Label: ADLARITY- donepezil hydrochloride patch








-
NDC Code(s):
65038-055-01,
65038-055-03,
65038-055-04,
65038-055-07, view more65038-055-10, 65038-056-01, 65038-056-03, 65038-056-04, 65038-056-07, 65038-056-10
- Packager: Corium, LLC.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated February 17, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ADLARITY safely and effectively. See full prescribing information for ADLARITY.
ADLARITY® (donepezil transdermal system)
Initial U.S. Approval: 1996INDICATIONS AND USAGE
ADLARITY is an acetylcholinesterase inhibitor indicated for the treatment of mild, moderate, and severe dementia of the Alzheimer’s type. (1)
DOSAGE AND ADMINISTRATION
- The recommended starting dosage is 5 mg/day. After 4 to 6 weeks, the dosage may be increased to the maximum recommended dosage of 10 mg/day. (2.1)
- If a patient has been on 5 mg/day oral donepezil for at least 4-6 weeks or on 10 mg/day of oral donepezil, the recommended starting dosage is 10 mg/day. (2.2)
- Administer ADLARITY as one transdermal system applied to the skin once weekly. (2.3)
DOSAGE FORMS AND STRENGTHS
- Transdermal System: 5 mg/day and 10 mg/day (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Application-site reactions have occurred with ADLARITY. Allergic contact dermatitis should be suspected if application-site reactions spread beyond the size of the transdermal system, if there is evidence of a more intense local reaction (e.g., increasing erythema, edema, papules, vesicles), and/or if symptoms do not significantly improve within 48 hours after transdermal system removal. (5.1)
- Cholinesterase inhibitors, including ADLARITY, are likely to exaggerate succinylcholine-type muscle relaxation during anesthesia. (5.2)
- Cholinesterase inhibitors, including ADLARITY, may have vagotonic effects on the sinoatrial and atrioventricular nodes manifesting as bradycardia or heart block. (5.3)
- ADLARITY can cause vomiting. Patients should be observed closely at initiation of treatment and after dose increases. (5.4)
- Patients should be monitored closely for symptoms of active or occult gastrointestinal (GI) bleeding, especially those at increased risk for developing ulcers. (5.5)
- Cholinomimetics, including ADLARITY, may cause bladder outflow obstructions. (5.6)
- Cholinomimetics, including ADLARITY, are believed to have some potential to cause generalized convulsions. (5.7)
- Cholinesterase inhibitors, including ADLARITY, should be prescribed with caution to patients with a history of asthma or obstructive pulmonary disease. (5.8)
ADVERSE REACTIONS
Most common adverse reactions (greater than 5% with donepezil tablets and twice the placebo rate) are nausea, diarrhea, insomnia, vomiting, muscle cramps, fatigue, and anorexia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Corium, Inc. at 1-800-910-8432 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
- Pregnancy: Based on animal data, may cause fetal harm (8.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Switching to ADLARITY from Donepezil Hydrochloride Tablets or Donepezil Hydrochloride ODT
2.3 Administration Information
2.4 Missed Dose
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Application Site Skin Reactions
5.2 Use During Anesthesia
5.3 Cardiovascular Conditions
5.4 Nausea and Vomiting
5.5 Peptic Ulcer Disease and GI Bleeding
5.6 Genitourinary Conditions
5.7 Neurological Conditions: Seizures
5.8 Pulmonary Conditions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Anticholinergics
7.2 Cholinomimetics and Other Cholinesterase Inhibitors
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Mild to Moderate Alzheimer’s Disease
14.2 Moderate to Severe Alzheimer’s Disease
14.3 Adhesion of ADLARITY Transdermal System
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended starting dosage of ADLARITY is 5 mg/day. After 4 to 6 weeks, the dosage may be increased to the maximum recommended dosage of 10 mg/day. Administer ADLARITY as one transdermal system applied to the skin once weekly [see Dosage and Administration (2.3)].
Doses of the transdermal system higher than the 10 mg/day equivalent have not been evaluated.2.2 Switching to ADLARITY from Donepezil Hydrochloride Tablets or Donepezil Hydrochloride ODT
Patients treated with donepezil hydrochloride 5 mg or 10 mg tablets may be switched to ADLARITY:
• A patient who is being treated with a total daily dose of 5 mg of oral donepezil hydrochloride can be switched to the once weekly 5 mg/day ADLARITY transdermal system. If a patient has been on 5 mg oral donepezil hydrochloride for at least 4-6 weeks, the patient may be switched immediately to the once weekly 10 mg/day transdermal system.
• A patient who is being treated with a total daily dose of 10 mg of oral donepezil hydrochloride can be switched to the once weekly 10 mg/day ADLARITY transdermal system.Instruct patients or caregivers to apply the first transdermal system with the last administered oral dose.
2.3 Administration Information
Each ADLARITY transdermal system delivers either 5 mg or 10 mg of donepezil daily for 7 days (one week cycle). At the end of 7 days, the used transdermal system is removed, and a new transdermal system is applied. Only one transdermal system should be applied at a time.
See the Instructions for Use for step-by-step instructions.
Preparation
• Remove one ADLARITY transdermal system from the refrigerator and allow the pouch to reach room temperature before opening.
o Do not use external heat sources to warm ADLARITY.
o Do not apply a cold transdermal system.
o Use within 24 hours of removing from the refrigerator.• Ensure the ADLARITY pouch seal has not been broken. Do not use ADLARITY if the transdermal system is damaged, cut, or altered in any way.
• Select application site:o The recommended application site is the back (avoiding the spine). If needed, the upper buttocks or the upper outer thigh may be used [see Clinical Studies (14.3)]. Use a location that will not be rubbed by tight clothing.
o Do not use the same location of an application site for at least 2 weeks (14 days) after removal of a transdermal system from that location.
o Do not apply to an area on skin where medication, cream, lotion, or powder has recently been applied.
o Do not apply to skin that is red, irritated, or cut.
o Do not shave the site.Application
• Apply ADLARITY to skin immediately after removing from the pouch.
• Apply to clean, dry, intact healthy skin with no to minimal hair (see Preparation).
• Press down firmly for 30 seconds to ensure good contact with skin at the edges of the transdermal system.
• ADLARITY use does not need to be interrupted due to bathing or hot weather. Avoid long exposure to external heat sources (e.g., excessive sunlight, saunas, solariums or heating pads).2.4 Missed Dose
If an ADLARITY transdermal system falls off, or if a dose is missed, apply a new transdermal system immediately and then replace this transdermal system 7 days later to start a new one-week cycle [see Dosage and Administration (2.3)].
-
3 DOSAGE FORMS AND STRENGTHS
ADLARITY is a rectangular transdermal system with rounded corners and a tan colored backing layer. ADLARITY is available in 2 strengths and labeled as either:
• ADLARITY (donepezil transdermal system) 5 mg/day (8.3 cm by 10.8 cm)
• ADLARITY (donepezil transdermal system) 10 mg/day (10.8 cm by 14.4 cm) -
4 CONTRAINDICATIONS
ADLARITY is contraindicated in:
• Patients with known hypersensitivity to donepezil or to piperidine derivatives
• Patients with a history of allergic contact dermatitis with use of ADLARITY [see Warnings and Precautions (5.1)] -
5 WARNINGS AND PRECAUTIONS
5.1 Application Site Skin Reactions
Skin application-site reactions have occurred with ADLARITY [see Adverse Reactions (6.1)]. These reactions are not in themselves an indication of sensitization. However, use of ADLARITY may lead to allergic contact dermatitis [see Contraindications (4)].
Allergic contact dermatitis should be suspected if application-site reactions spread beyond the size of the transdermal system, if there is evidence of a more intense local reaction (e.g., increasing erythema, edema, papules, vesicles), and if symptoms do not significantly improve within 48 hours after transdermal system removal.
5.2 Use During Anesthesia
ADLARITY, as a cholinesterase inhibitor, is likely to exaggerate succinylcholine-type muscle relaxation during anesthesia [see Drug Interactions (7.2)].
5.3 Cardiovascular Conditions
Because of their pharmacological action, cholinesterase inhibitors, including ADLARITY, may have vagotonic effects on the sinoatrial and atrioventricular nodes. This effect may manifest as bradycardia or heart block in patients both with and without known underlying cardiac conduction abnormalities. Syncopal episodes have been reported in association with the use of donepezil.
5.4 Nausea and Vomiting
Donepezil, as a predictable consequence of its pharmacological properties, has been shown to produce diarrhea, nausea, and vomiting [see Adverse Reactions (6.1)].
Although in most cases, these effects have been transient, sometimes lasting one to three weeks, and have resolved during continued use of donepezil, patients should be observed closely at the initiation of treatment and after dose increases.
5.5 Peptic Ulcer Disease and GI Bleeding
Through their primary action, cholinesterase inhibitors, including ADLARITY, may be expected to increase gastric acid secretion due to increased cholinergic activity. Therefore, patients should be monitored closely for symptoms of active or occult gastrointestinal bleeding, especially those at increased risk for developing ulcers (e.g., those with a history of ulcer disease or those receiving concurrent nonsteroidal anti-inflammatory drugs [NSAIDs]). Clinical studies of donepezil tablets in a dose of 5 mg/day to 10 mg/day have shown no increase, relative to placebo, in the incidence of either peptic ulcer disease or gastrointestinal bleeding.
