LAMIVUDINE AND ZIDOVUDINE - lamivudine and zidovudine tablet, film coated
Aurobindo Pharma Limited
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use LAMIVUDINE AND ZIDOVUDINE TABLETS safely and effectively. See full prescribing information for LAMIVUDINE AND ZIDOVUDINE TABLETS.
LAMIVUDINE and ZIDOVUDINE tablets, for oral use Initial U.S. Approval: 1997
WARNING: HEMATOLOGIC TOXICITY, MYOPATHY, LACTIC ACIDOSIS AND SEVERE HEPATOMEGALY WITH STEATOSIS, and EXACERBATIONS OF HEPATITIS B
|
FULL PRESCRIBING INFORMATION
WARNING: HEMATOLOGIC TOXICITY, MYOPATHY, LACTIC ACIDOSIS AND SEVERE HEPATOMEGALY WITH STEATOSIS, and EXACERBATIONS OF HEPATITIS B
Zidovudine, a component of lamivudine and zidovudine tablets, has been associated with hematologic toxicity including neutropenia and severe anemia, particularly in patients with advanced Human Immunodeficiency Virus (HIV-1) disease [see Warnings and Precautions (5.1)].
Prolonged use of zidovudine has been associated with symptomatic myopathy [see Warnings and Precautions (5.2)].
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues, including lamivudine and zidovudine. Discontinue lamivudine and zidovudine if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occur [see Warnings and Precautions (5.3)].
Severe acute exacerbations of hepatitis B have been reported in patients who are co-infected with hepatitis B virus (HBV) and HIV-1 and have discontinued lamivudine, which is one component of lamivudine and zidovudine. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who discontinue lamivudine and zidovudine and are co-infected with HIV-1 and HBV. If appropriate, initiation of anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.4)].
1 INDICATIONS AND USAGE
Lamivudine and Zidovudine tablets, a combination of 2 nucleoside analogues, are indicated in combination with other antiretrovirals for the treatment of human immunodeficiency virus type 1 (HIV-1) infection.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage for Adults and Adolescents
The recommended dosage of lamivudine and zidovudine tablets in HIV-1-infected adults and adolescents weighing greater than or equal to 30 kg is 1 tablet (containing 150 mg of lamivudine and 300 mg of zidovudine) taken orally twice daily.
2.2 Recommended Dosage for Pediatric Patients
The recommended dosage of scored lamivudine and zidovudine tablets for pediatric patients who weigh greater than or equal to 30 kg and for whom a solid oral dosage form is appropriate is 1 tablet administered orally twice daily.
Before prescribing lamivudine and zidovudine tablets, children should be assessed for the ability to swallow tablets. If a child is unable to reliably swallow a lamivudine and zidovudine tablet, the liquid oral formulations should be prescribed: EPIVIR (lamivudine) oral solution and RETROVIR (zidovudine) syrup.
2.3 Not Recommended Due to Lack of Dosage Adjustment
Because lamivudine and zidovudine is a fixed-dose tablet and cannot be dose adjusted, lamivudine and zidovudine tablets are not recommended for:
- pediatric patients weighing less than 30 kg [see Use in Specific Populations (8.4)].
- patients with creatinine clearance less than 50 mL per minute [see Use in Specific Populations (8.6)].
- patients with hepatic impairment [see Use in Specific Populations (8.7)].
- patients experiencing dose-limiting adverse reactions.
Liquid and solid oral formulations of the individual components of lamivudine and zidovudine tablets are available for these populations.
3 DOSAGE FORMS AND STRENGTHS
Lamivudine and Zidovudine Tablets USP, containing 150 mg lamivudine and 300 mg zidovudine, are white to off-white, modified capsule shaped, biconvex, film-coated tablets with deep breakline in between ‘J’ and ‘58’ on one side and deep breakline on the other side.
4 CONTRAINDICATIONS
Lamivudine and Zidovudine tablets are contraindicated in patients with a previous hypersensitivity reaction to lamivudine or zidovudine.
