Label: SIMULECT- basiliximab injection, powder, for solution


- NDC Code(s): 0078-0331-84, 0078-0393-61
- Packager: Novartis Pharmaceuticals Corporation
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated August 1, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
BOXED WARNING
(What is this?)
WARNING
Only physicians experienced in immunosuppression therapy and management of organ transplantation patients should prescribe Simulect® (basiliximab). The physician responsible for Simulect administration should have complete information requisite for the follow-up of the patient. Patients receiving the drug should be managed in facilities equipped and staffed with adequate laboratory and supportive medical resources.
-
DESCRIPTION
Basiliximab is a chimeric (murine/human) monoclonal antibody (IgG1к), produced by recombinant DNA technology, that functions as an immunosuppressive agent, specifically binding to and blocking the interleukin-2 receptor α-chain (IL-2Rα, also known as CD25 antigen) on the surface of activated T-lymphocytes. Based on the amino acid sequence, the calculated molecular weight of the protein is 144 kilodaltons. It is a glycoprotein obtained from fermentation of an established mouse myeloma cell line genetically engineered to express plasmids containing the human heavy and light chain constant region genes and mouse heavy and light chain variable region genes encoding the RFT5 antibody that binds selectively to the IL-2Rα.
Simulect® (basiliximab) for injection is a sterile, preservative-free lyophilisate, which is available in single-dose vials and is available in 10 mg and 20 mg strengths for intravenous administration after reconstitution.
Each 10-mg vial contains 10 mg of basiliximab, and dibasic sodium phosphate (anhydrous) (0.50 mg), glycine (20.3 mg), mannitol (40.6 mg), monobasic potassium phosphate (3.66 mg), sodium chloride (0.82 mg), and sucrose (10.1 mg) to be reconstituted in 2.5 mL of Sterile Water for Injection, USP.
Each 20-mg vial contains 20 mg of basiliximab, and dibasic sodium phosphate (anhydrous) (0.99 mg), glycine (40.1 mg), mannitol (80.1 mg), monobasic potassium phosphate (7.22 mg), sodium chloride (1.61 mg), and sucrose (20 mg) to be reconstituted in 5 mL of Sterile Water for Injection, USP.
-
CLINICAL PHARMACOLOGY
General
Mechanism of Action: Basiliximab functions as an IL-2 receptor antagonist by binding with high affinity (Ka = 1 x 1010 M-1) to the alpha chain of the high affinity IL-2 receptor complex and inhibiting IL-2 binding. Basiliximab is specifically targeted against IL-2Rα, which is selectively expressed on the surface of activated T-lymphocytes. This specific high affinity binding of Simulect® (basiliximab) to IL-2Rα competitively inhibits IL-2-mediated activation of lymphocytes, a critical pathway in the cellular immune response involved in allograft rejection.
While in the circulation, Simulect impairs the response of the immune system to antigenic challenges. Whether the ability to respond to repeated or ongoing challenges with those antigens returns to normal after Simulect is cleared is unknown (see PRECAUTIONS).
Pharmacokinetics
Adults: Single-dose and multiple-dose pharmacokinetic studies have been conducted in patients undergoing first kidney transplantation. Cumulative doses ranged from 15 mg up to 150 mg. Peak mean ± SD serum concentration following intravenous infusion of 20 mg over 30 minutes is 7.1 ± 5.1 mg/L. There is a dose-proportional increase in Cmax and area under the curve (AUC) up to the highest tested single dose of 60 mg. The volume of distribution at steady state is 8.6 ± 4.1 L. The extent and degree of distribution to various body compartments have not been fully studied. The terminal half-life is 7.2 ± 3.2 days. Total body clearance is 41 ± 19 mL/h. No clinically relevant influence of body weight or gender on distribution volume or clearance has been observed in adult patients. Elimination half-life was not influenced by age (20-69 years), gender or race (see DOSAGE AND ADMINISTRATION).