5.6 Genitourinary Conditions
Although not observed in clinical trials of ADLARITY or donepezil tablets, cholinomimetics, including ADLARITY, may cause bladder outflow obstruction.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described below and elsewhere in the labeling:
• Application Site Skin Reactions [see Warnings and Precautions (5.1)]
• Cardiovascular Conditions [see Warnings and Precautions (5.3)]
• Nausea and Vomiting [see Warnings and Precautions (5.4)]
• Peptic Ulcer Disease and GI Bleeding [see Warnings and Precautions (5.5)]
• Genitourinary Conditions [see Warnings and Precautions (5.6)]
• Neurological Conditions: Seizures [see Warnings and Precautions (5.7)]
• Pulmonary Conditions [see Warnings and Precautions (5.8)]6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Donepezil Tablet Studies [see Clinical Studies (14.1, 14.2)]
Donepezil tablets have been administered to over 1,700 individuals during clinical trials worldwide. Approximately 1,200 of these patients have been treated for at least 3 months and more than 1,000 patients have been treated for at least 6 months. Controlled and uncontrolled trials in the United States included approximately 900 patients. In regard to the highest dose of 10 mg/day, this population includes 650 patients treated for 3 months, 475 patients treated for 6 months, and 116 patients treated for over 1 year. The range of patient exposure is from 1 to 1,214 days.
Mild to Moderate Alzheimer’s Disease
Adverse Reactions Leading to Discontinuation
The rates of discontinuation from controlled clinical trials of donepezil tablets due to adverse reactions for the donepezil 5 mg/day treatment groups were comparable to those of placebo treatment groups at approximately 5%. The rate of discontinuation of patients who received 7-day escalations of donepezil tablets from 5 mg/day to 10 mg/day was higher at 13%.
The most common adverse reactions leading to discontinuation, defined as those occurring in at least 2% of patients and at twice or more the incidence seen in placebo patients, are shown in Table 2.
Table 2. Most Common Adverse Reactions Leading to Discontinuation in Patients with Mild to Moderate Alzheimer’s Disease Adverse Reaction
Placebo
(n=355)
%
5 mg/day donepezil tablet (n=350)
%
10 mg/day donepezil tablet (n=315)
%
Nausea
1
1
3
Diarrhea
0
<1
3
Vomiting
<1
<1
2
Most Common Adverse Reactions
The most common adverse reactions, defined as those occurring at a frequency of at least 5% in patients receiving donepezil tablets 10 mg/day and twice the placebo rate, are nausea, diarrhea, insomnia, vomiting, muscle cramp, fatigue, and anorexia. These adverse reactions were often transient, resolving during continued donepezil treatment without the need for dose modification.
There is evidence to suggest that the frequency of these common adverse reactions may be affected by the rate of titration. An open-label study was conducted with 269 patients who received placebo in the 15- and 30-week studies. These patients were titrated to a dose of 10 mg/day over a 6-week period. The rates of common adverse reactions were lower than those seen in patients titrated to donepezil tablets 10 mg/day over one week in the controlled clinical trials and were comparable to those seen in patients on 5 mg/day.
See Table 3 for a comparison of the most common adverse reactions following one- and six-week titration regimens.
Table 3. Comparison of Rates of Adverse Reactions in Mild to Moderate Patients Titrated to 10 mg/day over 1 and 6 Weeks No titration
One week titration
Six-week titration
Adverse
Reaction
Placebo
(n=315)
%
5 mg/day
donepezil tablet
(n=311)
%
10 mg/day
donepezil tablet
(n=315)
%
10 mg/day
donepezil tablet
(n=269)
%
Nausea
6
5
19
6
Diarrhea
5
8
15
9
Insomnia
6
6
14
6
Fatigue
3
4
8
3
Vomiting
3
3
8
5
Muscle cramps
2
6
8
3
Anorexia
2
3
7
3
Table 4 lists adverse reactions that occurred in at least 2% of patients in pooled placebo-controlled trials who received either donepezil tablets 5 mg or 10 mg and for which the rate of occurrence was greater for patients treated with donepezil than with placebo. In general, adverse reactions occurred more frequently in female patients and with advancing age.
Table 4. Adverse Reactions in Pooled Placebo-Controlled Clinical Trials in Mild to Moderate Alzheimer’s Disease Adverse Reaction
Placebo
(n=355)
%
Donepezil Tablet
(n=747)
%
Percent of Patients with any Adverse Reaction
72
74
Nausea
6
11
Diarrhea
5
10
Headache
9
10
Insomnia
6
9
Pain, various locations
8
9
Dizziness
6
8
Accident
6
7
Muscle Cramps
2
6
Fatigue
3
5
Vomiting
3
5
Anorexia
2
4
Ecchymosis
3
4
Abnormal Dreams
0
3
Depression
<1
3
Weight Loss
1
3
Arthritis
1
2
Frequent Urination
1
2
Somnolence
<1
2
Syncope
1
2
Severe Alzheimer’s Disease (Donepezil Tablets 5 mg/day and 10 mg/day)
Donepezil tablets have been administered to over 600 patients with severe Alzheimer’s disease during clinical trials of at least 6 months duration, including three double-blind, placebo-controlled trials, two of which had an open label extension.
Adverse Reactions Leading to Discontinuation
The rates of discontinuation from controlled clinical trials of donepezil tablets due to adverse reactions for the donepezil patients were approximately 12% compared to 7% for placebo patients. The most common adverse reactions leading to discontinuation, defined as those occurring in at least 2% of donepezil patients and at twice or more the incidence seen in placebo, were anorexia (2% vs. 1% placebo), nausea (2% vs. <1% placebo), diarrhea (2% vs. 0% placebo), and urinary tract infection (2% vs. 1% placebo).
Most Common Adverse Reactions
The most common adverse reactions, defined as those occurring at a frequency of at least 5% in patients receiving donepezil tablets and at twice or more the placebo rate, are diarrhea, anorexia, vomiting, nausea, and ecchymosis. These adverse reactions were often transient, resolving during continued donepezil treatment without the need for dose modification.
Table 5 lists adverse reactions that occurred in at least 2% of patients in pooled placebo-controlled trials who received donepezil tablets 5 mg or 10 mg and for which the rate of occurrence was greater for patients treated with donepezil than with placebo.
Table 5. Adverse Reactions in Pooled Controlled Clinical Trials in Severe Alzheimer’s Disease
Adverse Reaction
Placebo
(n=392)
%
Donepezil Tablet
(n=501)
%
Percent of Patients with any Adverse Reaction
73
81
Accident
12
13
Infection
9
11
Diarrhea
4
10
Anorexia
4
8
Vomiting
4
8
Nausea
2
6
Insomnia
4
5
Ecchymosis
2
5
Headache
3
4
Hypertension
2
3
Pain
2
3
Back Pain
2
3
Eczema
2
3
Hallucinations
1
3
Hostility
2
3
Increase in Creatine Phosphokinase
1
3
Nervousness
2
3
Fever
1
2
Chest Pain
<1
2
Confusion
1
2
Dehydration
1
2
Depression
1
2
Dizziness
1
2
Emotional Lability
1
2
Hemorrhage
1
2
Hyperlipemia
<1
2
Personality Disorder
1
2
Somnolence
1
2
Syncope
1
2
Urinary Incontinence
1
2
ADLARITY Transdermal System Study
The ADLARITY clinical development program included an open-label study in 60 healthy subjects who received ADLARITY 5 mg/day for 5 weeks in Period 1 as a titration dose. In Periods 2 and 3, the subjects received either ADLARITY 10 mg/day for 5 weeks or oral donepezil tablet 10 mg/day for 5 weeks in a randomized, crossover fashion. The most common adverse reactions (incidence >3%) occurring in healthy subjects receiving ADLARITY 10 mg/day were headache (15%), application site pruritus (9%), muscle spasms (9%), insomnia (7%), abdominal pain (6%), application site dermatitis (6%), constipation (6%), diarrhea (4%), application site pain (4%), dizziness (4%), abnormal dreams (4%), and skin laceration (4%).
Overall, the adverse reactions reported by healthy volunteers receiving the ADLARITY transdermal system were consistent with those reported by Alzheimer’s patients receiving oral donepezil tablets in clinical trials.
Application Site Reactions
Cases of skin irritation were captured after ADLARITY removal on an investigator-rated skin irritation scale. Skin irritation was observed, including erythema (64.6%), papules (16.0%), and edema (0.4%), following removal of 268 ADLARITY 10 mg/day transdermal systems; none of the ADLARITY transdermal systems were discontinued because of skin irritation. All application site adverse reactions were reported as mild.
In a clinical study investigating the skin sensitizing potential of ADLARITY in 229 healthy subjects, 4 cases of potential sensitization were observed [see Warnings and Precautions (5.1)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of donepezil. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders: hemolytic anemia
Cardiac Disorders: heart block (all types), QTc prolongation, and torsade de pointes
Gastrointestinal Disorders: abdominal pain
Hepatobiliary Disorders: cholecystitis, hepatitis, pancreatitis
Metabolism and Nutritional Disorders: hyponatremia
Musculoskeletal and Connective Tissue Disorders: rhabdomyolysis
Nervous System Disorders: convulsions, neuroleptic malignant syndrome
Psychiatric Disorder: agitation, aggression, confusion, hallucinations
Skin and Subcutaneous Tissue Disorders: rash
-
7 DRUG INTERACTIONS
7.1 Anticholinergics
Because of the mechanism of action, cholinesterase inhibitors, including ADLARITY, have the potential to interfere with the activity of anticholinergic medications.
7.2 Cholinomimetics and Other Cholinesterase Inhibitors
A synergistic effect may be expected when ADLARITY is given concurrently with succinylcholine, similar neuromuscular blocking agents, or cholinergic agonists (e.g., bethanechol) [see Warnings and Precautions (5.2)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risks associated with the use of ADLARITY in pregnant women.
