and Administration (2.11)].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide)
Patients or their caregivers should be advised of the potential benefits and risks of RENFLEXIS. Physicians should instruct their patients to read the Medication Guide before starting RENFLEXIS therapy and to reread it each time they receive an infusion. It is important that the patient's overall health be assessed at each treatment visit and that any questions resulting from the patient's or their caregiver's reading of the Medication Guide be discussed.
Immunosuppression
Inform patients that RENFLEXIS may lower the ability of their immune system to fight infections. Instruct patients of the importance of contacting their doctors if they develop any symptoms of an infection, including tuberculosis and reactivation of hepatitis B virus infections. Patients should be counseled about the risk of lymphoma and other malignancies while receiving RENFLEXIS.
-
SPL UNCLASSIFIED SECTION
Manufactured by: Samsung Bioepis Co., Ltd., 76, Songdogyoyuk-ro, Yeonsu-gu, Incheon, 21987, Republic of Korea
U.S. License No. 2046Manufactured for: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ 08889 USA
RENFLEXIS is a trademark of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
-
MEDICATION GUIDE
MEDICATION GUIDE
RENFLEXIS (Ren-FLEK-sis)
(infliximab-abda)
Lyophilized Concentrate for Injection, for Intravenous UseThis Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: February 2021 Read the Medication Guide that comes with RENFLEXIS before you receive the first treatment, and before each time you get a treatment of RENFLEXIS. This Medication Guide does not take the place of talking with your doctor about your medical condition or treatment.
What is the most important information I should know about RENFLEXIS?
RENFLEXIS may cause serious side effects, including:
1. Risk of infection
- RENFLEXIS is a medicine that affects your immune system. RENFLEXIS can lower the ability of your immune system to fight infections. Serious infections have happened in patients receiving RENFLEXIS. These infections include tuberculosis (TB) and infections caused by viruses, fungi or bacteria that have spread throughout the body. Some patients have died from these infections.
- •
- Your doctor should test you for TB before starting RENFLEXIS.
- •
- Your doctor should monitor you closely for signs and symptoms of TB during treatment with RENFLEXIS.
Before starting RENFLEXIS, tell your doctor if you:
- •
- think you have an infection. You should not start receiving RENFLEXIS if you have any kind of infection.
- •
- are being treated for an infection.
- •
- have signs of an infection, such as a fever, cough, flu-like symptoms.
- •
- have any open cuts or sores on your body.
- •
- get a lot of infections or have infections that keep coming back.
- •
- have diabetes or an immune system problem. People with these conditions have a higher chance for infections.
- •
- have TB, or have been in close contact with someone with TB.
- •
- live or have lived in certain parts of the country (such as the Ohio and Mississippi River valleys) where there is an increased risk for getting certain kinds of fungal infections (histoplasmosis, coccidioidomycosis, or blastomycosis). These infections may develop or become more severe if you receive RENFLEXIS. If you do not know if you have lived in an area where histoplasmosis, coccidioidomycosis, or blastomycosis is common, ask your doctor.
- •
- have or have had hepatitis B.
- •
- use the medicines KINERET (anakinra), ORENCIA (abatacept), ACTEMRA (tocilizumab), or other medicines called biologics used to treat the same conditions as RENFLEXIS.
After starting RENFLEXIS, if you have an infection, any sign of an infection including a fever, cough, flu-like symptoms, or have open cuts or sores on your body, call your doctor right away. RENFLEXIS can make you more likely to get infections or make any infection that you have worse.
2. Risk of Cancer
- •
- There have been cases of unusual cancers in children and teenage patients using tumor necrosis factor (TNF)-blocker medicines, such as RENFLEXIS.
- •
- For children and adults receiving TNF-blocker medicines, including RENFLEXIS, the chances of getting lymphoma or other cancers may increase.
- •
- Some people receiving TNF-blockers, including RENFLEXIS, developed a rare type of cancer called hepatosplenic T-cell lymphoma. This type of cancer often results in death. Most of these people were male teenagers or young men. Also, most people were being treated for Crohn's disease or ulcerative colitis with a TNF-blocker and another medicine called azathioprine or 6-mercaptopurine.
- •
- People who have been treated for rheumatoid arthritis, Crohn's disease, ulcerative colitis, ankylosing spondylitis, psoriatic arthritis and plaque psoriasis for a long time may be more likely to develop lymphoma. This is especially true for people with very active disease.
- •
- Some people treated with infliximab products, such as RENFLEXIS, have developed certain kinds of skin cancer. If any changes in the appearance of your skin or growths on your skin occur during or after your treatment with RENFLEXIS, tell your doctor.
- •
- Patients with Chronic Obstructive Pulmonary Disease (COPD), a specific type of lung disease, may have an increased risk for getting cancer while being treated with RENFLEXIS.
