ACEON- perindopril erbumine tablet
Symplmed, LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ACEON® safely and effectively. See full prescribing information for ACEON.
ACEON (perindopril erbumine) 2, 4 and 8 mg Tablets Initial U.S. Approval: 1993 INDICATIONS AND USAGEDOSAGE AND ADMINISTRATIONHypertension
Stable Coronary Artery Disease
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONSADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact Symplmed, LLC. at 1-888-985-7657 or FDA at 1-800-FDA-1088 or www.FDA.gov/Medwatch. DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION. Revised: 9/2017 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Hypertension
ACEON is indicated for the treatment of patients with essential hypertension. ACEON may be used alone or given with other classes of antihypertensives, especially thiazide diuretics.
1.2 Stable Coronary Artery Disease
ACEON is indicated for treatment of patients with stable coronary artery disease to reduce the risk of cardiovascular mortality or nonfatal myocardial infarction. ACEON can be used with conventional treatment for management of coronary artery disease, such as antiplatelet, antihypertensive or lipid-lowering therapy.
2 DOSAGE AND ADMINISTRATION
2.1 Hypertension
Use in Uncomplicated Hypertensive Patients: In patients with essential hypertension, the recommended initial dose is 4 mg once a day. The dose may be titrated, as needed to a maximum of 16 mg per day. The usual maintenance dose range is 4 mg to 8 mg administered as a single daily dose or in two divided doses.
Use in Elderly Patients: The recommended initial daily dosage of ACEON for the elderly is 4 mg daily, given in one or two divided doses. Experience with ACEON is limited in the elderly at doses exceeding 8 mg. Dosages above 8 mg should be administered with careful blood pressure monitoring and dose titration [see Use in Specific Populations (8.5)].
Use with Diuretics: In patients who are currently being treated with a diuretic, symptomatic hypotension can occur following the initial dose of ACEON. Consider reducing the dose of diuretic prior to starting ACEON [see Drug Interactions (7.1)].
2.2 Stable Coronary Artery Disease
In patients with stable coronary artery disease, ACEON should be given at an initial dose of 4 mg once daily for 2 weeks, and then increased as tolerated, to a maintenance dose of 8 mg once daily. In elderly patients (greater than 70 years), ACEON should be given as a 2 mg dose once daily in the first week, followed by 4 mg once daily in the second week and 8 mg once daily for maintenance dose if tolerated.
2.3 Dose Adjustment in Renal Impairment and Dialysis
Perindoprilat elimination is decreased in renally impaired patients. ACEON is not recommended in patients with creatinine clearance <30 mL/min. For patients with lesser degrees of impairment, the initial dosage should be 2 mg/day and dosage should not exceed 8 mg/day. During dialysis, perindopril is removed with the same clearance as in patients with normal renal function.
3 DOSAGE FORMS AND STRENGTHS
Tablets are oblong with a score on one side.
2 mg tablet is white and debossed on the unscored side with "ACN 2".
4 mg tablet is pink and debossed on the unscored side with "ACN 4".
8 mg tablet is salmon and debossed on the unscored side with "ACN 8".
4 CONTRAINDICATIONS
ACEON® (perindopril erbumine) is contraindicated in patients known to be hypersensitive (including angioedema) to this product or to any other ACE inhibitor. ACEON is also contraindicated in patients with hereditary or idiopathic angioedema.
Do not co-administer aliskiren with ACEON in patients with diabetes. [see Drug Interactions (7.8)]
ACEON is contraindicated in combination with neprilysin inhibitor (e.g., sacubitril). Do not administer ACEON within 36 hours of switching to or from sacubitril/valsartan, a neprilysin inhibitor [see Warnings and Precautions (5.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylactoid and Possibly Related Reactions
Presumably because angiotensin-converting enzyme inhibitors affect the metabolism of eicosanoids and polypeptides, including endogenous bradykinin, patients receiving ACE inhibitors (including ACEON) may be subject to a variety of adverse events, some of them serious. Black patients receiving ACE inhibitors have a higher incidence of angioedema compared to nonblacks.
Head and Neck Angioedema: Angioedema of the face, extremities, lips, tongue, glottis, or larynx has been reported in patients treated with ACE inhibitors, including ACEON (0.1% of patients treated with ACEON in U.S. clinical trials). Angioedema associated with involvement of the tongue, glottis or larynx may be fatal. In such cases, discontinue ACEON treatment immediately and observe until the swelling disappears. When involvement of the tongue, glottis, or larynx appears likely to cause airway obstruction, administer appropriate therapy, such as subcutaneous epinephrine solution 1:1000 (0.3 to 0.5 mL), promptly.