Adverse developmental effects (mortality and decreased body weight) were observed in the offspring of rats administered donepezil during pregnancy at a dose associated with minimal maternal toxicity. This dose was higher than the maximum recommended human dose (MRHD) of ADLARITY [see Data].
In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies are 2% to 4% and 15% to 20%, respectively. The background risks of major birth defects and miscarriage for the indicated population are unknown.
Data
Animal Data
Oral administration of donepezil to pregnant rats and rabbits during the period of organogenesis resulted in no adverse developmental effects. The highest doses (16 and 10 mg/kg/day, respectively) were approximately 16 and 19 times, respectively, the dose of donepezil at the MRHD of ADLARITY (10 mg/day) on a mg/m2 basis.
Oral administration of donepezil (1, 3, 10 mg/kg/day) to rats during late gestation and throughout lactation to weaning resulted in an increase in stillbirths and offspring mortality at the highest dose tested. The higher no-effect dose (3 mg/kg/day) is approximately 3 times the MRHD of ADLARITY on a mg/m2 basis.
8.2 Lactation
Risk Summary
There are no data on the presence of donepezil or its metabolites in human milk, the effects on the breastfed infant, or on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ADLARITY and any potential adverse effects on the breastfed infant from ADLARITY or from the underlying maternal condition.
8.5 Geriatric Use
Alzheimer’s disease is a disorder occurring primarily in individuals over 55 years of age. The mean age of patients enrolled in the clinical studies with donepezil tablets was 73 years; 80% of these patients were between 65 and 84 years old, and 49% of patients were at or above the age of 75. The efficacy and safety data presented in the clinical trials section were obtained from these patients. There were no clinically significant differences in most adverse reactions reported by patient groups ≥ 65 years old and < 65 years old [see Adverse Reactions (6.1)].
-
10 OVERDOSAGE
Because strategies for the management of overdose are continually evolving, it is advisable to remove the ADLARITY transdermal system and contact a Poison Control Center to determine the latest recommendations for the management of an overdose of any drug.
In case of overdose, general supportive measures should be utilized. Overdosage with cholinesterase inhibitors, including ADLARITY, can result in cholinergic crisis characterized by severe nausea, vomiting, salivation, sweating, bradycardia, hypotension, respiratory depression, collapse, and convulsions. Increasing muscle weakness is a possibility and may result in death if respiratory muscles are involved. Tertiary anticholinergics such as atropine may be used as an antidote for donepezil overdosage. Intravenous atropine sulfate titrated to effect is recommended: an initial dose of 1.0 to 2.0 mg IV with subsequent doses based upon clinical response. Atypical responses in blood pressure and heart rate have been reported with other cholinomimetics when co-administered with quaternary anticholinergics such as glycopyrrolate. It is not known whether donepezil and/or its metabolites can be removed by dialysis (hemodialysis, peritoneal dialysis, or hemofiltration).
Dose-related signs of toxicity in animals included reduced spontaneous movement, prone position, staggering gait, lacrimation, clonic convulsions, depressed respiration, salivation, miosis, tremors, fasciculation, and lower body surface temperature.
-
11 DESCRIPTION
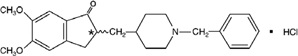
ADLARITY (donepezil transdermal system) contains donepezil hydrocholoride, which is a reversible inhibitor of the enzyme acetylcholinesterase, known chemically as (±)-2,3-dihydro-5,6-dimethoxy-2-[[1-(phenyl-methyl)-4-piperidinyl]methyl]-1H-inden-1-one hydrochloride. Donepezil hydrochloride is commonly referred to in the pharmacological literature as E2020. It has an empirical formula of C24H29NO3·HCl and a molecular weight of 415.96 g/mol. Donepezil hydrochloride is a white crystalline powder and is freely soluble in chloroform, soluble in water and in glacial acetic acid, slightly soluble in ethanol and in acetonitrile, and practically insoluble in ethyl acetate and in n-hexane.
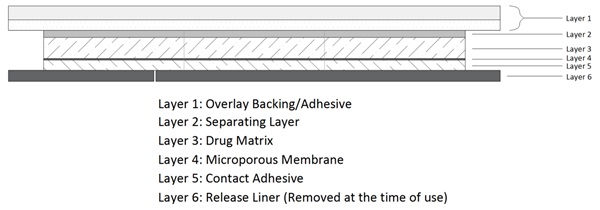
ADLARITY is for transdermal administration of donepezil over a 7-day period. The transdermal system is a rectangular 6-layer laminate containing a tan colored overlay backing/adhesive layer without donepezil, separating layer, drug matrix, membrane, contact adhesive, and a release liner.
Figure 1: Cross Section of ADLARITY
The ADLARITY transdermal system is available in two strengths (5 mg donepezil/day and 10 mg donepezil/day). Each system contains donepezil 88.4 mg or 176.7 mg donepezil, respectively, present as donepezil (15% to 35%) and donepezil HCl (65% to 85%). The inactive ingredients include acrylate copolymer, ascorbyl palmitate, crospovidone, glycerol, lauryl lactate, polypropylene membrane, sodium bicarbonate, sorbitan monolaurate, and triethyl citrate. The transdermal system contains a mixture of donepezil hydrochloride and donepezil base that delivers donepezil base to permeate the skin.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Donepezil is postulated to exert its therapeutic effect by enhancing cholinergic function. This is accomplished by increasing the concentration of acetylcholine through reversible inhibition of its hydrolysis by acetylcholinesterase. There is no evidence that donepezil alters the course of the underlying dementing process.
12.3 Pharmacokinetics
Absorption
In a relative bioavailability study following administration of multiple doses in 60 healthy volunteers, donepezil exposure (i.e., AUCtau and Cmax at steady state) from once weekly ADLARITY 10 mg/day was comparable with that from daily donepezil tablets 10 mg/day. A delay in absorption of donepezil is observed on initial transdermal system (TDS) application followed by a gradual increase in donepezil exposure over the course of the 7-day TDS application. With single-dose administration, maximum plasma donepezil concentrations are achieved in the latter part of the 7-day application period for ADLARITY 5 mg/day. Following multiple dose administration of ADLARITY, steady state is reached within 22 days (after three weeks). Donepezil exposure was dose proportional between ADLARITY 5 mg/day and 10 mg/day.
Donepezil exposure (AUCinf and Cmax) was 14-18% lower, and 21-24% higher, when the transdermal system was applied on the thigh and buttocks, respectively, when compared to application on the back [see Dosage and Administration (2.3)]. Mean plasma donepezil concentration profiles showed transitory increases (up to 60% increase in the partial AUC corresponding to the heat application periods) during heat sessions compared with control conditions [see Dosage and Administration (2.3)].
Distribution
Donepezil is approximately 96% bound to human plasma proteins, mainly to albumins (about 75%) and alpha1-acid glycoprotein (about 21%) over the concentration range of 2-1000 ng/mL. The steady state volume of distribution of donepezil is 12-16 L/kg.
Elimination
Metabolism
Donepezil is metabolized by CYP 450 isoenzymes 2D6 and 3A4 and undergoes glucuronidation. An active metabolite of donepezil, 6-O-desmethyl donepezil, has been reported to inhibit acetylcholinesterase to the same extent as donepezil in vitro. In the relative bioavailability study for ADLARITY, the systemic exposure to 6-O-desmethyl donepezil was less than 0.3% relative to donepezil exposure at steady state.
Excretion
Donepezil is both excreted in the urine intact and extensively metabolized to four major metabolites, two of which are known to be active, and a number of minor metabolites, not all of which have been identified. Following administration of 14C-labeled donepezil, approximately 57% and 15% of the total radioactivity was recovered in urine and feces, respectively, over a period of 10 days, while 28% remained unrecovered, with about 17% of the donepezil dose recovered in the urine as unchanged drug. The mean elimination half-life of donepezil following administration of ADLARITY is about 91 hours, and the mean apparent plasma clearance at steady state (CLss/F) is 0.12 L/hr/kg.
Specific Populations
Patients with Hepatic Impairment
In a study of 10 patients with stable alcoholic cirrhosis, the clearance of donepezil was decreased by 20% relative to 10 healthy age- and sex-matched subjects.
Patients with Renal Impairment
In a study of 11 patients with moderate to severe renal impairment the clearance of donepezil did not differ from 11 age- and sex-matched healthy subjects.
Age
No formal pharmacokinetic study was conducted to examine age-related differences in the pharmacokinetics of donepezil. Population pharmacokinetic analysis suggested that the clearance of donepezil in patients decreases with increasing age. When compared with 65-year old subjects, 90-year old subjects have a 17% decrease in clearance, while 40-year old subjects have a 33% increase in clearance. The effect of age on donepezil clearance may not be clinically significant.
Gender and Race
No specific pharmacokinetic study was conducted to investigate the effects of gender and race on the disposition of donepezil. However, retrospective pharmacokinetic analysis and population pharmacokinetic analysis of plasma donepezil concentrations measured in patients with Alzheimer’s disease indicates that gender and race (Japanese and Caucasians) did not affect the clearance of donepezil to an important degree.
Body Weight
There was a relationship noted between body weight and clearance for donepezil. Over the range of body weight from 50 kg to 110 kg, clearance increased from 7.77 L/h to 14.04 L/h, with a value of 10 L/hr for 70 kg individuals.
Drug Interactions
Effect of Donepezil on the Metabolism of Other Drugs
No in vivo clinical trials have investigated the effect of donepezil on the clearance of drugs metabolized by CYP3A4 (e.g., midazolam) or by CYP2D6 (e.g., imipramine). However, in vitro studies show a low rate of binding to these enzymes (mean Ki about 50-130 μM), that, given the therapeutic plasma concentrations of donepezil (164 nM), indicates little likelihood of interference. Based on in vitro studies, donepezil shows little or no evidence of direct inhibition of CYP2B6, CYP2C8, and CYP2C19 at clinically relevant concentrations.
