Label: DEFITELIO- defibrotide sodium injection, solution

- NDC Code(s): 68727-800-01, 68727-800-02
- Packager: Jazz Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated December 7, 2022
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DEFITELIO safely and effectively. See full prescribing information for DEFITELIO.
DEFITELIO (defibrotide sodium) injection, for intravenous use
Initial U.S. Approval: 2016INDICATIONS AND USAGE
DEFITELIO is indicated for the treatment of adult and pediatric patients with hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome (SOS), with renal or pulmonary dysfunction following hematopoietic stem-cell transplantation (HSCT). (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Injection: 200 mg/2.5 mL (80 mg/mL) in a single-patient-use vial. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- •
- Hemorrhage: Monitor patients for bleeding. Withhold or discontinue DEFITELIO if significant bleeding occurs. (2.3, 5.1)
- •
- Hypersensitivity Reactions: If severe or life threatening allergic reaction occurs, discontinue DEFITELIO, treat according to standard of care, and monitor until signs and symptoms resolve. (2.3, 5.2)
ADVERSE REACTIONS
The most common adverse reactions (incidence ≥10% and independent of causality) with DEFITELIO treatment were hypotension, diarrhea, vomiting, nausea, and epistaxis. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Jazz Pharmaceuticals, Inc. at 1-800-520-5568 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- •
- DEFITELIO may enhance the activity of antithrombotic/fibrinolytic drugs. (7)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Administration Instructions
2.3 Treatment Modification
2.4 Preparation Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hemorrhage
5.2 Hypersensitivity Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of DEFITELIO for adult and pediatric patients is 6.25 mg/kg every 6 hours given as a 2‑hour intravenous infusion. The dose should be based on patient’s baseline body weight, defined as the patient’s weight prior to the preparative regimen for HSCT.
Administer DEFITELIO for a minimum of 21 days. If after 21 days signs and symptoms of hepatic VOD have not resolved, continue DEFITELIO until resolution of VOD or up to a maximum of 60 days.
2.2 Administration Instructions
- •
- DEFITELIO must be diluted prior to infusion [see Dosage and Administration (2.4)].
- •
- Prior to administration of DEFITELIO, confirm that the patient is not experiencing clinically significant bleeding and is hemodynamically stable on no more than one vasopressor [see Warnings and Precautions (5.1)].
- •
- Administer DEFITELIO by constant intravenous infusion over a 2-hour period.
- •
- Administer the diluted DEFITELIO solution using an infusion set equipped with a 0.2 micron in-line filter. Flush the intravenous administration line (peripheral or central) with 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP immediately before and after administration.
- •
- Do not co‑administer DEFITELIO and other intravenous drugs concurrently within the same intravenous line.
2.3 Treatment Modification
Treatment modification, including temporary or permanent discontinuation of DEFITELIO, should follow the recommendations in Table 1.
Table 1: Treatment Modifications for Toxicity or Invasive Procedures
Event
Recommended Action
Hypersensitivity Reaction
Severe or life-threatening (anaphylaxis)
- 1.
- Discontinue DEFITELIO permanently; do not resume treatment.
Bleeding
Persistent, severe or potentially life-threatening
- 1.
- Withhold DEFITELIO.
- 2.
- Treat the cause of bleeding and give supportive care as clinically indicated.
- 3.
- Consider resuming treatment (at the same dose and infusion volume) when bleeding has stopped and the patient is hemodynamically stable.
Recurrent significant bleeding
- 1.
- Discontinue DEFITELIO permanently; do not resume treatment.
Invasive Procedures
- 1.
- There is no known reversal agent for the profibrinolytic effects of DEFITELIO. Discontinue DEFITELIO infusion at least 2 hours prior to an invasive procedure.
- 2.
- Resume DEFITELIO treatment after the procedure as soon as any procedure-related risk of bleeding is resolved.
2.4 Preparation Instructions
Dilute DEFITELIO in 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP to a concentration of 4 mg/mL to 20 mg/mL. Administer the diluted solution over 2 hours.