Patients taking concomitant mTOR inhibitor (e.g., temsirolimus) therapy or a neprilysin inhibitor may be at increased risk for angioedema [see Drug Interactions (7.9; 7.10)].
Intestinal Angioedema: Intestinal angioedema has been reported in patients treated with ACE inhibitors. These patients presented with abdominal pain (with or without nausea or vomiting); in some cases there was no prior history of facial angioedema and C-1 esterase levels were normal. The angioedema was diagnosed by procedures including abdominal CT scan or ultrasound, or at surgery, and symptoms resolved after stopping the ACE inhibitor. Intestinal angioedema should be included in the differential diagnosis of patients on ACE inhibitors presenting with abdominal pain.
5.2 Hypotension
ACEON can cause symptomatic hypotension. ACEON has been associated with hypotension in 0.3% of uncomplicated hypertensive patients in U.S. placebo-controlled trials. Symptoms related to orthostatic hypotension were reported in another 0.8% of patients.
Symptomatic hypotension is most likely to occur in patients who have been volume or salt-depleted as a result of prolonged diuretic therapy, dietary salt restriction, dialysis, diarrhea or vomiting [see Dosage and Administration (2.1)].
ACE inhibitors may cause excessive hypotension, and may be associated with oliguria or azotemia, and rarely with acute renal failure and death. In patients with ischemic heart disease or cerebrovascular disease, an excessive fall in blood pressure could result in a myocardial infarction or a cerebrovascular accident.
In patients at risk of excessive hypotension, ACEON therapy should be started under very close medical supervision. Patients should be followed closely for the first two weeks of treatment and whenever the dose of ACEON and/or diuretic is increased.
If excessive hypotension occurs, the patient should be placed immediately in a supine position and, if necessary, treated with an intravenous infusion of physiological saline. ACEON treatment can usually be continued following restoration of volume and blood pressure.
5.3 Neutropenia/Agranulocytosis
ACE inhibitors have been associated with agranulocytosis and bone marrow depression, most frequently in patients with renal impairment, especially patients with a collagen vascular disease such as systemic lupus erythematosus or scleroderma.
5.4 Fetal Toxicity
Pregnancy Category D
Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected discontinue ACEON as soon as possible [see Use in Specific Populations (8.1)].
5.5 Impaired Renal Function
As a consequence of inhibiting the renin-angiotensin-aldosterone system, changes in renal function may be anticipated in susceptible individuals. Renal function should be monitored periodically in patients receiving ACEON [see Dosage and Administration (2.3)], [see Drug Interactions (7)].
In patients with severe congestive heart failure, where renal function may depend on the activity of the renin-angiotensin-aldosterone system, treatment with ACE inhibitors, including ACEON, may be associated with oliguria, progressive azotemia, and, rarely, acute renal failure and death.
In hypertensive patients with unilateral or bilateral renal artery stenosis, increases in blood urea nitrogen and serum creatinine may occur; usually reversible upon discontinuation of the ACE inhibitor. In such patients, renal function should be monitored during the first few weeks of therapy.
Some ACEON-treated patients have developed minor and transient increases in blood urea nitrogen and serum creatinine especially in those concomitantly treated with a diuretic.
5.6 Hyperkalemia
Elevations of serum potassium have been observed in some patients treated with ACE inhibitors, including ACEON. Most cases were isolated single values that did not appear clinically relevant and were rarely a cause for withdrawal. Risk factors for the development of hyperkalemia include renal insufficiency, diabetes mellitus and the concomitant use of agents such as potassium-sparing diuretics, potassium supplements and/or potassium-containing salt substitutes [see Drug Interactions (7.2)].
Serum potassium should be monitored periodically in patients receiving ACEON.
5.7 Cough
Presumably because of the inhibition of the degradation of endogenous bradykinin, persistent nonproductive cough has been reported with all ACE inhibitors, generally resolving after discontinuation of therapy. Consider ACE inhibitor-induced cough in the differential diagnosis of cough.
5.8 Hepatic Failure
Rarely, ACE inhibitors have been associated with a syndrome that starts with cholestatic jaundice and progresses to fulminant hepatic necrosis and sometimes death. The mechanism of this syndrome is not understood. Patients receiving ACE inhibitors who develop jaundice or marked elevations of hepatic enzymes should discontinue the ACE inhibitor and receive appropriate medical follow-up.
