Label: CHOLESTYRAMINE powder, for suspension


-
Contains inactivated NDC Code(s)
NDC Code(s): 54868-4812-0, 54868-4812-1, 54868-5526-0 - Packager: Physicians Total Care, Inc.
- This is a repackaged label.
- Source NDC Code(s): 49884-465, 49884-466
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated April 28, 2010
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
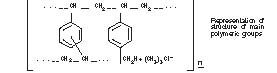
Cholestyramine for Oral Suspension USP, the chloride salt of a basic anion exchange resin, a cholesterol lowering agent, is intended for oral administration. Cholestyramine resin is quite hydrophilic, but insoluble in water. The cholestyramine resin in Cholestyramine is not absorbed from the digestive tract. Four grams of anhydrous cholestyramine resin is contained in 9 grams of Cholestyramine for Oral Suspension USP. Four grams of anhydrous cholestyramine resin is contained in 5 grams of Cholestyramine for Oral Suspension USP, Light. It is represented by the following structural formula:
Cholestyramine for Oral Suspension USP contains the following inactive ingredients: acacia, citric acid, D&C Yellow No. 10, FD&C Yellow No. 6, flavor (natural and artificial Orange), polysorbate 80, propylene glycol alginate and sucrose. Cholestyramine for Oral Suspension USP, Light contains the following inactive ingredients: aspartame, citric acid, colloidal silicon dioxide, D&C Yellow No. 10, FD&C Red No. 40, flavor (natural and artificial Orange), maltodextrin, propylene glycol alginate and xanthan gum.
-
CLINICAL PHARMACOLOGY
Cholesterol is probably the sole precursor of bile acids. During normal digestion, bile acids are secreted into the intestines. A major portion of the bile acids is absorbed from the intestinal tract and returned to the liver via the enterohepatic circulation. Only very small amounts of bile acids are found in normal serum.
Cholestyramine resin adsorbs and combines with the bile acids in the intestine to form an insoluble complex which is excreted in the feces. This results in a partial removal of bile acids from the enterohepatic circulation by preventing their absorption.
The increased fecal loss of bile acids due to Cholestyramine administration leads to an increased oxidation of cholesterol to bile acids, a decrease in beta lipoprotein or low density lipoprotein plasma levels and a decrease in serum cholesterol levels. Although in man, Cholestyramine produces an increase in hepatic synthesis of cholesterol, plasma cholesterol levels fall.
In patients with partial biliary obstruction, the reduction of serum bile acid levels by Cholestyramine reduces excess bile acids deposited in the dermal tissue with resultant decrease in pruritus.
-
INDICATIONS AND USAGE
1) Cholestyramine for Oral Suspension USP is indicated as adjunctive therapy to diet for the reduction of elevated serum cholesterol in patients with primary hypercholesterolemia (elevated low density lipoprotein [LDL] cholesterol) who do not respond adequately to diet. Cholestyramine may be useful to lower LDL cholesterol in patients who also have hypertriglyceridemia, but it is not indicated where hypertriglyceridemia is the abnormality of most concern.
Therapy with lipid-altering agents should be a component of multiple risk factor intervention in those individuals at significantly increased risk for atherosclerotic vascular disease due to hypercholesterolemia. Treatment should begin and continue with dietary therapy specific for the type of hyperlipoproteinemia determined prior to initiation of drug therapy. Excess body weight may be an important factor and caloric restriction for weight normalization should be addressed prior to drug therapy in the overweight.
Prior to initiating therapy with Cholestyramine, secondary causes of hypercholesterolemia (e.g., poorly controlled diabetes mellitus, hypothyroidism, nephrotic syndrome, dysproteinemias, obstructive liver disease, other drug therapy, alcoholism), should be excluded, and a lipid profile performed to assess Total cholesterol, HDL-C, and triglycerides (TG). For individuals with TG less than 400 mg/dL (less than 4.5 mmol/L), LDL-C can be estimated using the following equation:
LDL-C = Total cholesterol – [(TG/5) + HDL-C]
For TG levels >400 mg/dL, this equation is less accurate and LDL-C concentrations should be determined by ultracentrifugation. In hypertriglyceridemic patients, LDL-C may be low or normal despite elevated Total-C. In such cases Cholestyramine may not be indicated.