Vials contain no antimicrobial preservatives and are intended for a single-patient-use only. Partially used vials should be discarded. Use the diluted DEFITELIO solution within 4 hours if stored at room temperature or within 24 hours if stored under refrigeration. Up to four doses of DEFITELIO solution may be prepared at one time, if refrigerated.
Preparation Instructions:
- •
- Determine the dose (mg) and number of vials of DEFITELIO based on the individual patient’s baseline weight (weight prior to the preparative regimen for HSCT).
- •
- Calculate the volume of DEFITELIO needed, withdraw this amount from the vial(s) and add it to the infusion bag containing 0.9% Sodium Chloride Injection or 5% Dextrose Injection for each dose to make a final concentration of 4 mg/mL to 20 mg/mL.
- •
- Gently mix the solution for infusion.
- •
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Only clear solutions without visible particles should be used. Depending on the type and amount of diluent, the color of the diluted solution may vary from colorless to light yellow.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
The use of DEFITELIO is contraindicated in the following conditions:
- •
- Concomitant administration with systemic anticoagulant or fibrinolytic therapy [see Warnings and Precautions (5.1)]
- •
- Known hypersensitivity to DEFITELIO or to any of its excipients [see Warnings and Precautions (5.2)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Hemorrhage
DEFITELIO increased the activity of fibrinolytic enzymes in vitro, and it may increase the risk of bleeding in patients with VOD after hematopoietic stem-cell transplantation (HSCT). Do not initiate DEFITELIO in patients with active bleeding. Monitor patients for signs of bleeding. If patients on DEFITELIO develop bleeding, discontinue DEFITELIO, treat the underlying cause, and provide supportive care until the bleeding has stopped [see Dosage and Administration (2.3)].
Concomitant use of DEFITELIO and a systemic anticoagulant or fibrinolytic therapy (not including use for routine maintenance or reopening of central venous lines) may increase the risk of bleeding. Discontinue anticoagulants and fibrinolytic agents prior to DEFITELIO treatment, and consider delaying the start of DEFITELIO administration until the effects of the anticoagulant have abated [see Contraindications (4)].
5.2 Hypersensitivity Reactions
Hypersensitivity reactions have occurred in less than 2% of patients treated with DEFITELIO. These reactions include rash, urticaria and angioedema. One case of an anaphylactic reaction was reported in a patient who had previously received DEFITELIO. Monitor patients for hypersensitivity reactions, especially if there is a history of previous exposure. If a severe hypersensitivity reaction occurs, discontinue DEFITELIO, treat according to the standard of care, and monitor until symptoms resolve [see Dosage and Administration (2.3)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- •
- Hemorrhage [see Warnings and Precautions (5.1)]
- •
- Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of DEFITELIO was determined in 176 adult and pediatric patients with hepatic VOD with pulmonary and/or renal dysfunction following HSCT who were treated with DEFITELIO 6.25 mg/kg every 6 hours [see Clinical Studies (14)]. Patients were excluded from these trials if at time of study entry they had significant acute bleeding, active grades B-D graft-versus-host disease, or a requirement for multiple vasopressors to provide blood pressure support. For the purposes of adverse event recording in the clinical trials, events were not required to be reported if they were related to the hepatic VOD, or if they were expected to occur after hematopoietic stem-cell transplantation (HSCT), unless they were serious or Grade 4-5.
The median age of the safety population was 25 years (range: 1 month to 72 years), and 63% were ≥17 years of age. A total of 60% of patients were male, 78% were white, 89% had undergone allogeneic HSCT, and the underlying diagnosis was acute leukemia for 43%. At study entry, 13% were dialysis dependent and 18% were ventilator dependent. DEFITELIO was administered for a median of 21 days (range: 1 to 83 days).
Information about adverse reactions resulting in permanent discontinuation of DEFITELIO was available for 102 patients, and 35 (34%) of these patients had an adverse reaction with permanent discontinuation. Adverse reactions leading to permanent discontinuation included pulmonary alveolar hemorrhage in 5 (5%) patients; pulmonary hemorrhage, hypotension, catheter site hemorrhage, and multi-organ failure, each in 3 (3%) patients; and cerebral hemorrhage and sepsis, each in 2 (2%) patients.