5.9 Surgery/Anesthesia
In patients undergoing surgery or during anesthesia with agents that produce hypotension, ACEON may block angiotensin II formation that would otherwise occur secondary to compensatory renin release. Hypotension attributable to this mechanism can be corrected by volume expansion.
6 ADVERSE REACTIONS
Because clinical trials are conducted under widely varying conditions, adverse event rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
6.1 Clinical Trials Experience
The following adverse reactions are discussed elsewhere in labeling:
- Anaphylactoid reactions, including angioedema [see Warnings and Precautions (5.1)]
- Hypotension [see Warnings and Precautions (5.2)]
- Neutropenia and agranulocytosis [see Warnings and Precautions (5.3)]
- Impaired renal function [see Warnings and Precautions (5.5)]
- Hyperkalemia [see Warnings and Precautions (5.6)]
- Cough [see Warnings and Precautions (5.7)]
Hypertension
ACEON has been evaluated for safety in approximately 3,400 patients with hypertension in U.S. and foreign clinical trials. The data presented here are based on results from the 1,417 ACEON-treated patients who participated in the U.S. clinical trials. Over 220 of these patients were treated with ACEON® (perindopril erbumine) for at least one year.
In placebo-controlled U.S. clinical trials, the incidence of premature discontinuation of therapy due to adverse events was 6.5% in patients treated with ACEON and 6.7% in patients treated with placebo. The most common causes were cough, headache, asthenia and dizziness.
Among 1,012 patients in placebo-controlled U.S. trials, the overall frequency of reported adverse events was similar in patients treated with ACEON and in those treated with placebo (approximately 75% in each group). The only adverse events whose incidence on ACEON was at least 2% greater than on placebo were cough (12% vs. 4.5%) and back pain (5.8% vs. 3.1%).
Dizziness was not reported more frequently in the perindopril group (8.2%) than in the placebo group (8.5%), but its likelihood increased with dose, suggesting a causal relationship with perindopril.
Stable Coronary Artery Disease
Perindopril has been evaluated for safety in EUROPA, a double-blind, placebo-controlled study in 12,218 patients with stable coronary artery disease. The overall rate of discontinuation was about 22% on drug and placebo. The most common medical reasons for discontinuation that were more frequent on perindopril than placebo were cough, drug intolerance and hypotension.
6.2 Postmarketing Experience
Voluntary reports of adverse events in patients taking ACEON that have been received since market introduction and are of unknown causal relationship to ACEON include: cardiac arrest, eosinophilic pneumonitis, neutropenia/agranulocytosis, pancytopenia, anemia (including hemolytic and aplastic), thrombocytopenia, acute renal failure, nephritis, hepatic failure, jaundice (hepatocellular or cholestatic), symptomatic hyponatremia, bullous pemphigoid, pemphigus, acute pancreatitis, falls, psoriasis, exfoliative dermatitis and a syndrome which may include: arthralgia/arthritis, vasculitis, serositis, myalgia, fever, rash or other dermatologic manifestations, a positive antinuclear antibody (ANA), leukocytosis, eosinophilia or an elevated erythrocyte sedimentation rate (ESR).
6.3 Clinical Laboratory Test Findings
Hematology: Small decreases in hemoglobin and hematocrit occur frequently in hypertensive patients treated with ACEON, but are rarely of clinical importance. In controlled clinical trials, no patient was discontinued from therapy due to the development of anemia. Leukopenia (including neutropenia) was observed in 0.1% of patients in U.S. clinical trials [see Warnings and Precautions (5.3)].
Liver Function Tests: Elevations in ALT (1.6% ACEON versus 0.9% placebo) and AST (0.5% ACEON versus 0.4% placebo) have been observed in placebo-controlled clinical trials. The elevations were generally mild and transient and resolved after discontinuation of therapy.
7 DRUG INTERACTIONS
7.1 Diuretics
Patients on diuretics, and especially those started recently, may occasionally experience an excessive reduction of blood pressure after initiation of ACEON therapy. The possibility of hypotensive effects can be minimized by either decreasing the dose of or discontinuing the diuretic or increasing the salt intake prior to initiation of treatment with perindopril. If diuretic therapy cannot be altered, provide close medical supervision with the first dose of ACEON, for at least two hours and until blood pressure has stabilized for another hour [see Warnings and Precautions (5.2)].
The rate and extent of perindopril absorption and elimination are not affected by concomitant diuretics. The bioavailability of perindoprilat was reduced by diuretics, however, and this was associated with a decrease in plasma ACE inhibition.