Serum cholesterol and triglyceride levels should be determined periodically based on NCEP guidelines to confirm initial and adequate long-term response. A favorable trend in cholesterol reduction should occur during the first month of Cholestyramine therapy. The therapy should be continued to sustain cholesterol reduction. If adequate cholesterol reduction is not attained, increasing the dosage of Cholestyramine or adding other lipid-lowering agents in combination with Cholestyramine should be considered.
Since the goal of treatment is to lower LDL-C, the NCEP 4 recommends that LDL-C levels be used to initiate and assess treatment response. If LDL-C levels are not available then Total-C alone may be used to monitor long-term therapy. A lipoprotein analysis (including LDL-C determination) should be carried out once a year. The NCEP treatment guidelines are summarized below.
* Coronary heart disease or peripheral vascular disease (including symptomatic carotid artery disease).
LDL-Cholesterol mg/dL (mmol/L)
Definite Atherosclerotic
Disease*
Two or More Other Risk
Factors**
Initiation Level
Goal
NO
NO
Greater Than or Equal to 190
(Greater Than or Equal To 4.9)Less Than 160
(Less Than 4.1)NO
YES
Greater Than or Equal To 160
(Greater Than or Equal To 4.1)Less Than 130
(Less Than 3.4)YES
YES or NO
Greater Than or Equal To 130
(Greater Than or Equal To 3.4)Less Than or Equal To 100
(Less Than or Equal To 2.6)
** Other risk factors for coronary heart disease (CHD) include: age (males greater than or equal to 45 years; females greater than or equal to 55 years or premature menopause without estrogen replacement therapy); family history of premature CHD; current cigarette smoking; hypertension; confirmed HDL-C less than 35 mg/dL (less than 0.91 mmol/L); and diabetes mellitus. Subtract one risk factor if HDL-C is greater than or equal to 60 mg/dL (greater than or equal to 1.6 mmol/L).
Cholestyramine monotherapy has been demonstrated to retard the rate of progression 2,3 and increase the rate of regression3 of coronary atherosclerosis.
2) Cholestyramine for oral suspension is indicated for the relief of pruritus associated with partial biliary obstruction. Cholestyramine for oral suspension has been shown to have a variable effect on serum cholesterol in these patients. Patients with primary biliary cirrhosis may exhibit an elevated cholesterol as part of their disease.
- CONTRAINDICATIONS
- WARNINGS
-
PRECAUTIONS
General
Chronic use of cholestyramine resin may be associated with increased bleeding tendency due to hypoprothrombinemia associated with Vitamin K deficiency. This will usually respond promptly to parenteral Vitamin K1 and recurrences can be prevented by oral administration of Vitamin K1. Reduction of serum or red cell folate has been reported over long term administration of cholestyramine resin. Supplementation with folic acid should be considered in these cases.
There is a possibility that prolonged use of cholestyramine resin, since it is a chloride form of anion exchange resin, may produce hyperchloremic acidosis. This would especially be true in younger and smaller patients where the relative dosage may be higher. Caution should also be exercised in patients with renal insufficiency or volume depletion, and in patients receiving concomitant spironolactone.
Cholestyramine resin may produce or worsen pre-existing constipation. The dosage should be increased gradually in patients to minimize the risk of developing fecal impaction. In patients with pre-existing constipation, the starting dose should be 1 packet or 1 scoop once daily for 5–7 days, increasing to twice daily with monitoring of constipation and of serum lipoproteins, at least twice, 4–6 weeks apart. Increased fluid intake and fiber intake should be encouraged to alleviate constipation and a stool softener may occasionally be indicated. If the initial dose is well tolerated, the dose may be increased as needed by one dose/day (at monthly intervals) with periodic monitoring of serum lipoproteins. If constipation worsens or the desired therapeutic response is not achieved at one to six doses/day, combination therapy or alternate therapy should be considered. Particular effort should be made to avoid constipation in patients with symptomatic coronary artery disease. Constipation associated with cholestyramine resin may aggravate hemorrhoids.
Information for PatientsInform your physician if you are pregnant or plan to become pregnant or are breastfeeding. Drink plenty of fluids and mix each 9 gram dose of Cholestyramine for Oral Suspension USP in at least 2 to 6 ounces of fluid. Mix each 5 gram dose of Cholestyramine for Oral Suspension USP, Light in at least 2 to 6 ounces of fluid before taking. Sipping or holding the resin suspension in the mouth for prolonged periods may lead to changes in the surface of the teeth resulting in discoloration, erosion of enamel or decay; good oral hygiene should be maintained.