Information about adverse reactions of any grade was available for all 176 patients. The most common adverse reactions (incidence ≥10% and independent of causality) were hypotension, diarrhea, vomiting, nausea, and epistaxis. The most common serious adverse reactions (incidence ≥5% and independent of causality) were hypotension (11%) and pulmonary alveolar hemorrhage (7%). Hemorrhage events of any type and any grade were reported for 104 (59%) of the patients, and the events were grade 4-5 in 35 (20%).
Table 2 presents adverse reactions independent of causality ≥10% any grade or Grade 4/5 ≥2% reported in patients treated with DEFITELIO.
Table 2: Adverse Reactionsa ≥10% or Grade 4-5 Adverse Reactions ≥2%
DEFITELIO
(n=176)
Adverse Reaction a
Any grade
Grade 4-5b
Hypotension
65 (37%)
12 (7%)
Diarrhea
43 (24%)
0
Vomiting
31 (18%)
0
Nausea
28 (16%)
0
Epistaxis
24 (14%)
0
Pulmonary alveolar hemorrhage
15 (9%)
12 (7%)
Gastrointestinal hemorrhage
15 (9%)
5 (3%)
Sepsis
12 (7%)
9 (5%)
Graft versus host disease
11 (6%)
7 (4%)
Lung infiltration
10 (6%)
5 (3%)
Pneumonia
9 (5%)
5 (3%)
Pulmonary hemorrhage
7 (4%)
4 (2%)
Infection
6 (3%)
4 (2%)
Hemorrhage intracranial
5 (3%)
4 (2%)
Hyperuricemia
4 (2%)
4 (2%)
Cerebral hemorrhagec
3 (2%)
3 (2%)
a Excludes events considered to be due to the underlying disease: multi-organ failure, veno-occlusive disease, respiratory failure, renal failure, and hypoxia
b Adverse reactions considered life-threatening or fatal
c Cerebral hemorrhage has been included in the table due to clinical relevance
-
7 DRUG INTERACTIONS
Antithrombotic Agents
DEFITELIO may enhance the pharmacodynamic activity of antithrombotic/fibrinolytic drugs such as heparin or alteplase. Concomitant use of DEFITELIO with antithrombotic or fibrinolytic drugs is contraindicated because of an increased risk of hemorrhage [see Contraindications (4)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on DEFITELIO use in pregnant women. When administered to pregnant rabbits during the period of organogenesis at doses that were comparable to the recommended human dose based on body surface area, defibrotide sodium decreased the number of implantations and viable fetuses. Advise pregnant women of the potential risk of miscarriage.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies are 2-4% and 15-20%, respectively.
Data
Animal Data
Embryo-Fetal toxicity assessment was attempted in rats and rabbits, but was not possible because of high maternal mortality, abortion, and fetal resorption at all doses. Pregnant rats were administered defibrotide sodium from gestational day (GD) 6 to 15 at 0, 240, 1200, and 4800 mg/kg/day by continuous intravenous infusion over 24 hours or at 60, 120, and 240 mg/kg/day by 2-hour infusions 4 times per day. Pregnant rabbits were administered defibrotide sodium at 0, 30, 60, or 120 mg/kg/day from GD 6 to 18 by 2-hour infusions 4 times per day.
In another study in pregnant rabbits, 3 separate subgroups of animals were treated with doses of 80 mg/kg/day defibrotide sodium administered by 2-hour infusions 4 times per day for 5 days each in a staggered manner during the organogenesis period. The dose of 80 mg/kg/day is approximately equivalent to the recommended clinical dose on a mg/m2 basis. Subgroup 1 was dosed from GD 6 to 10, subgroup 2 was dosed from GD 10 to 14, and subgroup 3 was dosed from GD 14 to 18. An increased incidence of unilateral implantation was observed in defibrotide sodium-treated animals. Treatment with defibrotide sodium resulted in a decreased number of implantations and viable fetuses.