- •
- Some women being treated for rheumatoid arthritis with infliximab products have developed cervical cancer. For women receiving RENFLEXIS, including those over 60 years of age, your doctor may recommend that you continue to be regularly screened for cervical cancer.
- •
- Tell your doctor if you have ever had any type of cancer. Discuss with your doctor any need to adjust medicines you may be taking.
See the section "What are the possible side effects of RENFLEXIS?" below for more information.
What is RENFLEXIS?
- RENFLEXIS is a prescription medicine that is approved for patients with:
- •
- Rheumatoid Arthritis - adults with moderately to severely active rheumatoid arthritis, along with the medicine methotrexate.
- •
- Crohn's Disease - children 6 years and older and adults with Crohn's disease who have not responded well to other medicines.
- •
- Ankylosing Spondylitis
- •
- Psoriatic Arthritis
- •
- Plaque Psoriasis - adult patients with plaque psoriasis that is chronic (does not go away), severe, extensive, and/or disabling.
- •
- Ulcerative Colitis - children 6 years and older and adults with moderately to severely active ulcerative colitis who have not responded well to other medicines.
RENFLEXIS blocks the action of a protein in your body called tumor necrosis factor-alpha (TNF-alpha). TNF-alpha is made by your body's immune system. People with certain diseases have too much TNF-alpha that can cause the immune system to attack normal healthy parts of the body. RENFLEXIS can block the damage caused by too much TNF-alpha.
Who should not receive RENFLEXIS?
- You should not receive RENFLEXIS if you have:
- •
- heart failure, unless your doctor has examined you and decided that you are able to receive RENFLEXIS. Talk to your doctor about your heart failure.
- •
- had an allergic reaction to infliximab products, or any of the other ingredients in RENFLEXIS. See the end of this Medication Guide for a complete list of ingredients in RENFLEXIS.
What should I tell my doctor before starting treatment with RENFLEXIS?
Your doctor will assess your health before each treatment.
- Tell your doctor about all of your medical conditions, including if you:
- •
- have an infection (see "What is the most important information I should know about RENFLEXIS?").
- •
- have other liver problems including liver failure.
- •
- have heart failure or other heart conditions. If you have heart failure, it may get worse while you take RENFLEXIS.
- •
- have or have had any type of cancer.
- •
- have had phototherapy (treatment with ultraviolet light or sunlight along with a medicine to make your skin sensitive to light) for psoriasis. You may have a higher chance of getting skin cancer while receiving RENFLEXIS.
- •
- have COPD. Patients with COPD may have an increased risk of getting cancer while receiving RENFLEXIS.
- •
- have or have had a condition that affects your nervous system such as:
- •
- multiple sclerosis, or Guillain-Barré syndrome, or
- •
- if you experience any numbness or tingling, or
- •
- if you have had a seizure.
- •
- have recently received or are scheduled to receive a vaccine. Adults and children receiving RENFLEXIS should not receive live vaccines (for example, the Bacille Calmette-Guérin [BCG] vaccine) or treatment with a weakened bacteria (such as BCG for bladder cancer). Children should have all of their vaccines brought up to date before starting treatment with RENFLEXIS.
- •
- are pregnant or plan to become pregnant. It is not known if RENFLEXIS harms your unborn baby. RENFLEXIS should be given to a pregnant woman only if clearly needed. Talk to your doctor about stopping RENFLEXIS if you are pregnant or plan to become pregnant.
- •
- are breast-feeding or plan to breast-feed. It is not known whether RENFLEXIS passes into your breast milk. Talk to your doctor about the best way to feed your baby while receiving RENFLEXIS. You should not breast-feed while receiving RENFLEXIS.
If you have a baby and you were receiving RENFLEXIS during your pregnancy, it is important to tell your baby's doctor and other health care professionals about your RENFLEXIS use so they can decide when your baby should receive any vaccine. Certain vaccinations can cause infections.
If you received RENFLEXIS while you were pregnant, your baby may be at higher risk for getting an infection. If your baby receives a live vaccine within 6 months after birth, your baby may develop infections with serious complications that can lead to death. This includes live vaccines such as the BCG, rotavirus, or any other live vaccines. For other types of vaccines, talk with your doctor.
How should I receive RENFLEXIS?
- •
- You will be given RENFLEXIS through a needle placed in a vein (IV or intravenous infusion) in your arm.
- •
- Your doctor may decide to give you medicine before starting the RENFLEXIS infusion to prevent or lessen side effects.
- •
- Only a healthcare professional should prepare the medicine and administer it to you.
- •
- RENFLEXIS will be given to you over a period of about 2 hours.
- •
- If you have side effects from RENFLEXIS, the infusion may need to be adjusted or stopped. In addition, your healthcare professional may decide to treat your symptoms.