Pediatric: The pharmacokinetics of Simulect have been assessed in 39 pediatric patients undergoing renal transplantation. In infants and children (1-11 years of age, n = 25), the distribution volume and clearance were reduced by about 50% compared to adult renal transplantation patients. The volume of distribution at steady state was 4.8 ± 2.1 L, half-life was 9.5 ± 4.5 days and clearance was 17 ± 6 mL/h. Disposition parameters were not influenced to a clinically relevant extent by age (1-11 years of age), body weight (9-37 kg) or body surface area (0.44-1.20 m2) in this age group. In adolescents (12-16 years of age, n = 14), disposition was similar to that in adult renal transplantation patients. The volume of distribution at steady state was 7.8 ± 5.1 L, half-life was 9.1 ± 3.9 days and clearance was 31 ± 19 mL/h (see DOSAGE AND ADMINISTRATION).
Pharmacodynamics
Complete and consistent binding to IL-2Rα in adults is maintained as long as serum Simulect levels exceed 0.2 mcg/mL. As concentrations fall below this threshold, the IL-2Rα sites are no longer fully bound and the number of T-cells expressing unbound IL-2Rα returns to pretherapy values within 1-2 weeks. The relationship between serum concentration and receptor saturation was assessed in 13 pediatric patients and was similar to that characterized in adult renal transplantation patients. In vitro studies using human tissues indicate that Simulect binds only to lymphocytes.
The duration of clinically relevant IL-2 receptor blockade after the recommended course of Simulect is not known. When basiliximab was added to a regimen of cyclosporine, USP (MODIFIED) and corticosteroids in adult patients, the duration of IL-2Rα saturation was 36 ± 14 days (mean ± SD), similar to that observed in pediatric patients (36 ± 14 days) (see DOSAGE AND ADMINISTRATION). When basiliximab was added to a triple therapy regimen consisting of cyclosporine, USP (MODIFIED), corticosteroids, and azathioprine in adults, the duration was 50 ± 20 days and when added to cyclosporine, USP (MODIFIED), corticosteroids, and mycophenolate mofetil in adults, the duration was 59 ± 17 days (see PRECAUTIONS, Drug Interactions). No significant changes to circulating lymphocyte numbers or cell phenotypes were observed by flow cytometry.
-
CLINICAL STUDIES
The safety and efficacy of Simulect® (basiliximab) for the prophylaxis of acute organ rejection in adults following cadaveric- or living-donor renal transplantation were assessed in four randomized, double-blind, placebo-controlled clinical studies (1,184 patients). Of these four, two studies (Study 1 [EU/CAN] and Study 2 [US Study]) compared two 20-mg doses of Simulect with placebo, each administered intravenously as an infusion, as part of a standard immunosuppressive regimen comprised of cyclosporine, USP (MODIFIED) and corticosteroids. The other two controlled studies compared two 20-mg doses of Simulect with placebo, each administered intravenously as a bolus injection, as part of a standard triple-immunosuppressive regimen comprised of cyclosporine, USP (MODIFIED), corticosteroids and either azathioprine or mycophenolate mofetil (Study 3 and Study 4, respectively). The first dose of Simulect or placebo was administered within 2 hours prior to transplantation surgery (Day 0) and the second dose administered on Day 4 post-transplantation. The regimen of Simulect was chosen to provide 30-45 days of IL-2Rα saturation.
Seven hundred twenty-nine patients were enrolled in the two studies using a dual maintenance immunosuppressive regimen comprised of cyclosporine, USP (MODIFIED) and corticosteroids, of which 363 patients were treated with Simulect and 358 patients were placebo-treated. Study 1 was conducted at 21 sites in Europe and Canada (EU/CAN Study); Study 2 was conducted at 21 sites in the USA (US Study). Patients 18-75 years of age undergoing first cadaveric- (Study 1 and Study 2) or living-donor (Study 2 only) renal transplantation, with ≥ 1 HLA mismatch, were enrolled.1,2
The primary efficacy endpoint in both studies was the incidence of death, graft loss or an episode of acute rejection during the first 6 months post-transplantation. Secondary efficacy endpoints included the primary efficacy variable measured during the first 12 months post-transplantation, the incidence of biopsy-confirmed acute rejection during the first 6 and 12 months post-transplantation and patient survival and graft survival, each measured at 12 months post-transplantation. Table 1 summarizes the results of these studies. Figure 1 displays the Kaplan-Meier estimates of the percentage of patients by treatment group experiencing the primary efficacy endpoint during the first 12 months post-transplantation for Study 2. Patients in both studies receiving Simulect experienced a significantly lower incidence of biopsy-confirmed rejection episodes at both 6 and 12 months post-transplantation. There was no difference in the rate of delayed graft function, patient survival, or graft survival between Simulect-treated patients and placebo-treated patients in either study.