8.2 Lactation
Risk Summary
There is no information regarding the presence of DEFITELIO in human milk, the effects on the breastfed infant, or the effects on milk production. Because of the potential for serious adverse reactions, including bleeding in a breastfed infant, advise patients that breastfeeding is not recommended during treatment with DEFITELIO.
8.4 Pediatric Use
The safety and effectiveness of DEFITELIO have been established in pediatric patients. Use of DEFITELIO is supported by evidence from an adequate and well-controlled study and a dose finding study of DEFITELIO in adult and pediatric patients with VOD with evidence of renal or pulmonary dysfunction following HSCT. The clinical trials enrolled 66 pediatric patients in the following age groups: 22 infants (1 month up to less than 2 years), 30 children (2 years up to less than 12 years), and 14 adolescents (12 years to less than 17 years). The efficacy and safety outcomes were consistent across pediatric and adult patients in the clinical trials [see Adverse Reactions (6) and Clinical Studies (14)].
Juvenile Animal Toxicity Data
A juvenile toxicity study in 21-day-old rats was conducted with intravenous bolus administration of defibrotide sodium at 40, 150, or 320 mg/kg/day for 4 weeks. A delayed mean age of preputial separation was observed at all doses, suggesting a delay in onset of male puberty. The dose of 40 mg/kg/day is approximately 0.4 times the clinical dose on a mg/m2 basis for a child. The relevance of this finding for the onset of male puberty in humans is unknown.
8.5 Geriatric Use
Clinical studies of DEFITELIO did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
- 10 OVERDOSAGE
-
11 DESCRIPTION
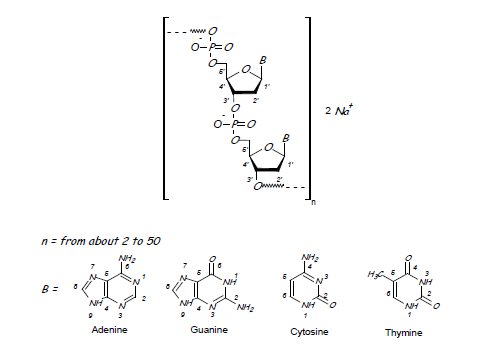
Defibrotide sodium is an oligonucleotide mixture with profibrinolytic properties. The chemical name of defibrotide sodium is polydeoxyribonucleotide, sodium salt. Defibrotide sodium is a polydisperse mixture of predominantly single-stranded (ss) polydeoxyribonucleotide sodium salts derived from porcine intestinal tissue having a mean weighted molecular weight of 14-19 kDa, and a potency of 27-39 and 28-38 biological units per mg as determined by two separate assays measuring the release of a product formed by contact between defibrotide sodium, plasmin and a plasmin substrate. The primary structure of defibrotide sodium is shown below.
DEFITELIO (defibrotide sodium) injection is a clear, light yellow to brown, sterile, preservative-free solution in a single-patient-use vial for intravenous use. Each milliliter of the injection contains 80 mg of defibrotide sodium and 10 mg of Sodium Citrate, USP, in Water for Injection, USP. Hydrochloric Acid, NF, and/or Sodium Hydroxide, NF, may have been used to adjust pH to 6.8-7.8.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism of action of defibrotide sodium has not been fully elucidated. In vitro, defibrotide sodium enhances the enzymatic activity of plasmin to hydrolyze fibrin clots. Studies evaluating the pharmacological effects of defibrotide sodium on endothelial cells (ECs) were conducted primarily in the human microvascular endothelial cell line. In vitro, defibrotide sodium increased tissue plasminogen activator (t-PA) and thrombomodulin expression, and decreased von Willebrand factor (vWF) and plasminogen activator inhibitor‑1 (PAI-1) expression, thereby reducing EC activation and increasing EC‑mediated fibrinolysis. Defibrotide sodium protected ECs from damage caused by chemotherapy, tumor necrosis factor-α (TNF-α), serum starvation, and perfusion.