- •
- A healthcare professional will monitor you during the RENFLEXIS infusion and for a period of time afterward for side effects. Your doctor may do certain tests while you are receiving RENFLEXIS to monitor you for side effects and to see how well you respond to the treatment.
- •
- Your doctor will determine the right dose of RENFLEXIS for you and how often you should receive it. Make sure to discuss with your doctor when you will receive infusions and to come in for all your infusions and follow-up appointments.
What should I avoid while receiving RENFLEXIS?
Do not take RENFLEXIS together with medicines such as KINERET (anakinra), ORENCIA (abatacept), ACTEMRA (tocilizumab), or other medicines called biologics that are used to treat the same conditions as RENFLEXIS.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. These include any other medicines to treat Crohn's disease, ulcerative colitis, rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis or psoriasis.
Know the medicines you take. Keep a list of your medicines and show them to your doctor and pharmacist when you get a new medicine.
What are the possible side effects of RENFLEXIS?
RENFLEXIS can cause serious side effects, including:
See "What is the most important information I should know about RENFLEXIS?".
Serious Infections
- •
- Some patients, especially those 65 years and older have had serious infections while receiving infliximab products, such as RENFLEXIS. These serious infections include TB and infections caused by viruses, fungi, or bacteria that have spread throughout the body. Some patients die from these infections. If you get an infection while receiving treatment with RENFLEXIS your doctor will treat your infection and may need to stop your RENFLEXIS treatment.
- •
- Tell your doctor right away if you have any of the following signs of an infection while receiving or after receiving RENFLEXIS:
- •
- a fever
- •
- feel very tired
- •
- have a cough
- •
- have flu-like symptoms warm, red, or painful skin
- •
- Your doctor will examine you for TB and perform a test to see if you have TB. If your doctor feels that you are at risk for TB, you may be treated with medicine for TB before you begin treatment with RENFLEXIS and during treatment with RENFLEXIS.
- •
- Even if your TB test is negative, your doctor should carefully monitor you for TB infections while you are receiving RENFLEXIS. Patients who had a negative TB skin test before receiving infliximab products have developed active TB.
- •
- If you are a chronic carrier of the hepatitis B virus, the virus can become active while you are being treated with RENFLEXIS. In some cases, patients have died as a result of hepatitis B virus being reactivated. Your doctor should do a blood test for hepatitis B virus before you start treatment with RENFLEXIS and occasionally while you are being treated. Tell your doctor if you have any of the following symptoms:
- •
- feel unwell
- •
- poor appetite
- •
- tiredness (fatigue)
- •
- fever, skin rash, or joint pain
Heart Failure
If you have a heart problem called congestive heart failure, your doctor should check you closely while you are receiving RENFLEXIS. Your congestive heart failure may get worse while you are receiving RENFLEXIS. Be sure to tell your doctor of any new or worse symptoms including:
- •
- shortness of breath
- •
- swelling of ankles or feet
- •
- sudden weight gain
Treatment with RENFLEXIS may need to be stopped if you get new or worse congestive heart failure.
Other Heart Problems
Some patients have experienced a heart attack (some of which led to death), low blood flow to the heart, or abnormal heart rhythm within 24 hours of beginning their infusion of an infliximab product. Symptoms may include chest discomfort or pain, arm pain, stomach pain, shortness of breath, anxiety, lightheadedness, dizziness, fainting, sweating, nausea, vomiting, fluttering or pounding in your chest, and/or a fast or a slow heartbeat. Tell your doctor right away if you have any of these symptoms.
Liver Injury
Some patients receiving infliximab products have developed serious liver problems. Tell your doctor if you have:
- •
- jaundice (skin and eyes turning yellow)
- •
- dark brown-colored urine
- •
- pain on the right side of your stomach area (right-sided abdominal pain)
- •
- fever
- •
- extreme tiredness (severe fatigue)
Blood Problems
In some patients receiving infliximab products, the body may not make enough of the blood cells that help fight infections or help stop bleeding. Tell your doctor if you:
- •
- have a fever that does not go away
- •
- bruise or bleed very easily
- •
- look very pale
Nervous System Disorders
Some patients receiving infliximab products have developed problems with their nervous system. Tell your doctor if you have:
- •
- changes in your vision
- •
- weakness in your arms or legs
- •
- numbness or tingling in any part of your body
- •
- seizures
Some patients have experienced a stroke within approximately 24 hours of their infusion with infliximab products. Tell your doctor right away if you have symptoms of a stroke which may include: numbness or weakness of the face, arm or leg, especially on one side of the body; sudden confusion, trouble speaking or understanding; sudden trouble seeing in one or both eyes, sudden trouble walking, dizziness, loss of balance or coordination or a sudden, severe headache.