There was no evidence that the clinical benefit of Simulect was limited to specific subpopulations based on age, gender, race, donor type (cadaveric or living donor allograft), or history of diabetes mellitus.
Table 1. Efficacy Parameters (Percentage of Patients) *USP (MODIFIED). Dual-therapy Regimen (cyclosporine* and corticosteroids) Study 1 Study 2 Placebo
(N = 185)Simulect®
(N = 190)p-value Placebo
(N = 173)Simulect®
(N = 173)p-value Primary endpoint
Death, graft loss or acute rejection episode (0-6 months)57% 42% 0.003 55% 38% 0.002 Secondary endpoints
Death, graft loss or acute rejection episode (0-12 months)60% 46% 0.007 58% 41% 0.001 Biopsy-confirmed rejection episode (0-6 months) 44% 30% 0.007 46% 33% 0.015 Biopsy-confirmed rejection episode (0-12 months) 46% 32% 0.005 49% 35% 0.009 Patient survival (12 months) 97% 95% 0.29 96% 97% 0.56 Patients with functioning graft (12 months) 87% 88% 0.70 93% 95% 0.50 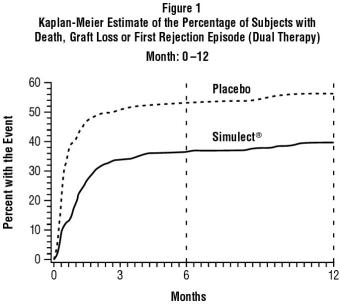
Two double-blind, randomized, placebo-controlled studies (Study 3 and Study 4) assessed the safety and efficacy of Simulect for the prophylaxis of acute renal transplant rejection in adults when used in combination with a triple immunosuppressive regimen. In Study 3, 340 patients were concomitantly treated with cyclosporine, USP (MODIFIED), corticosteroids and azathioprine (AZA), of which 168 patients were treated with Simulect and 172 patients were treated with placebo. In Study 4, 123 patients were concomitantly treated with cyclosporine, USP (MODIFIED), corticosteroids and mycophenolate mofetil (MMF), of which 59 patients were treated with Simulect and 64 patients were treated with placebo. Patients 18-70 years of age undergoing first or second cadaveric or living donor (related or unrelated) renal transplantation were enrolled in both studies.
The results of Study 3 are shown in Table 2. These results are consistent with the findings from Study 1 and Study 2.
Table 2. Efficacy Parameters (Percentage of Patients) *USP (MODIFIED). Study 3: Triple-therapy Regimen (cyclosporine*, corticosteroids, and azathioprine) Placebo Simulect® (N = 172) (N = 168) p-value Primary endpoint Acute rejection episode (0-6 months) 35% 21% 0.005 Secondary endpoints Death, graft loss or acute rejection episode (0-6 months) 40% 26% 0.008 Biopsy-confirmed rejection episode (0-6 months) 29% 18% 0.023 Patient survival (12 months) 97% 98% 1.000 Patients with functioning graft (12 months) 88% 90% 0.599 In Study 4, the percentage of patients experiencing biopsy-proven acute rejection by 6 months was 15% (9 of 59 patients) in the Simulect group and 27% (17 of 64 patients) in the placebo group. Although numerically lower, the difference in acute rejection was not significant.