12.2 Pharmacodynamics
Cardiac Electrophysiology
At a dose 2.4 times the maximum recommended dose, DEFITELIO does not prolong the QTc interval to any clinically relevant extent.
PAI-1 Inhibition
Plasma concentrations of PAI-1 were assessed on an exploratory basis as a potential pharmacodynamic marker for efficacy in Study 2. PAI-1 is an inhibitor of t-PA and therefore of fibrinolysis. Mean PAI-1 levels on Days 7 and 14 were lower than those at baseline in patients with complete response (CR) and in those who were alive at Day+100, but this trend did not reach statistical significance. There were no statistically significant differences in mean PAI-1 levels by treatment or outcome.
12.3 Pharmacokinetics
Absorption
After intravenous administration, peak plasma concentrations of defibrotide sodium occur approximately at the end of each infusion.
Distribution
Defibrotide sodium is highly bound to human plasma proteins (average 93%) and has a volume of distribution of 8.1 to 9.1 L.
Elimination
Metabolism followed by urinary excretion is likely the main route of elimination. The estimated total clearance was 3.4 to 6.1 L/h. The elimination half-life of defibrotide sodium is less than 2 hours. Similar plasma concentration profiles were observed in VOD patients after initial and multiple-dose administration of 6.25 mg/kg every 6 hours for 5 days. Therefore, no accumulation is expected following multiple-dose administration.
Metabolism
Though the precise pathway of defibrotide sodium degradation in plasma in vivo is largely unknown, it has been suggested that nucleases, nucleotidases, nucleosidases, deaminases, and phosphorylases metabolize polynucleotides progressively to oligonucleotides, nucleotides, nucleosides, and then to the free 2'-deoxyribose sugar, purine and pyrimidine bases.
The biotransformation of defibrotide sodium was investigated in vitro by incubation with human hepatocytes from donors of different ages and showed that defibrotide sodium does not undergo appreciable metabolism by human hepatocyte cells.
Excretion
After administration of 6.25 mg/kg to 15 mg/kg doses of DEFITELIO as 2-hour infusions, approximately 5-15% of the total dose was excreted in urine as defibrotide sodium, with the majority excreted during the first 4 hours.
Specific Populations
Age: Pediatric Population
Insufficient PK data were collected in pediatric patients to draw conclusions.
Renal Impairment
The safety, tolerability, and pharmacokinetics of 6.25 mg/kg as 2-hour intravenous infusions of DEFITELIO were evaluated in patients with Hemodialysis-dependent End Stage Renal Disease (ESRD) during hemodialysis and on days off dialysis, and in patients with severe renal disease or ESRD not requiring dialysis. Defibrotide sodium was not removed by hemodialysis, which had no notable effect on plasma clearance of defibrotide sodium. Terminal half-lives were consistently less than 2 hours, and there was no accumulation of defibrotide sodium following repeated dosing. Defibrotide sodium exposure (AUC) in patients with severe renal impairment or ESRD was 50% to 60% higher than that observed in matched healthy subjects. Peak concentration (Cmax) was 35% to 37% higher following single- and multiple-dose administration of defibrotide sodium.
Drug Interactions
Pharmacokinetic drug-drug interactions are unlikely at therapeutic dose. Data from in vitro studies using human biomaterial demonstrate that defibrotide sodium does not induce (CYP1A2, CYP2B6, CYP3A4, UGT1A1) or inhibit (CYP1A2, CYP2B6, CYP3A4, CYP2C8, CYP2C9, CYP2C19, CYP2D6, UGT1A1, UGT2B7) the major drug metabolizing enzymes and is not a substrate or inhibitor of the major drug uptake transporters (OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3) or efflux transporters (P-gp and BCRP).
There is some evidence (animal studies, ex vivo human plasma, and healthy volunteers) that defibrotide sodium may enhance the pharmacodynamic activity of heparin and alteplase [see Drug Interactions (7)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity studies have been conducted with intravenous administration of defibrotide sodium.