7.2 Potassium Supplements and Potassium-Sparing Diuretics
ACEON may increase serum potassium because of its potential to decrease aldosterone production. Use of potassium-sparing diuretics (spironolactone, amiloride, triamterene and others), potassium supplements or other drugs capable of increasing serum potassium (indomethacin, heparin, cyclosporine and others) can increase the risk of hyperkalemia. Therefore, if concomitant use of such agents is indicated, monitor the patient's serum potassium frequently.
7.3 Lithium
Increased serum lithium and symptoms of lithium toxicity have been reported in patients receiving concomitant lithium and ACE inhibitor therapy. Frequent monitoring of serum lithium concentration is recommended. Use of a diuretic may further increase the risk of lithium toxicity.
7.4 Gold
Nitritoid reactions (symptoms include facial flushing, nausea, vomiting and hypotension) have been reported rarely in patients on therapy with injectable gold (sodium aurothiomalate) and concomitant ACE Inhibitor therapy including ACEON.
7.5 Digoxin
A controlled pharmacokinetic study has shown no effect on plasma digoxin concentrations when coadministered with ACEON, but an effect of digoxin on the plasma concentration of perindopril/perindoprilat has not been excluded.
7.6 Gentamicin
Animal data have suggested the possibility of interaction between perindopril and gentamicin. However, this has not been investigated in human studies.
7.7 Non-Steroidal Anti-Inflammatory Agents including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors)
In patients who are elderly, volume-depleted (including those on diuretic therapy), or with compromised renal function, co-administration of NSAIDs, including selective COX-2 inhibitors, with ACE inhibitors, including perindopril, may result in deterioration of renal function, including possible acute renal failure. These effects are usually reversible. Monitor renal function periodically in patients receiving perindopril and NSAID therapy.
The antihypertensive effect of ACE inhibitors, including perindopril, may be attenuated by NSAIDs including selective COX-2 inhibitors.
7.8 Dual Blockade of the Renin-Angiotensin System (RAS)
Dual blockade of the RAS with angiotensin receptor blockers, ACE inhibitors, or aliskiren is associated with increased risks of hypotension, hyperkalemia, and changes in renal function (including acute renal failure) compared to monotherapy. Most patients receiving the combination of two RAS inhibitors do not obtain any additional benefit compared to monotherapy. In general, avoid combined use of RAS inhibitors. Closely monitor blood pressure, renal function and electrolytes in patients on ACEON and other agents that affect the RAS.
Do not co-administer aliskiren with ACEON in patients with diabetes. Avoid use of aliskiren with ACEON in patients with renal impairment (GFR <60 ml/min).
7.9 mTOR Inhibitors
Patients taking concomitant mTOR (mammalian target of rapamycin) inhibitor therapy may be at increased risk for angioedema [see Warnings and Precautions 5.1].
7.10 Neprilysin Inhibitor
Patients taking concomitant neprilysin inhibitors may be at increased risk for angioedema. [see Warnings and Precautions (5.1)]
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category D [see Boxed Warning and Warnings and Precautions (5.4)].
Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue ACEON as soon as possible. These adverse outcomes are usually associated with use of these drugs in the second and third trimester of pregnancy. Most epidemiologic studies examining fetal abnormalities after exposure to antihypertensive use in the first trimester have not distinguished drugs affecting the renin-angiotensin system from other antihypertensive agents. Appropriate management of maternal hypertension during pregnancy is important to optimize outcomes for both mother and fetus.
In the unusual case that there is no appropriate alternative to therapy with drugs affecting the renin-angiotensin system for a particular patient, apprise the mother of the potential risk to the fetus. Perform serial ultrasound examinations to assess the intra-amniotic environment. If oligohydramnios is observed, discontinue ACEON, unless it is considered lifesaving for the mother. Fetal testing may be appropriate, based on the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury. Closely observe infants with histories of in utero exposure to ACEON for hypotension, oliguria, and hyperkalemia [see Use in Specific Populations (8.4)].
Radioactivity was detectable in fetuses after administration of 14C-perindopril to pregnant rats.
8.3 Nursing Mothers
Milk of lactating rats contained radioactivity following administration of 14C-perindopril. It is not known whether perindopril is secreted in human milk. Because many drugs are secreted in human milk, caution should be exercised when ACEON is given to nursing mothers.