Whether donepezil has any potential for enzyme induction is not known. Formal pharmacokinetic studies evaluated the potential of donepezil for interaction with theophylline, cimetidine, warfarin, digoxin, and ketoconazole. No effects of donepezil on the pharmacokinetics of these drugs were observed.
Effect of Other Drugs on the Metabolism of Donepezil
Ketoconazole and quinidine, strong inhibitors of CYP450 3A and 2D6, respectively, inhibit donepezil metabolism in vitro. Whether there is a clinical effect of quinidine is not known. In a 7-day crossover study in 18 healthy volunteers, ketoconazole (200 mg once daily) increased mean donepezil (5 mg once daily) concentrations (AUC0-24 and Cmax) by 36%. The clinical relevance of this increase in concentration is unknown. Population pharmacokinetic analysis showed that in the presence of concomitant CYP2D6 inhibitors donepezil AUC was increased by approximately 17% to 20% in Alzheimer’s disease patients taking donepezil 10 mg and 23 mg tablets. This represented an average effect of weak, moderate, and strong CYP2D6 inhibitors.
Inducers of CYP3A (e.g., phenytoin, carbamazepine, dexamethasone, rifampin, and phenobarbital) could increase the rate of elimination of donepezil.
Formal pharmacokinetic studies demonstrated that the metabolism of donepezil is not significantly affected by concurrent administration of digoxin or cimetidine.
An in vitro study showed that donepezil was not a substrate of P-glycoprotein.
Drugs Highly Bound to Plasma Proteins
Drug displacement studies have been performed in vitro between this highly bound drug (96%) and other drugs such as furosemide, digoxin, and warfarin. Donepezil at concentrations of 0.3-10 μg/mL did not affect the binding of furosemide (5 μg/mL), digoxin (2 ng/mL), and warfarin (3 μg/mL) to human albumin. Similarly, the binding of donepezil to human albumin was not affected by furosemide, digoxin, and warfarin.
12.5 Pharmacogenomics
Examination of the effect of CYP2D6 genotype in Alzheimer’s patients showed differences in clearance values among CYP2D6 genotype subgroups. When compared to the extensive metabolizers, poor metabolizers had a 31.5% slower clearance and ultra-rapid metabolizers had a 24% faster clearance.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No evidence of carcinogenic potential was obtained in an 88-week carcinogenicity study of donepezil conducted in mice at oral doses up to 180 mg/kg/day (approximately 90 times the maximum recommended human dose [MRHD] of 10 mg/day on a mg/m2 basis), or in a 104-week carcinogenicity study in rats at oral doses up to 30 mg/kg/day (approximately 30 times the MRHD on a mg/m2 basis).
Mutagenesis
Donepezil was negative in in vitro (bacterial mutagenicity, mouse lymphoma tk, and chromosomal aberration) and in vivo (mouse micronucleus) assays.
Impairment of Fertility
Donepezil had no effect on fertility in rats at oral doses up to 10 mg/kg/day (approximately 10 times the MRHD of 10 mg/day on a mg/m2 basis) when administered to males and females prior to and during mating and continuing in females through implantation.
13.2 Animal Toxicology and/or Pharmacology
In an acute dose neurotoxicity study in female rats, oral administration of donepezil and memantine in combination resulted in increased incidence, severity, and distribution of neurodegeneration compared with memantine alone. The no-effect levels of the combination were associated with clinically relevant plasma donepezil and memantine levels.
The relevance of this finding to humans is unknown. -
14 CLINICAL STUDIES
The efficacy of ADLARITY is based on a relative bioavailability study in healthy subjects comparing ADLARITY transdermal system to ARICEPT tablets [see Clinical Pharmacology (12.3)]. The clinical studies described below were conducted using donepezil tablets.
14.1 Mild to Moderate Alzheimer’s Disease
The effectiveness of donepezil as a treatment for mild to moderate Alzheimer’s disease is demonstrated by the results of two randomized, double-blind, placebo-controlled clinical investigations of donepezil tablets in patients with Alzheimer’s disease (diagnosed by NINCDS and DSM III-R criteria, Mini-Mental State Examination ≥ 10 and ≤ 26 and Clinical Dementia Rating of 1 or 2). The mean age of patients participating in these donepezil trials was 73 years with a range of 50 to 94. Approximately 62% of patients were women and 38% were men. The racial distribution was white 95%, black 3%, and other races 2%.
The higher dose of 10 mg did not provide a statistically significantly greater clinical benefit than 5 mg. There is a suggestion, however, based upon order of group mean scores and dose trend analyses of data from these clinical trials, that a daily dose of 10 mg of donepezil might provide additional benefit for some patients. Accordingly, whether or not to employ a dose of 10 mg is a matter of prescriber and patient preference.
Study Outcome Measures
In each study, the effectiveness of treatment with donepezil was evaluated using a dual outcome assessment strategy.
The ability of donepezil to improve cognitive performance was assessed with the cognitive subscale of the Alzheimer’s Disease Assessment Scale (ADAS-cog), a multi-item instrument that has been extensively validated in longitudinal cohorts of Alzheimer’s disease patients. The ADAS-cog examines selected aspects of cognitive performance including elements of memory, orientation, attention, reasoning, language, and praxis. The ADAS-cog scoring range is from 0 to 70, with higher scores indicating greater cognitive impairment. Elderly normal adults may score as low as 0 or 1, but it is not unusual for non-demented adults to score slightly higher.
The patients recruited as participants in each study had mean scores on the ADAS-cog of approximately 26 points, with a range from 4 to 61. Experience based on longitudinal studies of ambulatory patients with mild to moderate Alzheimer’s disease suggest that scores on the ADAS-cog increase (worsen) by 6-12 points per year. However, smaller changes may be seen in patients with very mild or very advanced disease since the ADAS-cog is not uniformly sensitive to change over the course of the disease. The annualized rate of decline in the placebo patients participating in donepezil trials was approximately 2 to 4 points per year.
The ability of donepezil to produce an overall clinical effect was assessed using a Clinician’s Interview-Based Impression of Change that required the use of caregiver information, the CIBIC-plus. The CIBIC-plus is not a single instrument and is not a standardized instrument like the ADAS-cog. Clinical trials for investigational drugs have used a variety of CIBIC formats, each different in terms of depth and structure.
As such, results from a CIBIC-plus reflect clinical experience from the trial or trials in which it was used and cannot be compared directly with the results of CIBIC-plus evaluations from other clinical trials. The CIBIC- plus used in donepezil trials was a semi-structured instrument that was intended to examine four major areas of patient function: General, Cognitive, Behavioral, and Activities of Daily Living. It represents the assessment of a skilled clinician based upon his/her observations at an interview with the patient, in combination with information supplied by a caregiver familiar with the behavior of the patient over the interval rated. The CIBIC- plus is scored as a seven-point categorical rating, ranging from a score of 1, indicating “markedly improved,” to a score of 4, indicating “no change” to a score of 7, indicating “markedly worse.” The CIBIC-plus has not been systematically compared directly to assessments not using information from caregivers (CIBIC) or other global methods.
Thirty-Week Study
In a study of 30 weeks duration, 473 patients were randomized to receive single daily doses of placebo, 5 mg/day or 10 mg/day of donepezil tablets. The 30-week study was divided into a 24‑week double-blind active treatment phase followed by a 6-week single-blind placebo washout period. The study was designed to compare 5 mg/day or 10 mg/day fixed doses of donepezil tablets to placebo. However, to reduce the likelihood of cholinergic effects, the 10 mg/day treatment was started following an initial 7-day treatment with 5 mg/day doses.
Effects on the ADAS-cog
Figure 2 illustrates the time course for the change from baseline in ADAS-cog scores for all three dose groups over the 30 weeks of the study. After 24 weeks of treatment, the mean differences in the ADAS-cog change scores for donepezil treated patients compared to the patients on placebo were 2.8 and 3.1 points for the 5 mg/day and 10 mg/day treatments, respectively. These differences were statistically significant. While the treatment effect size may appear to be slightly greater for the 10 mg/day treatment, there was no statistically significant difference between the two active treatments.
Following 6 weeks of placebo washout, scores on the ADAS-cog for both the donepezil treatment groups were indistinguishable from those patients who had received only placebo for 30 weeks. This suggests that the beneficial effects of donepezil abate over 6 weeks following discontinuation of treatment and do not represent a change in the underlying disease. There was no evidence of a rebound effect 6 weeks after abrupt discontinuation of therapy.
Figure 2. Time-course of the Change from Baseline in ADAS-cog Score for Patients Completing 24 Weeks of Treatment
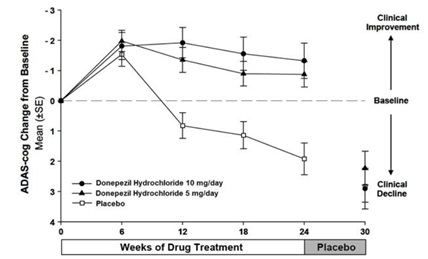
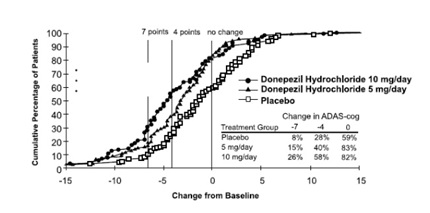
Figure 3 illustrates the cumulative percentages of patients from each of the three treatment groups who had attained the measure of improvement in ADAS-cog score shown on the X-axis. Three change scores (7-point and 4-point reductions from baseline or no change in score) have been identified for illustrative purposes, and the percent of patients in each group achieving that result is shown in the inset table.