Defibrotide sodium was not mutagenic in vitro in a bacterial reverse mutation assay (Ames assay). Defibrotide sodium was not clastogenic in an in vitro chromosomal aberrations assay in Chinese hamster ovary cells or an in vivo micronucleus assay conducted in bone marrow from rats administered defibrotide sodium by intravenous infusion.
Studies of fertility were not conducted with defibrotide sodium administered by the intravenous route. In repeat dose general toxicology studies, when defibrotide sodium was administered intravenously to rats and dogs for up to 13 weeks, there were no effects on male or female reproductive organs.
13.2 Animal Toxicology and/or Pharmacology
In the 13-week toxicity studies in rats and dogs, intravenous administration of defibrotide sodium transiently prolonged activated partial thromboplastin time (APTT) at 1200 and 4800 mg/kg/day administered as a continuous infusion in rats and at 300 and 1600 mg/kg/day administered in 2-hour infusions 4 times daily in dogs. Prothrombin time (PT) was also transiently increased at 4800 mg/kg/day in rats. These findings were observed at doses at least 6 times higher on a mg/m2 basis than the clinical dose of 25 mg/kg/day. The effects on APTT and PT may be due to direct effects on coagulation based on the dose-dependent response observed.
-
14 CLINICAL STUDIES
The efficacy of DEFITELIO was investigated in three studies: two prospective clinical trials (Study 1 and Study 2), and an expanded access study (Study 3).
Study 1 enrolled 102 adult and pediatric patients in the DEFITELIO treatment group with a diagnosis of VOD according to the following criteria (bilirubin of at least 2 mg/dL and at least two of the following findings: hepatomegaly, ascites, and weight gain greater than 5% by Day+21 post-HSCT) with an associated diagnosis of multi-organ dysfunction (pulmonary, renal, or both) by Day+28 post-HSCT. DEFITELIO was administered to the treatment group at a dose of 6.25 mg/kg infused every 6 hours for a minimum of 21 days and continued until patient was discharged from the hospital. Patients enrolled in the DEFITELIO treatment group were not permitted to receive concomitant medications such as heparin, warfarin, or alteplase because of an increased risk of bleeding.
Study 2 included adult and pediatric patients with a diagnosis of hepatic VOD and multi-organ dysfunction following HSCT, with 75 patients treated with DEFITELIO at a dose of 6.25 mg/kg infused every 6 hours. The planned minimum duration of treatment was 14 days. The treatment could be continued until signs of hepatic VOD resolved.
Study 3 is an expanded access program for DEFITELIO for the treatment of adult and pediatric patients with hepatic VOD. The efficacy of defibrotide was evaluated in 351 patients who had received a HSCT and developed hepatic VOD with renal or pulmonary dysfunction. All patients received DEFITELIO at a dose of 6.25 mg/kg infused every 6 hours.
Baseline demographic information and details for patients treated in these studies are provided below in Table 3.
Table 3: Baseline Demographics of Patients Treated with DEFITELIO at 6.25 mg/kg Every 6 Hours
Data Source Study 1 Study 2 Study 3 Design
Prospective
Prospective
Expanded Access Study
Number of patients
102
75
351
Median age (years)
(range)
21 years
(<1, 72)
32 years
(<1, 61)
15 years
(<1, 69)
Age, n(%)
< 17 years
≥ 17 years
44 (43%)
58 (57%)
22 (29%)
53 (71%)
189 (54%)
162 (46%)
Race, n(%)
White
Black/African American
Asian
Other
77 (75%)
6 (6%)
4 (4%)
15 (15%)
61 (81%)
6 (8%)
2 (3%)
6 (8%)
237 (68%)
21 (6%)
15 (4%)
78 (22%)
Gender, n(%)
Male
Female
64 (63%)
38 (37%)
41 (55%)
34 (45%)
184 (52%)
167 (48% )
Median number of days on treatment (days)
(range)
21.5 days(1,58)
19.5 days(3,83)
21.0 daysa(1,93)
Type of graft, n(%)
Allograft
Autograft
90 (88%)
12 (12%)
67 (89%)
8 (11%)
317 (90%)
34 (10%)
Ventilator or Dialysis Dependent at Study Entry, n(%)
34 (33%)
8 (11%)
149 (42%)
- a Duration of treatment from first dose to last dose is presented because days without treatment were not captured for the expanded access study.