8.4 Pediatric Use
Neonates with a history of in utero exposure to ACEON:
If oliguria or hypotension occurs, direct attention toward support of blood pressure and renal perfusion. Exchange transfusions or dialysis may be required as a means of reversing hypotension and/or substituting for disordered renal function. Perindopril, which crosses the placenta, can theoretically be removed from the neonatal circulation by these means, but limited experience has not shown that such removal is central to the treatment of these infants.
Safety and effectiveness of ACEON in pediatric patients have not been established.
8.5 Geriatric Use
The mean blood pressure effect of perindopril was somewhat smaller in patients over 60 than in younger patients, although the difference was not significant. Plasma concentrations of both perindopril and perindoprilat were increased in elderly patients compared to concentrations in younger patients. No adverse effects were clearly increased in older patients with the exception of dizziness and possibly rash.
Start at a low dose and titrate slowly as needed. Monitor for dizziness because of potential for falls.
Experience with ACEON in elderly patients at daily doses exceeding 8 mg is limited.
8.6 Renal Impairment
Dosage adjustment may be necessary in renally impaired patients [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
The bioavailability of perindoprilat is increased in patients with impaired hepatic function [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
In animals, doses of perindopril up to 2,500 mg/kg in mice, 3,000 mg/kg in rats and 1,600 mg/kg in dogs were non-lethal. Past experiences were scant but suggested that overdosage with other ACE inhibitors was also fairly well tolerated by humans. The most likely manifestation is hypotension, and treatment should be symptomatic and supportive. Therapy with the ACE inhibitor should be discontinued, and the patient should be observed. Dehydration, electrolyte imbalance and hypotension should be treated by established procedures.
Among the reported cases of perindopril overdosage, patients who were known to have ingested a dose of 80 mg to 120 mg required assisted ventilation and circulatory support. One additional patient developed hypothermia, circulatory arrest and died following ingestion of up to 180 mg of perindopril. The intervention for perindopril overdose may require vigorous support.
Laboratory determinations of serum levels of perindopril and its metabolites are not widely available, and such determinations have, in any event, no established role in the management of perindopril overdose.
No data are available to suggest physiological maneuvers (e.g., maneuvers to change the pH of the urine) that might accelerate elimination of perindopril and its metabolites. Perindopril can be removed by hemodialysis, with clearance of 52 mL/min for perindopril and 67 mL/min for perindoprilat.
Angiotensin II could presumably serve as a specific antagonist-antidote in the settling of perindopril overdose, but angiotensin II is essentially unavailable outside of scattered research facilities. Because the hypotensive effect of perindopril is achieved through vasodilation and effective hypovolemia, it is reasonable to treat perindopril overdose by infusion of normal saline solution.
11 DESCRIPTION
ACEON® (perindopril erbumine) Tablets contain the tert-butylamine salt of perindopril, the ethyl ester of a non-sulfhydryl angiotensin-converting enzyme (ACE) inhibitor. Perindopril erbumine is chemically described as (2S,3∝S,7∝S)-1-[(S)-N-[(S)-1-Carboxy-butyl]alanyl]hexahydro-2-indolinecarboxylic acid, 1-ethyl ester, compound with tert-butylamine (1:1). Its molecular formula is C19H32N2O5C4H11N. Its structural formula is:
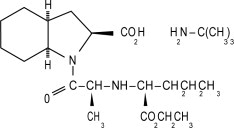
Perindopril erbumine is a white, crystalline powder with a molecular weight of 368.47 (free acid) or 441.61 (salt form). It is freely soluble in water (60% w/w), alcohol and chloroform.
Perindopril is the free acid form of perindopril erbumine, is a pro-drug and metabolized in vivo by hydrolysis of the ester group to form perindoprilat, the biologically active metabolite.
ACEON is available in 2 mg, 4 mg and 8 mg strengths for oral administration. In addition to perindopril erbumine, each tablet contains the following inactive ingredients: colloidal silica (hydrophobic), lactose, magnesium stearate and microcrystalline cellulose. The 4 mg and 8 mg tablets also contain iron oxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
ACEON® (perindopril erbumine) is a pro-drug for perindoprilat, which inhibits ACE in human subjects and animals. The mechanism through which perindoprilat lowers blood pressure is believed to be primarily inhibition of ACE activity. ACE is a peptidyl dipeptidase that catalyzes conversion of the inactive decapeptide, angiotensin I, to the vasoconstrictor, angiotensin II. Angiotensin II is a potent peripheral vasoconstrictor, which stimulates aldosterone secretion by the adrenal cortex, and provides negative feedback on renin secretion. Inhibition of ACE results in decreased plasma angiotensin II, leading to decreased vasoconstriction, increased plasma renin activity and decreased aldosterone secretion. The latter results in diuresis and natriuresis and may be associated with a small increase of serum potassium.