The curves demonstrate that both patients assigned to placebo and donepezil have a wide range of responses, but that the active treatment groups are more likely to show greater improvements. A curve for an effective treatment would be shifted to the left of the curve for placebo, while an ineffective or deleterious treatment would be superimposed upon or shifted to the right of the curve for placebo.
Figure 3. Cumulative Percentage of Patients Completing 24 Weeks of Double-blind Treatment with Specified Changes from Baseline ADAS-cog Scores. The Percentages of Randomized Patients who Completed the Study were: Placebo 80%, 5 mg/day 85%, and 10 mg/day 68%.
Effects on the CIBIC-plus
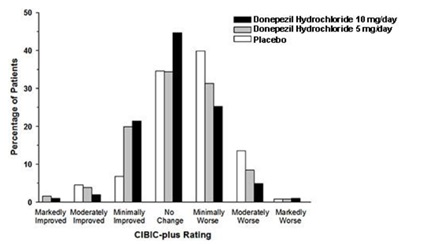
Figure 4 is a histogram of the frequency distribution of CIBIC-plus scores attained by patients assigned to each of the three treatment groups who completed 24 weeks of treatment. The mean drug-placebo differences for these groups of patients were 0.35 points and 0.39 points for 5 mg/day and 10 mg/day of donepezil, respectively. These differences were statistically significant. There was no statistically significant difference between the two active treatments.
Figure 4. Frequency Distribution of CIBIC-plus Scores at Week 24
Fifteen-Week Study
In a study of 15 weeks duration, patients were randomized to receive single daily doses of placebo or either 5 mg/day or 10 mg/day of donepezil tablets for 12 weeks, followed by a 3-week placebo washout period. As in the 30-week study, to avoid acute cholinergic effects, the 10 mg/day treatment followed an initial 7-day treatment with 5 mg/day doses.
Effects on the ADAS-cog
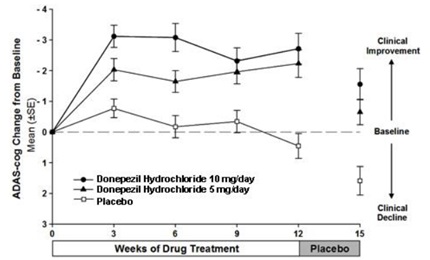
Figure 5 illustrates the time course of the change from baseline in ADAS-cog scores for all three dose groups over the 15 weeks of the study. After 12 weeks of treatment, the differences in mean ADAS-cog change scores for the donepezil treated patients compared to the patients on placebo were 2.7 and 3.0 points each, for the 5 and 10 mg/day donepezil treatment groups, respectively. These differences were statistically significant. The effect size for the 10 mg/day group may appear to be slightly larger than that for 5 mg/day. However, the differences between active treatments were not statistically significant.
Figure 5. Time-course of the Change from Baseline in ADAS-cog Score for Patients Completing the 15-week Study
Following 3 weeks of placebo washout, scores on the ADAS-cog for both the donepezil treatment groups increased, indicating that discontinuation of donepezil resulted in a loss of its treatment effect. The duration of this placebo washout period was not sufficient to characterize the rate of loss of the treatment effect, but the 30-week study (see above) demonstrated that treatment effects associated with the use of donepezil abate within 6 weeks of treatment discontinuation.
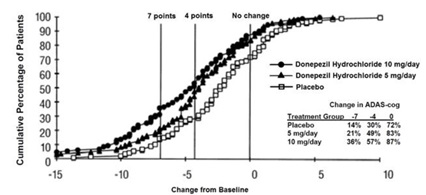
Figure 6 illustrates the cumulative percentages of patients from each of the three treatment groups who attained the measure of improvement in ADAS-cog score shown on the X-axis. The same three change scores (7-point and 4-point reductions from baseline or no change in score) as selected for the 30-week study have been used for this illustration. The percentages of patients achieving those results are shown in the inset table.
As observed in the 30-week study, the curves demonstrate that patients assigned to either placebo or to donepezil have a wide range of responses, but that the donepezil treated patients are more likely to show greater improvements in cognitive performance.
Figure 6. Cumulative Percentage of Patients with Specified Changes from Baseline ADAS-cog Scores. The Percentages of Randomized Patients Within Each Treatment Group Who Completed the Study Were: Placebo 93%, 5 mg/day 90%, and 10 mg/day 82%.
Effects on the CIBIC-plus
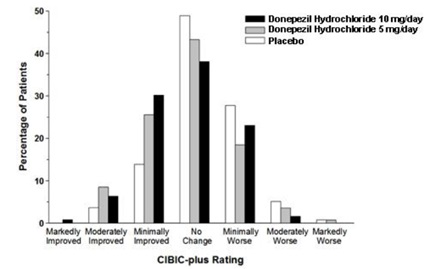
Figure 7 is a histogram of the frequency distribution of CIBIC-plus scores attained by patients assigned to each of the three treatment groups who completed 12 weeks of treatment. The differences in mean scores for donepezil treated patients compared to the patients on placebo at Week 12 were 0.36 and 0.38 points for the 5 mg/day and 10 mg/day treatment groups, respectively. These differences were statistically significant.
Figure 7. Frequency Distribution of CIBIC-plus Scores at Week 12
In both studies, patient age, sex, and race were not found to predict the clinical outcome of donepezil treatment.
14.2 Moderate to Severe Alzheimer’s Disease
The effectiveness of donepezil in the treatment of patients with moderate to severe Alzheimer’s disease was established in studies employing doses of 10 mg/day.
Swedish 6 Month Study (10 mg/day)
The effectiveness of donepezil as a treatment for severe Alzheimer’s disease is demonstrated by the results of a randomized, double-blind, placebo-controlled clinical study conducted in Sweden (6-month study) in patients with probable or possible Alzheimer’s disease diagnosed by NINCDS-ADRDA and DSM-IV criteria, MMSE: range of 1-10. Two hundred and forty-eight (248) patients with severe Alzheimer’s disease were randomized to donepezil tablets or placebo. For patients randomized to donepezil, treatment was initiated at 5 mg once daily for 28 days and then increased to 10 mg once daily. At the end of the 6-month treatment period, 90.5% of the donepezil treated patients were receiving the 10 mg/day dose. The mean age of patients was 84.9 years, with a range of 59 to 99. Approximately 77% of patients were women, and 23% were men. Almost all patients were Caucasian. Probable Alzheimer’s disease was diagnosed in the majority of the patients (83.6% of donepezil treated patients and 84.2% of placebo treated patients).
Study Outcome Measures
The effectiveness of treatment with donepezil was determined using a dual outcome assessment strategy that evaluated cognitive function using an instrument designed for more impaired patients and overall function through caregiver-rated assessment. This study showed that patients on donepezil experienced significant improvement on both measures compared to placebo.
The ability of donepezil to improve cognitive performance was assessed with the Severe Impairment Battery (SIB). The SIB, a multi-item instrument, has been validated for the evaluation of cognitive function in patients with moderate to severe dementia. The SIB evaluates selective aspects of cognitive performance, including elements of memory, language, orientation, attention, praxis, visuospatial ability, construction, and social interaction. The SIB scoring range is from 0 to 100, with lower scores indicating greater cognitive impairment.
Daily function was assessed using the Modified Alzheimer’s Disease Cooperative Study Activities of Daily Living Inventory for Severe Alzheimer’s Disease (ADCS-ADL-severe). The ADCS-ADL-severe is derived from the Alzheimer’s Disease Cooperative Study Activities of Daily Living Inventory, which is a comprehensive battery of ADL questions used to measure the functional capabilities of patients. Each ADL item is rated from the highest level of independent performance to complete loss. The ADCS-ADL-severe is a subset of 19 items, including ratings of the patient’s ability to eat, dress, bathe, use the telephone, get around (or travel), and perform other activities of daily living; it has been validated for the assessment of patients with moderate to severe dementia. The ADCS-ADL-severe has a scoring range of 0 to 54, with the lower scores indicating greater functional impairment. The investigator performs the inventory by interviewing a caregiver, in this study a nurse staff member, familiar with the functioning of the patient.
Effects on the SIB
Figure 8 shows the time course for the change from baseline in SIB score for the two treatment groups over the 6 months of the study. At 6 months of treatment, the mean difference in the SIB change scores for donepezil treated patients compared to patients on placebo was 5.9 points. Donepezil treatment was statistically significantly superior to placebo.
Figure 8. Time Course of the Change from Baseline in SIB Score for Patients Completing 6 Months of Treatment
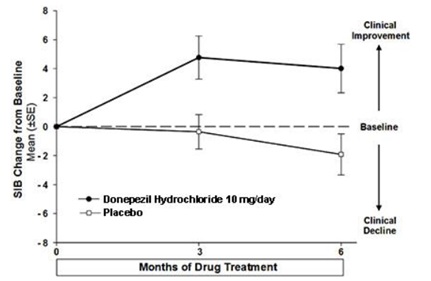
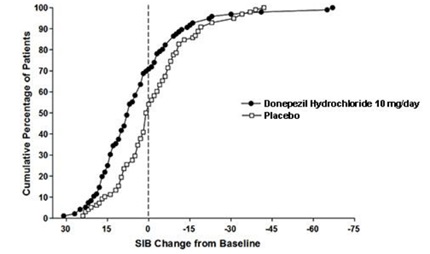
Figure 9 illustrates the cumulative percentages of patients from each of the two treatment groups who attained the measure of improvement in SIB score shown on the X-axis. While patients assigned both to donepezil and to placebo have a wide range of responses, the curves show that the donepezil group is more likely to show a greater improvement in cognitive performance.