In a multicenter, randomized, double-blind, placebo-controlled trial of Simulect for the prevention of allograft rejection in liver transplant recipients (n = 381) receiving concomitant cyclosporine, USP (MODIFIED) and steroids, the incidence of the combined endpoint of death, graft loss, or first biopsy-confirmed rejection episode at either 6 or 12 months was similar between patients randomized to receive Simulect and those randomized to receive placebo.
The efficacy of Simulect for the prophylaxis of acute rejection in recipients of a second renal allograft has not been demonstrated.
Long-Term Followup
Five-year patient survival and graft survival data were provided by 71% and 58% of the original subjects of Study 1 and Study 2, respectively. Subjects in both studies continued to receive a dual-therapy regimen with cyclosporine, USP (MODIFIED) and corticosteroid. No difference was observed between groups in the 5-year graft survival in either Study 1 (91% Simulect group, 92% placebo group) or Study 2 (85% Simulect group, 86% placebo group). In Study 1, patient survival was lower in the Simulect-treated patients compared to the placebo-treated patients (142/163 [87%] vs. 156/164 [95%], respectively). The cause of this difference in survival is unknown. The data do not indicate an increase in malignancy- or infection-related mortality. In Study 2, patient survival in the placebo group (90%) was the same compared to Simulect group (90%).
-
INDICATIONS AND USAGE
Simulect® (basiliximab) is indicated for the prophylaxis of acute organ rejection in patients receiving renal transplantation when used as part of an immunosuppressive regimen that includes cyclosporine, USP (MODIFIED), and corticosteroids.
The efficacy of Simulect for the prophylaxis of acute rejection in recipients of other solid organ allografts has not been demonstrated.
-
CONTRAINDICATIONS
Simulect® (basiliximab) is contraindicated in patients with known hypersensitivity to basiliximab or any other component of the formulation. See composition of Simulect under DESCRIPTION.
General
Simulect® (basiliximab) should be administered under qualified medical supervision. Patients should be informed of the potential benefits of therapy and the risks associated with administration of immunosuppressive therapy.
While neither the incidence of lymphoproliferative disorders nor opportunistic infections was higher in Simulect-treated patients than in placebo-treated patients, patients on immunosuppressive therapy are at increased risk for developing these complications and should be monitored accordingly.
Hypersensitivity
Severe acute (onset within 24 hours) hypersensitivity reactions, including anaphylaxis have been observed both on initial exposure to Simulect and/or following re-exposure after several months. These reactions may include hypotension, tachycardia, cardiac failure, dyspnea, wheezing, bronchospasm, pulmonary edema, respiratory failure, urticaria, rash, pruritus, and/or sneezing. Extreme caution should be exercised in all patients previously given Simulect when being administered a subsequent course of Simulect. A subgroup of patients may be particularly at risk of developing severe hypersensitivity reactions on re-administration. These are patients in whom concomitant immunosuppression was discontinued prematurely (e.g., due to abandoned transplantation or early loss of the graft) following the initial administration of Simulect. If a severe hypersensitivity reaction occurs, therapy with Simulect should be permanently discontinued. Medications for the treatment of severe hypersensitivity reactions, including anaphylaxis should be available for immediate use.
-
PRECAUTIONS
General
It is not known whether Simulect® (basiliximab) use will have a long-term effect on the ability of the immune system to respond to antigens first encountered during Simulect-induced immunosuppression.
Immunogenicity
Of renal transplantation patients treated with Simulect and tested for anti-idiotype antibodies, 4/339 developed an anti-idiotype antibody response, with no deleterious clinical effect upon the patient. In none of these cases was there evidence that the presence of anti-idiotype antibody accelerated Simulect clearance or decreased the period of receptor saturation. In Study 2, the incidence of human anti-murine antibody (HAMA) in renal transplantation patients treated with Simulect was 2/138 in patients not exposed to muromonab-CD3 and 4/34 in patients who subsequently received muromonab-CD3. The available clinical data on the use of muromonab-CD3 in patients previously treated with Simulect suggest that subsequent use of muromonab-CD3 or other murine anti-lymphocytic antibody preparations is not precluded.