ACE is identical to kininase II, an enzyme that degrades bradykinin. Whether increased levels of bradykinin, a potent vasodepressor peptide, play a role in the therapeutic effects of ACEON remains to be elucidated.
While the principal mechanism of perindopril in blood pressure reduction is believed to be through the renin-angiotensin-aldosterone system, ACE inhibitors have some effect even in apparent low-renin hypertension. Perindopril has been studied in relatively few black patients, usually a low-renin population, and the average response of diastolic blood pressure to perindopril was about half the response seen in nonblack patients, a finding consistent with previous experience of other ACE inhibitors.
12.2 Pharmacodynamics
After administration of perindopril, ACE is inhibited in a dose and blood concentration-related fashion, with the maximal inhibition of 80 to 90% attained by 8 mg persisting for 10 to 12 hours. Twenty-four hour ACE inhibition is about 60% after these doses. The degree of ACE inhibition achieved by a given dose appears to diminish over time (the ID50 increases). The pressor response to an angiotensin I infusion is reduced by perindopril, but this effect is not as persistent as the effect on ACE; there is about 35% inhibition at 24 hours after a 12 mg dose.
12.3 Pharmacokinetics
Absorption: Oral administration of ACEON results in peak plasma concentrations that occur at approximately 1 hour. The absolute oral bioavailability of perindopril is about 75%. Following absorption, approximately 30 to 50% of systemically available perindopril is hydrolyzed to its active metabolite, perindoprilat, which has a mean bioavailability of about 25%. Peak plasma concentrations of perindoprilat are attained 3 to 7 hours after perindopril administration. Oral administration of ACEON with food does not significantly lower the rate or extent of perindopril absorption relative to the fasted state. However, the extent of biotransformation of perindopril to the active metabolite, perindoprilat, is reduced approximately 43%, resulting in a reduction in the plasma ACE inhibition curve of approximately 20%, probably clinically insignificant. In clinical trials, perindopril was generally administered in a non-fasting state.
With 4 mg, 8 mg and 16 mg doses of ACEON, Cmax and AUC of perindopril and perindoprilat increase in a dose-proportional manner following both single oral dosing and at steady state during a once-a-day multiple dosing regimen.
Distribution: Approximately 60% of circulating perindopril is bound to plasma proteins, and only 10 to 20% of perindoprilat is bound. Therefore, drug interactions mediated through effects on protein binding are not anticipated.
Metabolism and Elimination: Following oral administration perindopril exhibits multicompartment pharmacokinetics including a deep tissue compartment (ACE binding sites). The mean half-life of perindopril associated with most of its elimination is approximately 0.8 to 1 hours.
Perindopril is extensively metabolized following oral administration, with only 4 to 12% of the dose recovered unchanged in the urine. Six metabolites resulting from hydrolysis, glucuronidation and cyclization via dehydration have been identified. These include the active ACE inhibitor, perindoprilat (hydrolyzed perindopril), perindopril and perindoprilat glucuronides, dehydrated perindopril and the diastereoisomers of dehydrated perindoprilat. In humans, hepatic esterase appears to be responsible for the hydrolysis of perindopril.
The active metabolite, perindoprilat, also exhibits multicompartment pharmacokinetics following the oral administration of ACEON. Formation of perindoprilat is gradual with peak plasma concentrations occurring between 3 and 7 hours. The subsequent decline in plasma concentration shows an apparent mean half-life of 3 to 10 hours for the majority of the elimination, with a prolonged terminal elimination half-life of 30 to 120 hours resulting from slow dissociation of perindoprilat from plasma/tissue ACE binding sites. During repeated oral once daily dosing with perindopril, perindoprilat accumulates about 1.5 to 2 fold and attains steady state plasma levels in 3 to 6 days. The clearance of perindoprilat and its metabolites is almost exclusively renal.
Elderly: Plasma concentrations of both perindopril and perindoprilat in elderly patients (greater than 70 years) are approximately twice those observed in younger patients, reflecting both increased conversion of perindopril to perindoprilat and decreased renal excretion of perindoprilat [see Dosage and Administration (2.1) and Use In Specific Populations (8.5)].
Heart Failure: Perindoprilat clearance is reduced in congestive heart failure patients, resulting in a 40% higher dose interval AUC.