Figure 9. Cumulative Percentage of Patients Completing 6 Months of Double-blind Treatment with Particular Changes from Baseline in SIB Scores
Effects on the ADCS-ADL-severe
Figure 10 illustrates the time course for the change from baseline in ADCS-ADL-severe scores for patients in the two treatment groups over the 6 months of the study. After 6 months of treatment, the mean difference in the ADCS-ADL-severe change scores for donepezil treated patients compared to patients on placebo was 1.8 points. Donepezil treatment was statistically significantly superior to placebo.
Figure 10. Time Course of the Change from Baseline in ADCS-ADL-Severe Score for Patients Completing 6 Months of Treatment
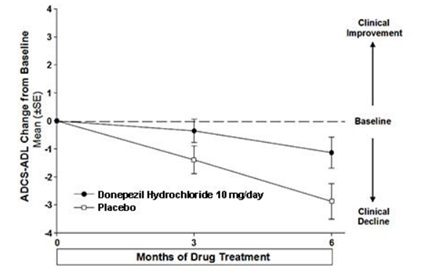
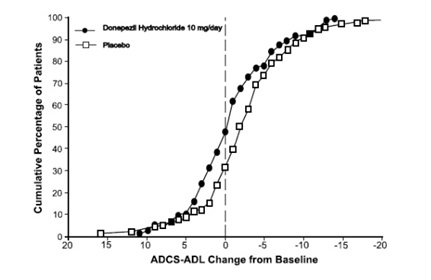
Figure 11 shows the cumulative percentages of patients from each treatment group with specified changes from baseline ADCS-ADL-severe scores. While both patients assigned to donepezil and placebo have a wide range of responses, the curves demonstrate that the donepezil group is more likely to show a smaller decline or an improvement.
Figure 11. Cumulative Percentage of Patients Completing 6 Months of Double-blind Treatment with Particular Changes from Baseline in ADCS-ADL-Severe Scores.
Japanese 24-Week Study (10 mg/day)
In a study of 24 weeks duration conducted in Japan, 325 patients with severe Alzheimer’s disease were randomized to doses of 5 mg/day or 10 mg/day of donepezil tablets, administered once daily, or placebo. Patients randomized to treatment with donepezil were to achieve their assigned doses by titration, beginning at 3 mg/day, and extending over a maximum of 6 weeks. Two hundred and forty-eight (248) patients completed the study, with similar proportions of patients completing the study in each treatment group. The primary efficacy measures for this study were the SIB and CIBIC-plus.
At 24 weeks of treatment, statistically significant treatment differences were observed between the 10 mg/day dose of donepezil and placebo on both the SIB and CIBIC-plus. The 5 mg/day dose of donepezil showed a statistically significant superiority to placebo on the SIB, but not on the CIBIC-plus.
14.3 Adhesion of ADLARITY Transdermal System
Based on a clinical study in 85 subjects, each wearing one ADLARITY 5 mg/day on the back, 80 transdermal systems (94%) exhibited 80% or greater surface area adhesion at all timepoints evaluated (every 12 hours) throughout the 168-hour wear period. Based on a clinical study in 85 subjects, each wearing an ADLARITY 10 mg/day for 168 hours on the back for 4 consecutive weeks, 307 of 338 transdermal systems (91%) exhibited 80% or greater surface area adhesion at all timepoints evaluated. No full detachments were seen for any transdermal system applied. In a separate study, which assessed wear at different application sites (back, thigh, and buttock), at least 85% of the transdermal systems applied at every site exhibited 80% or greater surface area adhesion for the duration of wear. One full detachment on the back was noted in the study.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
ADLARITY is supplied in cartons, with each carton containing four transdermal systems that are individually packaged in sealed pouches.
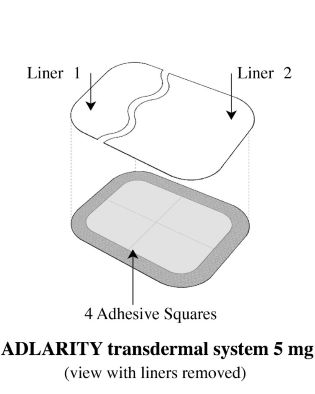
• The ADLARITY 5 mg/day adhesive transdermal system is rectangular in shape, with rounded corners, measuring 8.3 cm by 10.8 cm, and containing 97 mg of donepezil hydrochloride. Each transdermal system has a tan colored backing with the ADLARITY (donepezil transdermal system) logo and 5 mg/day printed in black on the non-adhesive side. ADLARITY 5 mg/day is available in a carton of 4 (NDC 65038-055-03).
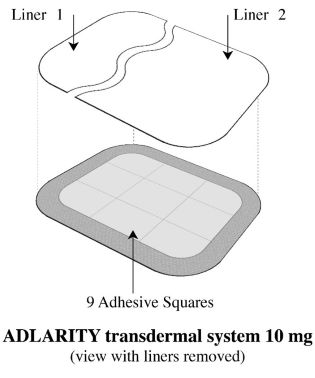
• The ADLARITY 10 mg/day adhesive transdermal system is rectangular in shape, with rounded corners, measuring 10.8 cm by 14.4 cm, and containing 194 mg of donepezil hydrochloride. Each transdermal system has a tan colored backing with the ADLARITY (donepezil transdermal system) logo and 10 mg/day printed in black on the non-adhesive side. ADLARITY 10 mg/day is available in a carton of 4 (NDC 65038-056-03).
16.2 Storage and Handling
Store ADLARITY in the refrigerator between 2°C to 8°C (36°F to 46°F). Do not freeze. Allow the pouch to reach room temperature before opening and removing the new transdermal system for application [see Dosage and Administration (2.3)]. Keep ADLARITY in the individually sealed pouch until use. Used transdermal systems should be folded with the adhesive surfaces pressed together and discarded in the trash. Do not flush used transdermal systems down the toilet.
-
17 PATIENT COUNSELING INFORMATION
Advise patient or caregiver to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Administration Instructions
Inform patients or caregivers of the importance of applying the correct dose on the correct body site. Instruct patients or caregivers to apply the transdermal system to clean, dry, intact, healthy skin with minimal hair (sparse, fine hair is okay) and to rotate the application site in order to minimize skin irritation. The same exact body location should not be used within 14 days, although another part of the same anatomic site (e.g., the back) may be used in consecutive weeks. The previous week’s transdermal system must be removed before applying a new transdermal system to a different skin location. Prior to application, allow the pouch to reach room temperature before opening. ADLARITY should be replaced every 7 days. Only 1 transdermal system should be worn at a time.
Instruct patients or caregivers to avoid exposing the transdermal system to external heat sources (excessive sunlight, saunas, solariums, heat pads) for long periods of time [see Dosage and Administration (2.3)].
Missed Dose
Instruct patients who have missed a dose to apply a new transdermal system immediately and then replace the new transdermal system 7 days later at the usual application time to start a new cycle [see Dosage and Administration (2.4)]. Instruct patients to not apply 2 transdermal systems to make up for a missed application.
Inform the patients or caregivers to contact a physician for titration instructions if treatment has been interrupted.
Discarding Used Transdermal Systems
Instruct patients or caregivers to fold the transdermal system in half after use and discard it in the trash, out of the reach and sight of children and pets. Inform patients or caregivers that drug still remains in the transdermal system after 7-day usage and that used transdermal systems should not be flushed down the toilet. Instruct patients or caregivers to avoid eye contact and to wash their hands after handling the transdermal system. In case of accidental contact with the eyes, or if their eyes become red after handling the transdermal system, advise to rinse immediately with plenty of water and to seek medical advice if symptoms do not resolve.
Skin Reactions
Inform patients or caregivers about the potential for allergic contact dermatitis reactions to occur. Instruct patients or caregivers to inform a healthcare provider if site reactions spread beyond the application site of the transdermal system, if there is evidence of an intense local reaction (e.g., increasing erythema, edema, papules, vesicles), and/or if symptoms do not significantly improve within 48 hours after transdermal system removal [see Warnings and Precautions (5.1)].
Adverse Reactions
Advise patients and caregivers that ADLARITY may cause nausea, diarrhea, insomnia, vomiting, muscle cramps, fatigue, and decreased appetite [see Warnings and Precautions (5.4) and Adverse Reactions (6.1)].
Pregnancy
Advise patients to notify their healthcare provider if they are pregnant or plan to become pregnant [see Use in Specific Populations (8.1)].
Manufactured and distributed by: Corium, Inc., Grand Rapids, MI 49512
-
PATIENT INFORMATION
ADLARITY® (Ad-lare-it-ee)
(donepezil transdermal system)
What is ADLARITY?
- ADLARITY is a prescription medicine used to treat mild, moderate, and severe dementia of the Alzheimer’s type.
- It is not known if ADLARITY is safe and effective in children.
Do not use ADLARITY if you:
- are allergic to donepezil or to medicines that contain piperidines. See the end of this Patient Information leaflet for a complete list of ingredients in ADLARITY.
- have had a skin reaction called allergic contact dermatitis to ADLARITY
Ask your healthcare provider if you are not sure if you should use the ADLARITY transdermal system.
Before using ADLARITY, tell your healthcare provider about all of your medical conditions, including if you:
- have any heart problems, including problems with irregular, slow, or fast heartbeats.
- have stomach ulcers.
- have problems passing urine.
- have seizures.
- have asthma or other lung problems.
- are pregnant or plan to become pregnant. It is not known if ADLARITY will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if ADLARITY passes into your breast milk.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. ADLARITY may affect the way other medicines work and other medicines may affect how ADLARITY works.