These data reflect the percentage of patients whose test results were considered positive for antibodies to Simulect in an ELISA assay, and are highly dependent on the sensitivity and specificity of the assay. Additionally the observed incidence of antibody positivity in an assay may be influenced by several factors, including sample handling, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to Simulect with the incidence of antibodies to other products may be misleading.
Drug Interactions
No dose adjustment is necessary when Simulect is added to triple-immunosuppression regimens, including cyclosporine, corticosteroids, and either azathioprine or mycophenolate mofetil. Three clinical trials have investigated Simulect use in combination with triple-therapy regimens. Pharmacokinetics were assessed in two of these trials. Total body clearance of Simulect was reduced by an average 22% and 51% when azathioprine and mycophenolate mofetil, respectively, were added to a regimen consisting of cyclosporine, USP (MODIFIED) and corticosteroids. Nonetheless, the range of individual Simulect clearance values in the presence of azathioprine (12-57 mL/h) or mycophenolate mofetil (7-54 mL/h) did not extend outside the range observed with dual therapy (10-78 mL/h). The following medications have been administered in clinical trials with Simulect with no increase in adverse reactions: ATG/ALG, azathioprine, corticosteroids, cyclosporine, mycophenolate mofetil, and muromonab-CD3.
Carcinogenesis/Mutagenesis/Impairment of Fertility
No mutagenic potential of Simulect was observed in the in vitro assays with Salmonella (Ames) and V79 Chinese hamster cells. No long-term or fertility studies in laboratory animals have been performed to evaluate the potential of Simulect to produce carcinogenicity or fertility impairment, respectively.
There are no adequate and well-controlled studies in pregnant women. No maternal toxicity, embryotoxicity, or teratogenicity was observed in cynomolgus monkeys 100 days post coitum following dosing with basiliximab during the organogenesis period; blood levels in pregnant monkeys were 13-fold higher than those seen in human patients. Immunotoxicology studies have not been performed in the offspring. Because IgG molecules are known to cross the placental barrier, because the IL-2 receptor may play an important role in development of the immune system, and because animal reproduction studies are not always predictive of human response, Simulect should only be used in pregnant women when the potential benefit justifies the potential risk to the fetus. Women of childbearing potential should use effective contraception before beginning Simulect therapy, during therapy, and for 4 months after completion of Simulect therapy.
Nursing Mothers
It is not known whether Simulect is excreted in human milk. Because many drugs, including human antibodies are excreted in human milk, and because of the potential for adverse reactions, a decision should be made to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
No randomized, placebo-controlled studies have been completed in pediatric patients. In a safety and pharmacokinetic study, 41 pediatric patients (1-11 years of age [n = 27], 12-16 years of age [n = 14], median age 8.1 years) were treated with Simulect via intravenous bolus injection in addition to standard immunosuppressive agents, including cyclosporine, USP (MODIFIED), corticosteroids, azathioprine, and mycophenolate mofetil. The acute rejection rate at 6 months was comparable to that in adults in the triple-therapy trials. The most frequently reported adverse events were hypertension, hypertrichosis, and rhinitis (49% each), urinary tract infections (46%), and fever (39%). Overall, the adverse event profile was consistent with general clinical experience in the pediatric renal transplantation population and with the profile in the controlled adult renal transplantation studies. The available pharmacokinetic data in children and adolescents are described in CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION.
It is not known whether the immune response to vaccines, infection, and other antigenic stimuli administered or encountered during Simulect therapy is impaired or whether such response will remain impaired after Simulect therapy.
Geriatric Use
Controlled clinical studies of Simulect have included a small number of patients 65 years and older (Simulect 28; placebo 32). From the available data comparing Simulect and placebo-treated patients, the adverse event profile in patients ≥ 65 years of age is not different from patients < 65 years of age and no age-related dosing adjustment is required. Caution must be used in giving immunosuppressive drugs to elderly patients.
-
ADVERSE REACTIONS
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The adverse reaction information from clinical trials does, however, provide a basis for identifying the adverse events that appear to be related to drug use and for approximating rates.