Laboratory TestsSerum cholesterol levels should be determined frequently during the first few months of therapy and periodically thereafter. Serum triglyceride levels should be measured periodically to detect whether significant changes have occurred.
The LRC-CPPT showed a dose-related increase in serum triglycerides of 10.7%–17.1% in the cholestyramine-treated group, compared with an increase of 7.9%–11.7% in the placebo group. Based on the mean values and adjusting for the placebo group, the cholestyramine-treated group showed an increase of 5% over pre-entry levels the first year of the study and an increase of 4.3% the seventh year.
Drug InteractionsCholestyramine for Oral Suspension USP may delay or reduce the absorption of concomitant oral medication such as phenylbutazone, warfarin, thiazide diuretics (acidic), or propranolol (basic), as well as tetracycline, penicillin G, phenobarbital, thyroid and thyroxine preparations, estrogens and progestins, and digitalis. Interference with the absorption of oral phosphate supplements has been observed with another positively-charged bile acid sequestrant. Cholestyramine may interfere with the pharmacokinetics of drugs that undergo enterohepatic circulation. The discontinuance of Cholestyramine could pose a hazard to health if a potentially toxic drug such as digitalis has been titrated to a maintenance level while the patient was taking Cholestyramine.
Because cholestyramine binds bile acids, Cholestyramine may interfere with normal fat digestion and absorption and thus may prevent absorption of fat-soluble vitamins such as A, D, E and K. When Cholestyramine is given for long periods of time, concomitant supplementation with water-miscible (or parenteral) forms of fat-soluble vitamins should be considered.
SINCE CHOLESTYRAMINE MAY BIND OTHER DRUGS GIVEN CONCURRENTLY, IT IS RECOMMENDED THAT PATIENTS TAKE OTHER DRUGS AT LEAST ONE HOUR BEFORE OR 4 TO 6 HOURS AFTER CHOLESTYRAMINE (OR AT AS GREAT AN INTERVAL AS POSSIBLE) TO AVOID IMPEDING THEIR ABSORPTION.
Carcinogenesis and Mutagenesis and Impairment of FertilityIn studies conducted in rats in which cholestyramine resin was used as a tool to investigate the role of various intestinal factors, such as fat, bile salts and microbial flora, in the development of intestinal tumors induced by potent carcinogens, the incidence of such tumors was observed to be greater in cholestyramine resin-treated rats than in control rats.
The relevance of this laboratory observation from studies in rats to the clinical use of Cholestyramine is not known. In the LRC-CPPT study referred to above, the total incidence of fatal and nonfatal neoplasms was similar in both treatment groups. When the many different categories of tumors are examined, various alimentary system cancers were somewhat more prevalent in the cholestyramine group. The small numbers and the multiple categories prevent conclusions from being drawn. However, in view of the fact that cholestyramine resin is confined to the GI tract and not absorbed, and in light of the animal experiments referred to above, a six-year post-trial follow-up of the LRC-CPPT 5 patient population has been completed (a total of 13.4 years of in-trial plus post-trial follow-up) and revealed no significant difference in the incidence of cause-specific mortality or cancer morbidity between cholestyramine and placebo treated patients.
PregnancyPregnancy Category CThere are no adequate and well controlled studies in pregnant women. The use of Cholestyramine in pregnancy or lactation or by women of childbearing age requires that the potential benefits of drug therapy be weighed against the possible hazards to the mother and child. Cholestyramine is not absorbed systemically, however, it is known to interfere with absorption of fat-soluble vitamins; accordingly, regular prenatal supplementation may not be adequate (see PRECAUTIONS: Drug Interactions).
Nursing MothersCaution should be exercised when Cholestyramine is administered to a nursing mother. The possible lack of proper vitamin absorption described in the “Pregnancy” section
Pediatric UseAlthough an optimal dosage schedule has not been established, standard texts (6,7) list a usual pediatric dose of 240 mg/kg/day of anhydrous cholestyramine resin in two to three divided doses, normally not to exceed 8 gm/day with dose titration based on response and tolerance.