Renal Impairment: With perindopril doses of 2 mg to 4 mg, perindoprilat AUC increases with decreasing renal function. At creatinine clearances of 30 to 80 mL/min, AUC is about double that at 100 mL/min. When creatinine clearance drops below 30 mL/min, AUC increases more markedly.
In a limited number of patients studied, perindopril clearance by dialysis ranged from about 40 to 80 mL/min. Perindoprilat clearance by dialysis ranged from about 40 to 90 mL/min [see Dosage and Administration (2.3)].
Hepatic Impairment: The bioavailability of perindoprilat is increased in patients with impaired hepatic function. Plasma concentrations of perindoprilat in patients with impaired liver function were about 50% higher than those observed in healthy subjects or hypertensive patients with normal liver function.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity: No evidence of carcinogenic effect was observed in studies in rats and mice when perindopril was administered at dosages up to 20 times (mg/kg) or 2 to 4 times (mg/m2) the maximum proposed clinical doses (16 mg/day) for 104 weeks.
Mutagenesis: No genotoxic potential was detected for ACEON, perindoprilat and other metabolites in various in vitro and in vivo investigations, including the Ames test, the Saccharomyces cerevisiae D4 test, cultured human lymphocytes, TK ± mouse lymphoma assay, mouse and rat micronucleus tests and Chinese hamster bone marrow assay.
Impairment of Fertility: There was no meaningful effect on reproductive performance or fertility in the rat given up to 30 times (mg/kg) or 6 times (mg/m2) the proposed maximum clinical dosage of ACEON during the period of spermatogenesis in males or oogenesis and gestation in females.
14 CLINICAL STUDIES
14.1 Hypertension
In placebo-controlled studies of perindopril monotherapy (2 mg to 16 mg once daily) in patients with a mean blood pressure of about 150/100 mm Hg, 2 mg had little effect, but doses of 4 mg to 16 mg lowered blood pressure. The 8 mg and 16 mg doses were indistinguishable, and both had a greater effect than the 4 mg dose. In these studies, doses of 8 mg and 16 mg per day gave supine, trough blood pressure reductions of 9 to 15/5 to 6 mm Hg. When once daily and twice daily dosing were compared, the twice daily dosing regimen was generally slightly superior, but by not more than about 0.5 mm Hg to 1 mm Hg. After 2 mg to 16 mg doses of perindopril, the trough mean systolic and diastolic blood pressure effects were about 75 to 100% of peak effects.
Perindopril's effects on blood pressure were similar when given alone or on a background of 25 mg hydrochlorothiazide In general, the effect of perindopril occurred promptly, with effects increasing slightly over several weeks.
Formal interaction studies of ACEON® (perindopril erbumine) have not been carried out with antihypertensive agents other than thiazides. Limited experience in controlled and uncontrolled trials coadministering ACEON with a calcium channel blocker, a loop diuretic or triple therapy (beta-blocker, vasodilator and a diuretic), does not suggest any unexpected interactions. In general, ACE inhibitors have less than additive effects when given with beta-adrenergic blockers, presumably because both work in part through the renin angiotensin system.
In uncontrolled studies in patients with insulin-dependent diabetes, perindopril did not appear to affect glycemic control. In long-term use, no effect on urinary protein excretion was seen in these patients.
The effectiveness of ACEON was not influenced by sex and it was less effective in black patients than in nonblack patients. In elderly patients (greater than or equal to 60 years), the mean blood pressure effect was somewhat smaller than in younger patients, although the difference was not significant.
14.2 Stable Coronary Artery Disease
The EURopean trial On reduction of cardiac events with Perindopril in stable coronary Artery disease (EUROPA) was a multicenter, randomized, double-blind and placebo-controlled study conducted in 12,218 patients who had evidence of stable coronary artery disease without clinical heart failure. Patients had evidence of coronary artery disease documented by previous myocardial infarction more than 3 months before screening, coronary revascularization more than 6 months before screening, angiographic evidence of stenosis (at least 70% narrowing of one or more major coronary arteries), or positive stress test in men with a history of chest pain. After a run-in period of 4 weeks during which all patients received perindopril 2 mg to 8 mg, the patients were randomly assigned to perindopril 8 mg once daily (n=6,110) or matching placebo (n=6,108). The mean follow-up was 4.2 years. The study examined the long-term effects of perindopril on time to first event of cardiovascular mortality, nonfatal myocardial infarction, or cardiac arrest in patients with stable coronary artery disease.