The incidence of adverse events for Simulect® (basiliximab) was determined in four randomized, double-blind, placebo-controlled clinical trials for the prevention of renal allograft rejection. Two of the studies (Study 1 and Study 2), used a dual maintenance immunosuppressive regimen comprised of cyclosporine, USP (MODIFIED) and corticosteroids, whereas the other two studies (Study 3 and Study 4) used a triple-immunosuppressive regimen comprised of cyclosporine, USP (MODIFIED), corticosteroids, and either azathioprine or mycophenolate mofetil.
Simulect did not appear to add to the background of adverse events seen in organ transplantation patients as a consequence of their underlying disease and the concurrent administration of immunosuppressants and other medications. Adverse events were reported by 96% of the patients in the placebo-treated group and 96% of the patients in the Simulect-treated group. In the four placebo-controlled studies, the pattern of adverse events in 590 patients treated with the recommended dose of Simulect was similar to that in 594 patients treated with placebo. Simulect did not increase the incidence of serious adverse events observed compared with placebo.
The most frequently reported adverse events were gastrointestinal disorders, reported in 69% of Simulect-treated patients and 67% of placebo-treated patients.
The incidence and types of adverse events were similar in Simulect-treated and placebo-treated patients. The following adverse events occurred in ≥ 10% of Simulect-treated patients: Gastrointestinal System: constipation, nausea, abdominal pain, vomiting, diarrhea, dyspepsia; Body as a Whole-General: pain, peripheral edema, fever, viral infection; Metabolic and Nutritional: hyperkalemia, hypokalemia, hyperglycemia, hypercholesterolemia, hypophosphatemia, hyperuricemia; Urinary System: urinary tract infection; Respiratory System: dyspnea, upper respiratory tract infection; Skin and Appendages: surgical wound complications, acne; Cardiovascular Disorders-General: hypertension; Central and Peripheral Nervous System: headache, tremor; Psychiatric: insomnia; Red Blood Cell: anemia.
The following adverse events, not mentioned above, were reported with an incidence of ≥ 3% and < 10% in pooled analysis of patients treated with Simulect in the four controlled clinical trials, or in an analysis of the two dual-therapy trials: Body as a Whole-General: accidental trauma, asthenia, chest pain, increased drug level, infection, face edema, fatigue, dependent edema, generalized edema, leg edema, malaise, rigors, sepsis; Cardiovascular: abnormal heart sounds, aggravated hypertension, angina pectoris, cardiac failure, chest pain, hypotension; Endocrine: increased glucocorticoids; Gastrointestinal: enlarged abdomen, esophagitis, flatulence, gastrointestinal disorder, gastroenteritis, GI hemorrhage, gum hyperplasia, melena, moniliasis, ulcerative stomatitis; Heart Rate and Rhythm: arrhythmia, atrial fibrillation, tachycardia; Metabolic and Nutritional: acidosis, dehydration, diabetes mellitus, fluid overload, hypercalcemia, hyperlipemia, hypertriglyceridemia, hypocalcemia, hypoglycemia, hypomagnesemia, hypoproteinemia, weight increase; Musculoskeletal: arthralgia, arthropathy, back pain, bone fracture, cramps, hernia, myalgia, leg pain; Nervous System: dizziness, neuropathy, paraesthesia, hypoesthesia; Platelet and Bleeding: hematoma, hemorrhage, purpura, thrombocytopenia, thrombosis; Psychiatric: agitation, anxiety, depression; Red Blood Cell: polycythemia; Reproductive Disorders, Male: genital edema, impotence; Respiratory: bronchitis, bronchospasm, abnormal chest sounds, coughing, pharyngitis, pneumonia, pulmonary disorder, pulmonary edema, rhinitis, sinusitis; Skin and Appendages: cyst, herpes simplex, herpes zoster, hypertrichosis, pruritus, rash, skin disorder, skin ulceration; Urinary: albuminuria, bladder disorder, dysuria, frequent micturition, hematuria, increased non-protein nitrogen, oliguria, abnormal renal function, renal tubular necrosis, surgery, ureteral disorder, urinary retention; Vascular Disorders: vascular disorder; Vision Disorders: cataract, conjunctivitis, abnormal vision; White Blood Cell: leucopenia. Among these events, leucopenia and hypertriglyceridemia occurred more frequently in the two triple-therapy studies using azathioprine and mycophenolate mofetil than in the dual-therapy studies.