Allergic Reactions
Some patients have had allergic reactions to infliximab products. Some of these reactions were severe. These reactions can happen while you are getting your RENFLEXIS treatment or shortly afterward. Your doctor may need to stop or pause your treatment with RENFLEXIS and may give you medicines to treat the allergic reaction. Signs of an allergic reaction can include:
- •
- hives (red, raised, itchy patches of skin)
- •
- difficulty breathing
- •
- chest pain
- •
- high or low blood pressure
- •
- fever
- •
- chills
Some patients treated with infliximab products have had delayed allergic reactions. The delayed reactions occurred 3 to 12 days after receiving treatment with an infliximab product. Tell your doctor right away if you have any of these signs of delayed allergic reaction to RENFLEXIS:
- •
- fever
- •
- rash
- •
- headache
- •
- sore throat
- •
- muscle or joint pain
- •
- swelling of the face and hands
- •
- difficulty swallowing
Lupus-like Syndrome
Some patients have developed symptoms that are like the symptoms of Lupus. If you develop any of the following symptoms, your doctor may decide to stop your treatment with RENFLEXIS.
- •
- chest discomfort or pain that does not go away
- •
- shortness of breath
- •
- joint pain
- •
- rash on the cheeks or arms that gets worse in the sun
Psoriasis
Some people receiving infliximab products had new psoriasis or worsening of psoriasis they already had. Tell your doctor if you develop red scaly patches or raised bumps on the skin that are filled with pus. Your doctor may decide to stop your treatment with RENFLEXIS.
The most common side effects of infliximab products include:
- •
- respiratory infections, such as sinus infections and sore throat
- •
- headache
- •
- coughing
- •
- stomach pain
Infusion reactions can happen up to 2 hours after your infusion of RENFLEXIS. Symptoms of infusion reactions may include:
- •
- fever
- •
- chills
- •
- chest pain
- •
- low blood pressure or high blood pressure
- •
- shortness of breath
- •
- rash
- •
- itching
Children with Crohn's disease showed some differences in side effects of treatment compared with adults with Crohn's disease. The side effects that happened more in children were: anemia (low red blood cells), leukopenia (low white blood cells), flushing (redness or blushing), viral infections, neutropenia (low neutrophils, the white blood cells that fight infection), bone fracture, bacterial infection and allergic reactions of the breathing tract. Among patients who took infliximab for ulcerative colitis in clinical studies, more children had infections as compared with adults.
Tell your doctor about any side effect that bothers you or does not go away.
These are not all of the side effects with RENFLEXIS. Ask your doctor or pharmacist for more information.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA1088.
General information about RENFLEXIS
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use RENFLEXIS for a condition for which it was not prescribed. Do not give RENFLEXIS to other people, even if they have the same symptoms that you have. It may harm them.
You can ask your doctor or pharmacist for information about RENFLEXIS that is written for health professionals.
What are the ingredients in RENFLEXIS?
The active ingredient is infliximab-abda.
The inactive ingredients in RENFLEXIS include: dibasic sodium phosphate heptahydrate, monobasic sodium phosphate monohydrate, polysorbate 80, and sucrose. No preservatives are present.
Manufactured by: Samsung Bioepis Co., Ltd., 76, Songdogyoyuk-ro, Yeonsu-gu, Incheon, 21987, Republic of Korea
U.S. License No. 2046
Manufactured for: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ 08889 USA
RENFLEXIS is a trademark of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
For more information, go to www.RENFLEXIS.com or call 1-877-888-4231.
-
PRINCIPAL DISPLAY PANEL - 100 mg Vial Carton
NDC 0006-4305-02
100
mg/vialMERCK
RENFLEXIS™
(infliximab-abda)
for injection100 mg / vial
For Intravenous Infusion Only
Dispense the enclosed
Medication Guide to each patient.Must reconstitute and dilute before
intravenous infusion. Must infuse
over at least 2 hours with
an in-line filter.Single-dose vial.
Discard unused portion.Rx only
SAMSUNG BIOEPIS
-
INGREDIENTS AND APPEARANCE
RENFLEXIS
infliximab injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0006-4305 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength infliximab (UNII: B72HH48FLU) (infliximab - UNII:B72HH48FLU) infliximab 100 mg Inactive Ingredients Ingredient Name Strength sucrose (UNII: C151H8M554) 500 mg polysorbate 80 (UNII: 6OZP39ZG8H) 0.5 mg sodium phosphate, monobasic, monohydrate (UNII: 593YOG76RN) 6.3 mg sodium phosphate, dibasic, heptahydrate (UNII: 70WT22SF4B) 1.2 mg Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0006-4305-02 1 in 1 CARTON 07/01/2017 1 NDC:0006-4305-01 1 in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761054 07/01/2017 Labeler - Merck Sharp & Dohme LLC (118446553) Registrant - Samsung Bioepis Co., Ltd. (557822226)