In calculating pediatric dosages, 44.4 mg of anhydrous cholestyramine resin are contained in 100 mg of Cholestyramine for Oral Suspension USP and 80 mg of anhydrous cholestyramine resin are contained in 100 mg of Cholestyramine for Oral Suspension USP, Light.
The effects of long-term administration, as well as its effect in maintaining lowered cholesterol levels in pediatric patients, are unknown. (Also see ADVERSE REACTIONS.)
-
ADVERSE REACTIONS
The most common adverse reaction is constipation. When used as a cholesterol-lowering agent predisposing factors for most complaints of constipation are high dose and increased age (more than 60 years old). Most instances of constipation are mild, transient, and controlled with conventional therapy. Some patients require a temporary decrease in dosage or discontinuation of therapy.
Less Frequent Adverse Reactions: Abdominal discomfort and/or pain, flatulence, nausea, vomiting, diarrhea, eructation, anorexia, and steatorrhea, bleeding tendencies due to hypoprothrombinemia (Vitamin K deficiency) as well as Vitamin A (one case of night blindness reported) and D deficiencies, hyperchloremic acidosis in children, osteoporosis, rash and irritation of the skin, tongue and perianal area. Rare reports of intestinal obstruction, including two deaths, have been reported in pediatric patients.
Occasional calcified material has been observed in the biliary tree, including calcification of the gallbladder, in patients to whom cholestyramine resin has been given. However, this may be a manifestation of the liver disease and not drug related.
One patient experienced biliary colic on each of three occasions on which he took cholestyramine resin. One patient diagnosed as acute abdominal symptom complex was found to have a “pasty mass” in the transverse colon on x-ray.
Other events (not necessarily drug related) reported in patients taking cholestyramine resin include:
Gastrointestinal—GI-rectal bleeding, black stools, hemorrhoidal bleeding, bleeding from known duodenal ulcer, dysphagia, hiccups, ulcer attack, sour taste, pancreatitis, rectal pain, diverticulitis.
Laboratory test changes—Liver function abnormalities.
Hematologic—Prolonged prothrombin time, ecchymosis, anemia
Hypersensitivity—Urticaria, asthma, wheezing, shortness of breath.
Musculoskeletal—Backache, muscle and joint pains, arthritis.
Neurologic—Headache, anxiety, vertigo, dizziness, fatigue, tinnitus, syncope, drowsiness, femoral nerve pain, paresthesia.
Renal—Hematuria, dysuria, burnt odor to urine, diuresis.
Miscellaneous—Weight loss, weight gain, increased libido, swollen glands, edema, dental bleeding, dental caries, erosion of tooth enamel, tooth discoloration.
-
OVERDOSAGE
Overdosage with Cholestyramine has been reported in a patient taking 150% of the maximum recommended daily dosage for a period of several weeks. No ill effects were reported. Should an overdosage occur, the chief potential harm would be obstruction of the gastrointestinal tract. The location of such potential obstruction, the degree of obstruction, and the presence or absence of normal gut motility would determine treatment.
-
DOSAGE AND ADMINISTRATION
The recommended starting adult dose for all cholestyramine for oral suspension powdered products (Cholestyramine for Oral Suspension USP and Cholestyramine for Oral Suspension USP, Light) is one packet or one level scoopful once or twice a day. The recommended maintenance dose for all cholestyramine for oral suspension powdered products is 2 to 4 packets or scoopfuls daily (8-16 grams anhydrous cholestyramine resin) divided into two doses. Four grams of anhydrous cholestyramine resin is contained in each measured dose of Cholestyramine as follows:
Cholestyramine for Oral Suspension USP 9 grams Cholestyramine for Oral Suspension USP, Light 5 grams It is recommended that increases in dose be gradual with periodic assessment of lipid/lipoprotein levels at intervals of not less than 4 weeks. The maximum recommended daily dose is six packets or scoopfuls of cholestyramine for oral suspension (24 grams of anhydrous cholestyramine resin). The suggested time of administration is at mealtime but may be modified to avoid interference with absorption of other medications. Although the recommended dosing schedule is twice daily, cholestyramine for oral suspension may be administered in 1–6 doses per day.
Cholestyramine should not be taken in its dry form. Always mix Cholestyramine with water or other fluids before ingesting. See Preparation Instructions.