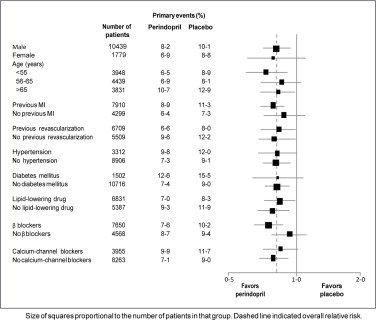
The mean age of patients was 60 years; 85% were male, 92% were taking platelet inhibitors, 63% were taking β blockers, and 56% were taking lipid-lowering therapy. The EUROPA study showed that perindopril significantly reduced the relative risk for the primary endpoint events (Table 1). This beneficial effect is largely attributable to a reduction in the risk of nonfatal myocardial infarction. This beneficial effect of perindopril on the primary outcome was evident after about one year of treatment (Figure 1). The outcome was similar across all predefined subgroups by age, underlying disease or concomitant medication (Figure 2).
| CI=confidence interval; RRR: relative risk reduction; MI: myocardial infarction | ||||
| Perindopril
(N = 6,110) | Placebo
(N = 6,108) | RRR
(95% CI) | P | |
| Combined Endpoint | ||||
| Cardiovascular mortality, nonfatal MI or cardiac arrest | 488 (8%) | 603 (9.9%) | 20% (9 to 29) | 0.0003 |
| Component Endpoint | ||||
| Cardiovascular mortality | 215 (3.5%) | 249 (4.1%) | 14% (-3 to 28) | 0.107 |
| Nonfatal MI | 295 (4.8%) | 378 (6.2%) | 22% (10 to 33) | 0.001 |
| Cardiac arrest | 6 (0.1%) | 11 (0.2%) | 46% (-47 to 80) | 0.22 |
Figure 1. Time to First Occurrence of Primary Endpoint
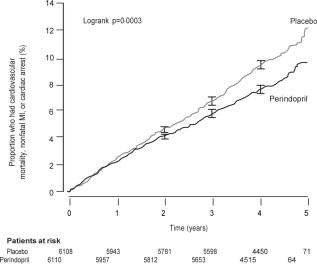
Figure 2. Beneficial Effect of Perindopril Treatment on Primary Endpoint in Predefined Subgroups
16 HOW SUPPLIED/STORAGE AND HANDLING
Tablets are oblong with a score on one side.
| Tablets | Appearance | NDC (Bottles of 100) |
| 2 mg | White, debossed "ACN 2" on unscored side | NDC 61894-000-02 |
| 4 mg | Pink, debossed "ACN 4" on unscored side | NDC 61894-001-02 |
| 8 mg | Salmon-colored, debossed "ACN 8" on unscored side | NDC 61894-002-02 |
Keep out of the reach of children.
Store at controlled room temperature 20° to 25°C (68° to 77°F) [see USP]. Protect from moisture.
 For further information, please call our medical communications department toll-free at 888-985-7657.
For further information, please call our medical communications department toll-free at 888-985-7657.
17 PATIENT COUNSELING INFORMATION
Female patients of childbearing age should be told about the consequences of exposure to ACEON during pregnancy. Discuss treatment options with women planning to become pregnant. Patients should be asked to report pregnancies to their physicians as soon as possible.
Tell patients to report promptly any indication of infection (e.g., sore throat, fever) which could be a sign of neutropenia.
Manufactured by:
Patheon Pharmaceuticals, Inc.
Cincinnati, OH 45237 USA
Marketed by:
Symplmed, LLC.
Cincinnati, OH 45227 USA
3-030615.9
Rev Jan 2015
70029867
Revised: 03, 2015
symplmed™
PRINCIPAL DISPLAY PANEL –2mg 100 Tablet Bottle Label
100 Tablets
NDC 61894-000-02
Aceon®
perindopril erbumine Tablets
2mg
Rx ONLY
Keep out of reach of children
symplmed™



| ACEON
perindopril erbumine tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| ACEON
perindopril erbumine tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| ACEON
perindopril erbumine tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Symplmed, LLC (079198402) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Oril Industrie | 501890719 | API MANUFACTURE(61894-000, 61894-001, 61894-002) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Patheon Pharmaceuticals Inc. | 005286822 | MANUFACTURE(61894-000, 61894-001, 61894-002) , ANALYSIS(61894-000, 61894-001, 61894-002) , PACK(61894-000, 61894-001, 61894-002) , LABEL(61894-000, 61894-001, 61894-002) | |