Malignancies
The incidence of malignancies in the controlled clinical trials of renal transplant was not significantly different between groups at 1 year (9/590 Simulect-treated patients vs. 12/594 placebo-treated patients) or among patients with 5-year follow-up from Studies 1 and 2 (21/295 Simulect-treated patients vs. 21/291 placebo-treated patients). The incidence of lymphoproliferative disease was not significantly different between groups, and less than 1% in the Simulect-treated patients.
Infections
The overall incidence of cytomegalovirus infection was similar in Simulect- and placebo-treated patients (15% vs. 17%) receiving a dual- or triple-immunosuppression regimen. However, in patients receiving a triple-immunosuppression regimen, the incidence of serious cytomegalovirus infection was higher in Simulect-treated patients compared to placebo-treated patients (11% vs. 5%). The rates of infections, serious infections, and infectious organisms were similar in the Simulect- and placebo-treatment groups among dual- and triple-therapy treated patients.
Postmarketing Experience
Severe acute hypersensitivity reactions, including anaphylaxis characterized by hypotension, tachycardia, cardiac failure, dyspnea, wheezing, bronchospasm, pulmonary edema, respiratory failure, urticaria, rash, pruritus, and/or sneezing, as well as capillary leak syndrome and cytokine release syndrome, have been reported during post-marketing experience with Simulect.
-
OVERDOSAGE
A maximum tolerated dose of Simulect® (basiliximab) has not been determined in patients. During the course of clinical studies, Simulect has been administered to adult renal transplantation patients in single doses of up to 60 mg, or in divided doses over 3-5 days of up to 120 mg, without any associated serious adverse events. There has been one spontaneous report of a pediatric renal transplantation patient who received a single 20-mg dose (2.3 mg/kg) without adverse events.
-
DOSAGE AND ADMINISTRATION
Simulect® (basiliximab) is used as part of an immunosuppressive regimen that includes cyclosporine, USP (MODIFIED) and corticosteroids. Simulect is for central or peripheral intravenous administration only. Reconstituted Simulect should be given either as a bolus injection or diluted to a volume of 25 mL (10-mg vial) or 50 mL (20-mg vial) with 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP and administered as an intravenous infusion over 20 to 30 minutes. Bolus administration may be associated with nausea, vomiting and local reactions, including pain.
Simulect should only be administered once it has been determined that the patient will receive the graft and concomitant immunosuppression. Patients previously administered Simulect should only be re-exposed to a subsequent course of therapy with extreme caution due to the potential risk of hypersensitivity (see WARNINGS).
Parenteral drug products should be inspected visually for particulate matter and discoloration before administration. After reconstitution, Simulect should be a clear-to-opalescent, colorless solution. If particulate matter is present or the solution is colored, do not use.
Care must be taken to assure sterility of the prepared solution because the drug product does not contain any antimicrobial preservatives or bacteriostatic agents.
It is recommended that after reconstitution, the solution should be used immediately. If not used immediately, it can be stored at 2ºC to 8ºC (36ºF to 46ºF) for 24 hours or at room temperature for 4 hours. Discard the reconstituted solution if not used within 24 hours.
No incompatibility between Simulect and polyvinyl chloride bags or infusion sets has been observed. No data are available on the compatibility of Simulect with other intravenous substances. Other drug substances should not be added or infused simultaneously through the same intravenous line.
Adults
In adult patients, the recommended regimen is two doses of 20 mg each. The first 20-mg dose should be given within 2 hours prior to transplantation surgery. The recommended second 20-mg dose should be given 4 days after transplantation. The second dose should be withheld if complications, such as severe hypersensitivity reactions to Simulect or graft loss occur.