Especially tell your healthcare provider if you take medicines called nonsteroidal anti-inflammatory drugs (NSAIDs). Ask your healthcare provider if you are not sure if any of your medicines are NSAIDs. Taking NSAIDs and ADLARITY together may make you more likely to get stomach ulcers.
ADLARITY taken with certain medicines used for anesthesia may cause side effects. Tell your healthcare provider or dentist that you use the ADLARITY transdermal system before you have:
- surgery
- medical procedures
- dental surgery or procedures
Know the medicines that you take. Keep a list of all of your medicines. Show it to your healthcare provider before you start a new medicine.
How should I use ADLARITY?
See the Instructions for Use at the end of this Patient Information leaflet for step-by-step instructions on how to apply, remove, and throw away (dispose of) ADLARITY.
- Use ADLARITY exactly how your healthcare provider tells you to use it.
- ADLARITY is for skin use only.
- Apply 1 ADLARITY transdermal system at a time to your skin 1 time weekly (every 7 days).
- Apply to clean, dry, intact skin with little to no hair.
- If your ADLARITY transdermal system falls off, or if you miss a dose of ADLARITY, apply a new transdermal system right away. You should remove your transdermal system 7 days later.
- If you stop using ADLARITY, call your healthcare provider for instructions before you start using ADLARITY again.
- In case of overdose, get medical help or contact a live poison help line right away.
What should I avoid while using ADLARITY?
- Do not touch your eyes after you touch the ADLARITY transdermal system. In case of accidental contact with your eyes, or if your eyes become red after handling the transdermal system, rinse your eyes right away with water and get medical help if symptoms do not go away.
- Avoid exposure to heat sources, such as excessive sunlight, saunas, sunrooms, or heating pads, for long periods of time. Too much medicine could be absorbed into your body.
What are the possible side effects of ADLARITY?
ADLARITY may cause serious side effects, including:
-
Skin reactions. Skin reactions that include redness and itching may happen at the application site when using ADLARITY. Stop using ADLARITY and call your healthcare provider if you get any of these skin reactions and they do not get better within 2 days (48 hours) after the transdermal system is removed:
- increased redness or swelling
- peeling or blistering of the skin
- spreading beyond the application site
- Slow heartbeat and fainting. Call your healthcare provider right away if you feel faint or lightheaded while using ADLARITY.
-
More stomach acid. This increases the chance of ulcers and bleeding. The risk is higher for some people, such as those who have had ulcers or take NSAIDs. Call your healthcare provider right away if you have any of these symptoms:
- heartburn or stomach pain that is new or does not go away.
- nausea or vomiting, blood in your vomit, or dark vomit that looks like coffee grounds.
- bowel movements or stools that look like black tar.
- Problems passing urine. Call your healthcare provider right away if you have problems passing urine while using ADLARITY.
- Seizures. Call your healthcare provider right away if you have seizures while using ADLARITY.
- Worsening of lung problems in people with asthma or other lung disease. Call your healthcare provider if you have new or worsening lung problems.
The most common side effects of donepezil, the medicine in ADLARITY, are:
- nausea
- diarrhea
- not sleeping well
- vomiting
- muscle cramps
- feeling tired
- not wanting to eat
These are not all of the possible side effects of ADLARITY.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA- 1088.
How should I store ADLARITY?
- Store ADLARITY in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Do not store ADLARITY in the freezer.
- Keep ADLARITY in the sealed pouch until ready for use.
Keep ADLARITY and all medicines out of the reach of children.
General information about the safe and effective use of ADLARITY.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ADLARITY for a condition for which it was not prescribed. Do not give ADLARITY to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider for information about ADLARITY that is written for health professionals.
What are the ingredients in ADLARITY?
Active ingredient: donepezil hydrochloride
Inactive ingredients:acrylate copolymer, ascorbyl palmitate, crospovidone, glycerol, lauryl lactate, polypropylene membrane, sodium bicarbonate, sorbitan monolaurate, and triethyl citrate.
ADLARITY is a registered trademark of Corium, Inc.
Manufactured and distributed by: Corium, Inc., Grand Rapids, MI 49512
Rx Only
© 2022 Corium, Inc.
For more information, go to www.ADLARITY.com, or call 1-800-910-8432.
This Patent Information has been approved by the U.S. Food and Drug Administration Issued: 3/2022
-
INSTRUCTIONS FOR USE
ADLARITY® (Ad-lare-it-ee)
(donepezil transdermal system)
Read this Instructions for Use before you use ADLARITY for the first time and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition and treatment. Talk to your healthcare provider or pharmacist about how to use the ADLARITY transdermal system.
Important information you need to know before using ADLARITY:
- Use the ADLARITY transdermal system exactly as your doctor tells you to.
- Wear only 1 transdermal system at a time.
- Each ADLARITY transdermal system should be worn continuously for 7 days.
- The ADLARITY transdermal system should be worn during showers or baths.
- Each ADLARITY transdermal system should be replaced after 7 days. Choose a convenient time on a specific day of the week that works best for the regular application of the ADLARITY transdermal system, such as every Saturday morning.
- Do not apply a new transdermal system to the same skin area for at least 2 weeks (14 days) after removal to decrease the chance of skin irritation. For example, a new transdermal system can be placed on the same body location, such as the back, but should not be placed on the same area of the back for at least 2 weeks.
- Remove the old transdermal system before applying the new transdermal system to a different skin site. You can write down the date and site of application to remind you when to remove and replace the transdermal system.
- Avoid exposure to heat sources such as excessive sunlight, saunas, sunrooms, or heating pads for long periods of time. Too much medicine could be absorbed into your body.
- Use ADLARITY within 24 hours after removal from the refrigerator.
- Do not apply a cold transdermal system. Do not use external heat sources, such as a microwave, hair dryer, heating pad or direct sunlight, to warm the transdermal system.
The ADLARITY transdermal system
- ADLARITY is a tan rectangular transdermal system that comes in 2 strengths: 5 mg and 10 mg.
- ADLARITY transdermal system is supplied in a carton containing 4 individual transdermal systems of the same strength (4 week supply). Each transdermal system is sealed inside a pouch protecting it until you are ready to apply it.
- ADLARITY is applied 1 time weekly (every 7 days).

How to store ADLARITY
- Store ADLARITY in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Do not store ADLARITY in the freezer.
- Keep ADLARITY in the sealed pouch until ready for use.
- Keep ADLARITY and all medicines out of the reach of children.
How to use ADLARITY
Step 1: Where to apply the ADLARITY transdermal system
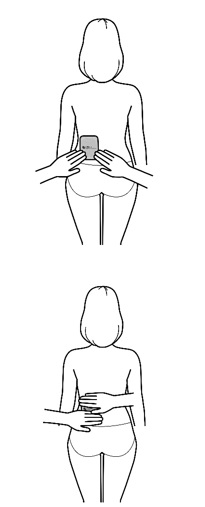
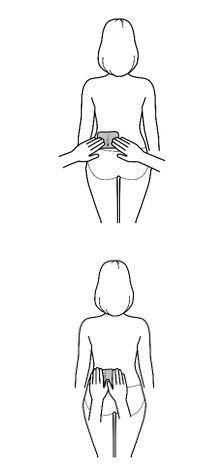
Important: Wear only 1 transdermal system at a time.
- Check the expiration date.
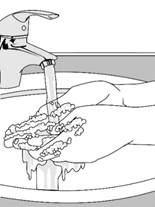
- Wash your hands with soap and water before handling the transdermal system.
- Choose a clean and dry area of skin, where no cream, lotion, or powder has recently been applied.
- Choose skin without cuts or scrapes and hairless or nearly hairless. If there is hair at the skin site, do not shave as this may increase risk of skin irritation. Use scissors to clip hair as close to the skin as possible, before applying the transdermal system.
- Apply the transdermal system to the skin in areas shown with grey shading. The recommended application site is the back. If self-applying the transdermal system or if the transdermal system is not expected to be removed by the patient before the full 7 days, ADLARITY can be placed on the upper buttocks or upper outer thigh.
- Apply the transdermal system in either a vertical (short side of the transdermal system at the top and bottom) or horizontal (short side of the transdermal system on the left and right) direction. If the transdermal system is placed on the buttocks, it should be positioned in a horizontal direction.
- Do not apply the transdermal system to skin that has cream, lotion, or powder on it.
- Do not apply the transdermal system to an area where it can be rubbed off by tight clothing or undergarments.
- Do not apply the transdermal system over skin folds or across the spine.
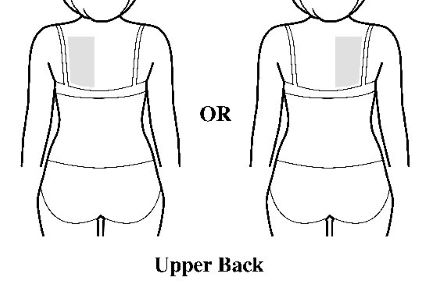
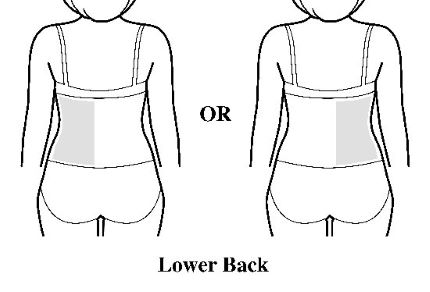
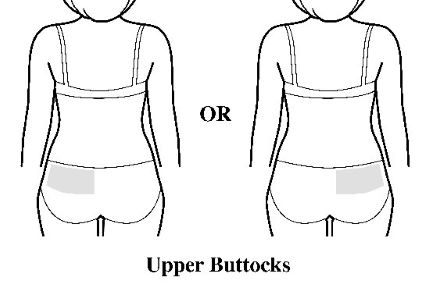
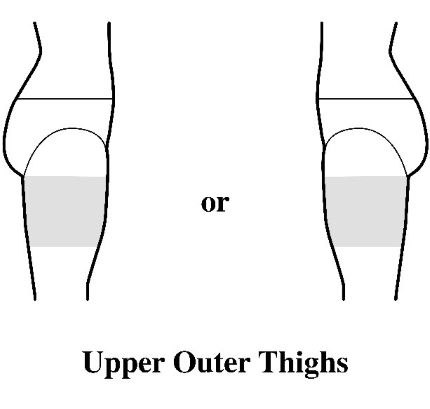
Step 2: Removing the ADLARITY transdermal system from the pouch
- Allow the pouch to reach room temperature before opening and applying.