Concomitant TherapyPreliminary evidence suggests that the lipid-lowering effects of Cholestyramine on total and LDL-cholesterol are enhanced when combined with a HMG-CoA reductase inhibitor, e.g., pravastatin, lovastatin, simvastatin, and fluvastatin. Additive effects on LDL-cholesterol are also seen with combined nicotinic acid/Cholestyramine therapy. See the Drug Interactions subsection of the PRECAUTIONS section for recommendations on administering concomitant therapy.
PREPARATIONThe color of Cholestyramine may vary somewhat from batch to batch but this variation does not affect the performance of the product. Place the contents of one single-dose packet or one level scoopful of Cholestyramine in a glass or cup. Add an amount of water or other noncarbonated beverage of your choice depending on the product being used:
Product Formula Amount of Water or other Non-Carbonated Liquid Cholestyramine for Oral Suspension USP 2-6 ounces per dose Cholestyramine for Oral Suspension USP, Light 2-6 ounces per dose Stir to a uniform consistency and drink.
Cholestyramine may also be mixed with highly fluid soups or pulpy fruits with a high moisture content such as applesauce or crushed pineapple.
-
HOW SUPPLIED
Cholestyramine for Oral Suspension USP is available in cans containing 378 grams and in cartons of sixty 9 gram packets. Four grams of anhydrous cholestyramine resin are contained in 9 grams of Cholestyramine for Oral Suspension USP. The 378 g can includes a 15 cc scoop. The scoop is not interchangeable with scoops from other products.
NDC 54868-4812-0 Can, 378 g NDC 54868-4812-1 Carton of 60, 9 g packets Cholestyramine for Oral Suspension USP, Light is available in cans containing 210 grams and in cartons of sixty 5 gram packets. Four grams of anhydrous cholestyramine resin are contained in 5 grams of Cholestyramine for Oral Suspension USP, Light. The 210 g can includes a 9 cc scoop. The scoop is not interchangeable with scoops from other products.
Storage
NDC 54868-5526-0 Carton of 60, 5 g packets Store between 20º-25ºC (68º-77ºF). [See USP Controlled Room Temperature]. Excursions permitted to 15º-30ºC (59º-86ºF).
-
CLINICAL STUDIES
In a large, placebo-controlled, multi-clinic study, LRC-CPPT 1, hypercholesterolemic subjects treated with Cholestyramine had mean reductions in total and low-density lipoprotein cholesterol (LDL-C) which exceeded those for diet and placebo treatment by 7.2% and 10.4%, respectively. Over the seven-year study period the Cholestyramine group experienced a 19% reduction (relative to the incidence in the placebo group) in the combined rate of coronary heart disease death plus non-fatal myocardial infarction (cumulative incidences of 7% Cholestyramine and 8.6% placebo). The subjects included in the study were men aged 35–59 with serum cholesterol levels above 265 mg/dL and no previous history of heart disease. It is not clear to what extent these findings can be extrapolated to females and other segments of the hypercholesterolemic population. (See also PRECAUTIONS: Carcinogenesis, Mutagenesis, Impairment of Fertility.)
Two controlled clinical trials have examined the effects of Cholestyramine monotherapy upon coronary atherosclerotic lesions using coronary arteriography. In the NHLBI Type II Coronary Intervention Trial 2, 116 patients (80% male) with coronary artery disease (CAD) documented by arteriography were randomized to Cholestyramine or placebo for five years of treatment. Final study arteriography revealed progression of coronary artery disease in 49% of placebo patients compared to 32% of the Cholestyramine group (p less than 0.05).
In the St. Thomas Atherosclerosis Regression Study (STARS) 3, 90 hypercholesterolemic men with CAD were randomized to three blinded treatments: usual care, lipid-lowering diet, and lipid-lowering diet plus Cholestyramine. After 36 months, follow-up coronary arteriography revealed progression of disease in 46% of usual care patients, 15% of patients on lipid-lowering diet and 12% of those receiving diet plus Cholestyramine (p less than 0.02). The mean absolute width of coronary segments decreased in the usual care group, increased slightly (0.003mm) in the diet group and increased by 0.103mm in the diet plus Cholestyramine group (p less than 0.05). Thus in these randomized controlled clinical trials using coronary arteriography, Cholestyramine monotherapy has been demonstrated to slow progression 2,3 and promote regression3 of atherosclerotic lesions in the coronary arteries of patients with coronary artery disease.