Pediatric
In pediatric patients weighing less than 35 kg, the recommended regimen is two doses of 10 mg each. In pediatric patients weighing 35 kg or more, the recommended regimen is two doses of 20 mg each. The first dose should be given within 2 hours prior to transplantation surgery. The recommended second dose should be given 4 days after transplantation. The second dose should be withheld if complications, such as severe hypersensitivity reactions to Simulect or graft loss occur.
Reconstitution of 10 mg Simulect® Vial
To prepare the reconstituted solution, add 2.5 mL of Sterile Water for Injection, USP, using aseptic technique, to the vial containing the Simulect powder. Shake the vial gently to dissolve the powder.
The reconstituted solution is isotonic and may be given either as a bolus injection or diluted to a volume of 25 mL with 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP for infusion. When mixing the solution, gently invert the bag in order to avoid foaming; DO NOT SHAKE.
Reconstitution of 20 mg Simulect® Vial
To prepare the reconstituted solution, add 5 mL of Sterile Water for Injection, USP, using aseptic technique, to the vial containing the Simulect powder. Shake the vial gently to dissolve the powder.
The reconstituted solution is isotonic and may be given either as a bolus injection or diluted to a volume of 50 mL with 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP for infusion. When mixing the solution, gently invert the bag in order to avoid foaming; DO NOT SHAKE.
-
HOW SUPPLIED
Simulect® (basiliximab) is supplied in a single-dose glass vial.
Each carton contains one of the following
1 Simulect 10 mg vial…………………………………………………….NDC 0078-0393-61
1 Simulect 20 mg vial…………………………………………………….NDC 0078-0331-84
Store lyophilized Simulect under refrigerated conditions at 2ºC to 8ºC (36ºF to 46ºF).
Do not use beyond the expiration date stamped on the vial.
-
REFERENCES
- Kahan, B.D., Rajagopalan P.R. and Hall M., Transplantation, 67, 276-284 (1999).
- Nashan, B., Moore R., Amlot P., Schmidt A.-G., Abeywickrama K. and Soulillou J.-P., Lancet 350, 1193-1198 (1997).
Manufactured by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936US License No. 1244
© Novartis
T2020-125
Revised: August 2020
- Kahan, B.D., Rajagopalan P.R. and Hall M., Transplantation, 67, 276-284 (1999).
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
SIMULECT
basiliximab injection, powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0078-0393 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BASILIXIMAB (UNII: 9927MT646M) (BASILIXIMAB - UNII:9927MT646M) BASILIXIMAB 10 mg in 2.5 mL Inactive Ingredients Ingredient Name Strength SODIUM PHOSPHATE, DIBASIC, ANHYDROUS (UNII: 22ADO53M6F) 0.50 mg in 2.5 mL GLYCINE (UNII: TE7660XO1C) 20 mg in 2.5 mL MANNITOL (UNII: 3OWL53L36A) 41 mg in 2.5 mL POTASSIUM PHOSPHATE, MONOBASIC (UNII: 4J9FJ0HL51) 3.66 mg in 2.5 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 0.82 mg in 2.5 mL SUCROSE (UNII: C151H8M554) 10 mg in 2.5 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0078-0393-61 1 in 1 CARTON 05/12/1998 1 2.5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103764 05/12/1998 SIMULECT
basiliximab injection, powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0078-0331 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BASILIXIMAB (UNII: 9927MT646M) (BASILIXIMAB - UNII:9927MT646M) BASILIXIMAB 20 mg in 5 mL Inactive Ingredients Ingredient Name Strength SODIUM PHOSPHATE, DIBASIC, ANHYDROUS (UNII: 22ADO53M6F) 0.99 mg in 5 mL GLYCINE (UNII: TE7660XO1C) 40 mg in 5 mL MANNITOL (UNII: 3OWL53L36A) 80 mg in 5 mL POTASSIUM PHOSPHATE, MONOBASIC (UNII: 4J9FJ0HL51) 7.22 mg in 5 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 1.61 mg in 5 mL SUCROSE (UNII: C151H8M554) 20 mg in 5 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0078-0331-84 1 in 1 CARTON 05/12/1998 1 5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103764 05/12/1998 Labeler - Novartis Pharmaceuticals Corporation (002147023)