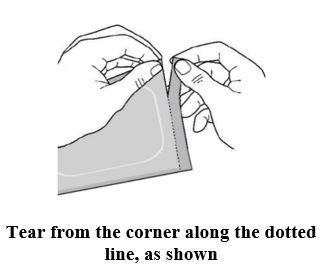
- Make sure seal is not broken.
- Open the pouch by carefully tearing along the dotted line from the corner.
- Remove the ADLARITY transdermal system from the pouch. Apply to skin right away.
- Do not cut, damage, or use parts of the transdermal system. Use the full transdermal system to get required dose.
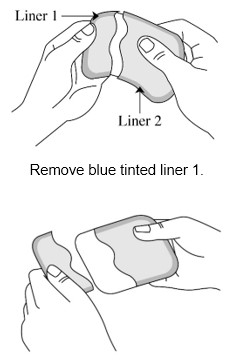
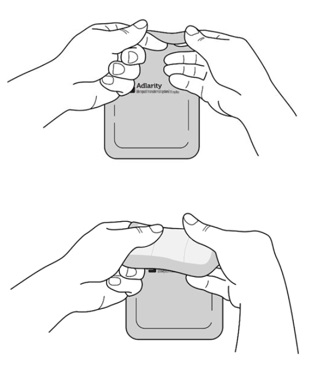
Step 3: Removing Liner 1 from the ADLARITY transdermal system
- Hold the transdermal system so the liner side (blue tinted transparent film) is facing you.
- Bend the transdermal system at the wavy cut in the middle to separate Liner 1 from Liner 2.
- Remove Liner 1. Keep Liner 2attached to the transdermal system (you will remove it in Step 4).
- Avoid touching the sticky (adhesive) side of the transdermal system with your fingers as that can keep the transdermal system from staying on for the full 7 days. Avoid sticking the transdermal system to itself.
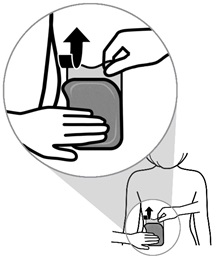
Step 4: Applying the ADLARITY transdermal system and removing Liner 2
- Place the short, sticky (adhesive) side of the transdermal system to the application site against the patient’s skin. Slowly peel away blue tinted liner 2 while applying gentle pressure with the fingers of your other hand to smooth the transdermal system in place.
Vertical Application
Horizaontal Application
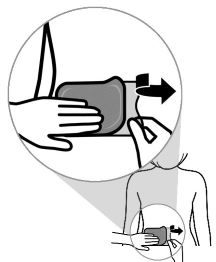
- Smooth the entire transdermal system with your fingers or palm of your hand to avoid creating folds. Press down on the transdermal system firmly for 30 seconds to make sure that the edges stick to the skin.
Remove blue tinted liner 2.
Remove blue tinted liner 2.
- Wash your hands with soap and water after applying the transdermal system.
Step 5: Removing the ADLARITY transdermal system
- Follow the steps below to remove and safely throw away (dispose of) a used ADLARITY transdermal system after 7 days.
- Using the thumbs of both hands, slowly and evenly peel off the ADLARITY transdermal system from the top to the bottom.
- Following this step may lower the chance that pieces of the transdermal system will be left on the skin and cause irritation at the application site.
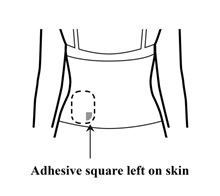
Step 6: Checking the skin for any remaining adhesive squares
- Look at the transdermal system application site for any remaining adhesive squares left on the skin.
- If you see any adhesive squares left on the skin, you can use your fingers to remove them and place them on the sticky side of the used transdermal system.
- Mineral oil or baby oil may be used to wipe off any remaining adhesive.
- Do not use alcohol or nail polish remover to remove remaining adhesive.
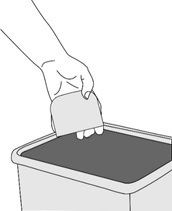
Step 7: Disposing of the used ADLARITY transdermal system
- Fold the transdermal system in half with the sticky (adhesive) side pressed together.
- Throw away (dispose of) the used transdermal system in the household trash.
- Do not reuse transdermal systems.
- Do not flush used transdermal systems down the toilet.
- Wash your hands with soap and water after you throw away (dispose of) the used transdermal system.
Note: Some medicine stays in the transdermal system after the 7 days of use. The used transdermal system should be folded together and safely thrown away.
NOTE: If the transdermal system falls off before it is time to change it, do not try to reapply that transdermal system. Instead, choose a new application site and repeat Step 2 through Step 4 to apply a new transdermal system to the skin. Replace the new transdermal system 7 days later to start a new 1 week cycle.
For example, if you usually replace the ADLARITY transdermal system on Saturdays, but during one of the weeks the transdermal system falls off on Wednesday, apply a new ADLARITY transdermal system right away and replace it the following Wednesday.
ADLARITY is a trademark of Corium, Inc.
Manufactured and distributed by: Corium, Inc., Grand Rapids, MI 49512
This Instructions for Use has been approved by the U.S. Food and Drug Administration.Approved: 3/2022
- Primary Panel - Pouch (Contains 1 transdermal system) 5 mg
- Primary Panel - Carton (Contains 4 transdermal systems) 5 mg
- Primary Panel - Sample Pouch (Contains 1 transdermal system) 5 mg
- Primary Panel - Sample Carton (Contains 1 transdermal systems) 5 mg
- Primary Panel - Pouch (Contains 1 transdermal system) 10 mg
- Primary Panel - Carton (Contains 4 transdermal systems) 10 mg
- Primary Panel - Sample Pouch (Contains 1 transdermal system) 10 mg
- Primary Panel - Carton (Contains 1 transdermal systems) 10 mg
-
INGREDIENTS AND APPEARANCE
ADLARITY
donepezil hydrochloride patchProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:65038-055 Route of Administration TRANSDERMAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Donepezil hydrochloride (UNII: 3O2T2PJ89D) (DONEPEZIL - UNII:8SSC91326P) Donepezil hydrochloride 5 mg Inactive Ingredients Ingredient Name Strength Glycerin (UNII: PDC6A3C0OX) Triethyl Citrate (UNII: 8Z96QXD6UM) Sorbitan Monolaurate (UNII: 6W9PS8B71J) Lauryl Lactate (UNII: G5SU0BFK7O) Sodium Bicarbonate (UNII: 8MDF5V39QO) Crospovidone (UNII: 2S7830E561) Ascorbyl Palmitate (UNII: QN83US2B0N) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:65038-055-03 4 in 1 CARTON 03/11/2022 1 NDC:65038-055-01 1 in 1 POUCH; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 2 NDC:65038-055-01 1 in 1 POUCH; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 09/07/2022 3 NDC:65038-055-07 10 in 1 CARTON 03/11/2022 03/12/2022 3 NDC:65038-055-04 1 in 1 CARTON 3 NDC:65038-055-10 1 in 1 POUCH; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 4 NDC:65038-055-04 1 in 1 CARTON 03/11/2022 4 NDC:65038-055-10 1 in 1 POUCH; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 5 NDC:65038-055-10 1 in 1 POUCH; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 09/07/2022 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212304 03/11/2022 ADLARITY
donepezil hydrochloride patchProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:65038-056 Route of Administration TRANSDERMAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Donepezil hydrochloride (UNII: 3O2T2PJ89D) (DONEPEZIL - UNII:8SSC91326P) Donepezil hydrochloride 10 mg Inactive Ingredients Ingredient Name Strength Glycerin (UNII: PDC6A3C0OX) Triethyl Citrate (UNII: 8Z96QXD6UM) Sorbitan Monolaurate (UNII: 6W9PS8B71J) Lauryl Lactate (UNII: G5SU0BFK7O) Sodium Bicarbonate (UNII: 8MDF5V39QO) Crospovidone (UNII: 2S7830E561) Ascorbyl Palmitate (UNII: QN83US2B0N) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:65038-056-03 4 in 1 CARTON 03/11/2022 1 NDC:65038-056-01 1 in 1 POUCH; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 2 NDC:65038-056-01 1 in 1 POUCH; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 09/07/2022 3 NDC:65038-056-07 10 in 1 CARTON 03/11/2022 03/12/2022 3 NDC:65038-056-04 1 in 1 CARTON 3 NDC:65038-056-10 1 in 1 POUCH; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 4 NDC:65038-056-04 1 in 1 CARTON 03/11/2022 4 NDC:65038-056-10 1 in 1 POUCH; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 5 NDC:65038-056-10 1 in 1 POUCH; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 09/07/2022 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212304 03/11/2022 Labeler - Corium, LLC. (839674207)