The effect of intensive lipid-lowering therapy on coronary atherosclerosis has been assessed by arteriography in hyperlipidemic patients. In these randomized, controlled clinical trials, patients were treated for two to four years by either conventional measures (diet, placebo, or in some cases low dose resin), or intensive combination therapy using diet plus colestipol (an anion exchange resin with a mechanism of action and an effect on serum lipids similar to that of Cholestyramine for Oral Suspension USP and Cholestyramine for Oral Suspension USP, Light) plus either nicotinic acid or lovastatin. When compared to conventional measures, intensive lipid-lowering combination therapy significantly reduced the frequency of progression and increased the frequency of regression of coronary atherosclerotic lesions in patients with or at risk for coronary artery disease.
-
REFERENCES
- The Lipid Research Clinics Coronary Primary Prevention Trial Results: (I) Reduction in Incidence of Coronary Heart Disease; (II) The Relationship of Reduction in Incidence of Coronary Heart Disease to Cholesterol Lowering. JAMA 1984; 251:351-374.
- Brensike JF, Levy RI, Kelsey SF, et al. Effects of therapy with cholestyramine on progression of coronary arteriosclerosis: results of the NHLBI type II coronary intervention study. Circulation 1984;69:313-24.
- Watts, GF, Lewis B, Brunt JNH, Lewis ES, et al. Effects on coronary artery disease of lipid-lowering diet, or diet plus cholestyramine, in the St Thomas Atherosclerosis Regression Study (STARS). Lancet 1992;339:563-69.
- National Cholesterol Education Program. Second Report of the Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel II). Circulation 1994 Mar; 89(3):1333-445.
- The Lipid Research Clinics Investigators. The Lipid Research Clinics Coronary Primary Prevention Trial: Results of 6 Years of Post-Trial Follow-up. Arch Intern Med 1992; 152:1399-1410.
- Behrman RE et al (eds): Nelson, Textbook of Pediatrics, ed 15. Philadelphia, PA, WB Saunders Company, 1996.
- Takemoto CK et al (eds): Pediatric Dosage Handbook, ed 3. Cleveland/Akron, OH, Lexi-Comp, Inc., 1996-1997.
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
CHOLESTYRAMINE
cholestyramine powder, for suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54868-4812(NDC:49884-465) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CHOLESTYRAMINE (UNII: 4B33BGI082) (CHOLESTYRAMINE - UNII:4B33BGI082) CHOLESTYRAMINE 4 g in 9 g Inactive Ingredients Ingredient Name Strength ACACIA (UNII: 5C5403N26O) CITRIC ACID ANHYDROUS (UNII: XF417D3PSL) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) POLYSORBATE 80 (UNII: 6OZP39ZG8H) PROPYLENE GLYCOL ALGINATE (UNII: 26CD3J2R0C) SUCROSE (UNII: C151H8M554) Product Characteristics Color Score Shape Size Flavor ORANGE Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54868-4812-0 378 g in 1 CAN 2 NDC:54868-4812-1 60 in 1 CARTON 2 9 g in 1 PACKET Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077204 03/22/2010 CHOLESTYRAMINE
cholestyramine powder, for suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54868-5526(NDC:49884-466) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CHOLESTYRAMINE (UNII: 4B33BGI082) (CHOLESTYRAMINE - UNII:4B33BGI082) CHOLESTYRAMINE 4 g in 5 g Inactive Ingredients Ingredient Name Strength ASPARTAME (UNII: Z0H242BBR1) CITRIC ACID ANHYDROUS (UNII: XF417D3PSL) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C RED NO. 40 (UNII: WZB9127XOA) MALTODEXTRIN (UNII: 7CVR7L4A2D) PROPYLENE GLYCOL ALGINATE (UNII: 26CD3J2R0C) XANTHAN GUM (UNII: TTV12P4NEE) Product Characteristics Color Score Shape Size Flavor ORANGE Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54868-5526-0 60 in 1 CARTON 1 5 g in 1 PACKET Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077203 02/08/2006 Labeler - Physicians Total Care, Inc. (194123980) Establishment Name Address ID/FEI Business Operations Physicians Total Care, Inc. 194123980 relabel