The efficacy of DEFITELIO was based on survival at Day + 100 after HSCT. In Study 1, the survival rate was 38% (95% CI: 29%, 48%) at 100 days after transplantation. In Study 2 the survival rate was 44% (95% CI: 33%, 55%) at 100 days after transplantation. In Study 3, the Day + 100 survival was 45% (95% CI: 40%, 51%).
Based on published reports and analyses of patient level data for individuals with hepatic VOD with renal or pulmonary dysfunction who received supportive care or interventions other than DEFITELIO, the expected Day +100 survival rates are 21% to 31%.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
DEFITELIO (defibrotide sodium) injection is supplied in a single-patient-use, clear glass vial as a clear, light yellow to brown, sterile, preservative-free solution for intravenous infusion. Each vial (NDC 68727-800-01) contains 200 mg/2.5 mL (at a concentration of 80 mg/mL) of defibrotide sodium.
Each carton of DEFITELIO (defibrotide sodium) injection (NDC 68727-800-02) contains 10 vials.
Store DEFITELIO (defibrotide sodium) injection at 20°C-25°C (68°F-77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) (see USP controlled room temperature).
-
17 PATIENT COUNSELING INFORMATION
- •
- Hemorrhage: Advise patients and caregivers that DEFITELIO may increase the risk of bleeding (hemorrhage). Instruct patients to immediately report any signs or symptoms suggestive of hemorrhage (unusual bleeding, easy bruising, blood in urine or stool, headache, confusion, slurred speech, or altered vision) [see Warnings and Precautions (5.1)].
- •
- Hypersensitivity Reactions: Ask patients if they have been treated with defibrotide sodium previously. Instruct patients on the risk of allergic reactions, including anaphylaxis. Describe the symptoms of allergic reactions, including anaphylaxis, and instruct the patient to seek medical attention immediately if they experience such symptoms [see Warnings and Precautions (5.2)].
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
This product's label may have been updated. For full prescribing information, please visit labels.fda.gov.
Distributed by:
Jazz Pharmaceuticals, Inc.
Palo Alto, CA 94304
Protected by U.S. Patent Nos. 11,085,043 and 11,236,328
DEFITELIO® is a registered trademark of Jazz Pharmaceuticals plc or its subsidiaries.
© 2022 Jazz Pharmaceuticals -
INGREDIENTS AND APPEARANCE
DEFITELIO
defibrotide sodium injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:68727-800 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEFIBROTIDE SODIUM (UNII: L7CHH2B2J0) (DEFIBROTIDE FREE ACID - UNII:568FY5I1YI) DEFIBROTIDE SODIUM 80 mg in 1 mL Inactive Ingredients Ingredient Name Strength TRISODIUM CITRATE DIHYDRATE (UNII: B22547B95K) WATER (UNII: 059QF0KO0R) HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:68727-800-01 2.5 mL in 1 VIAL; Type 0: Not a Combination Product 03/30/2016 2 NDC:68727-800-02 25 mL in 1 CARTON; Type 0: Not a Combination Product 03/30/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208114 03/30/2016 Labeler - Jazz Pharmaceuticals, Inc. (135926363) Registrant - Jazz Pharmaceuticals, Inc. (135926363) Establishment Name Address ID/FEI Business Operations Gentium S.r.L 432729114 API MANUFACTURE(68727-800) , ANALYSIS(68727-800) Establishment Name Address ID/FEI Business Operations Patheon Italia S.p.A. 434078638 MANUFACTURE(68727-800) Establishment Name Address ID/FEI Business Operations Jazz Pharmaceuticals Ireland Limited 896650210 MANUFACTURE(68727-800)