5 WARNINGS AND PRECAUTIONS
5.1 Hematologic Toxicity/Bone Marrow Suppression
Zidovudine, a component of lamivudine and zidovudine, has been associated with hematologic toxicity including neutropenia and anemia, particularly in patients with advanced HIV-1 disease. Lamivudine and Zidovudine should be used with caution in patients who have bone marrow compromise evidenced by granulocyte count less than 1,000 cells per mm3 or hemoglobin less than 9.5 grams per dL [see Adverse Reactions (6.1)].
Frequent blood counts are strongly recommended in patients with advanced HIV-1 disease who are treated with lamivudine and zidovudine. Periodic blood counts are recommended for other HIV-1-infected patients. If anemia or neutropenia develops, dosage interruption may be needed.
5.2 Myopathy
Myopathy and myositis, with pathological changes similar to that produced by HIV-1 disease, have been associated with prolonged use of zidovudine, and therefore may occur with therapy with lamivudine and zidovudine.
5.3 Lactic Acidosis and Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues, including lamivudine and zidovudine. A majority of these cases have been in women. Female sex and obesity may be risk factors for the development of lactic acidosis and severe hepatomegaly with steatosis in patients treated with antiretroviral nucleoside analogues. See full prescribing information for EPIVIR (lamivudine) and RETROVIR (zidovudine). Treatment with lamivudine and zidovudine should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity, which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations.
5.4 Patients with Hepatitis B Virus Co-infection
Posttreatment Exacerbations of Hepatitis
Clinical and laboratory evidence of exacerbations of hepatitis have occurred after discontinuation of lamivudine. See full prescribing information for EPIVIR (lamivudine). Patients should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment.
Emergence of Lamivudine-Resistant HBV
Safety and efficacy of lamivudine have not been established for treatment of chronic hepatitis B in subjects dually infected with HIV-1 and HBV. Emergence of hepatitis B virus variants associated with resistance to lamivudine has been reported in HIV-1-infected subjects who have received lamivudine-containing antiretroviral regimens in the presence of concurrent infection with hepatitis B virus. See full prescribing information for EPIVIR (lamivudine).
5.5 Use with Interferon- and Ribavirin-Based Regimens
Patients receiving interferon alfa with or without ribavirin and lamivudine and zidovudine should be closely monitored for treatment-associated toxicities, especially hepatic decompensation, neutropenia, and anemia. See full prescribing information for RETROVIR (zidovudine). Discontinuation of lamivudine and zidovudine should be considered as medically appropriate. Dose reduction or discontinuation of interferon alfa, ribavirin, or both should also be considered if worsening clinical toxicities are observed, including hepatic decompensation (e.g., Child-Pugh greater than 6) (see full prescribing information for interferon and ribavirin).
Exacerbation of anemia has been reported in HIV-1/HCV co-infected patients receiving ribavirin and zidovudine. Coadministration of ribavirin and lamivudine and zidovudine is not advised.
5.6 Pancreatitis
Lamivudine and Zidovudine should be used with caution in patients with a history of pancreatitis or other significant risk factors for the development of pancreatitis. Treatment with lamivudine and zidovudine should be stopped immediately if clinical signs, symptoms, or laboratory abnormalities suggestive of pancreatitis occur [see Adverse Reactions (6.1)].
5.7 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including lamivudine and zidovudine. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.8 Lipoatrophy
Treatment with zidovudine, a component of lamivudine and zidovudine, has been associated with loss of subcutaneous fat. The incidence and severity of lipoatrophy are related to cumulative exposure. This fat loss, which is most evident in the face, limbs, and buttocks, may be only partially reversible and improvement may take months to years after switching to a non-zidovudine-containing regimen. Patients should be regularly assessed for signs of lipoatrophy during therapy with zidovudine-containing products, and if feasible, therapy should be switched to an alternative regimen if there is suspicion of lipoatrophy.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Hematologic toxicity, including neutropenia and anemia [see Boxed Warning, Warnings and Precautions (5.1)].
- Symptomatic myopathy [see Boxed Warning, Warnings and Precautions (5.2)].
- Lactic acidosis and severe hepatomegaly with steatosis [see Boxed Warning, Warnings and Precautions (5.3)].
- Exacerbations of hepatitis B [see Boxed Warning, Warnings and Precautions (5.4)].
- Hepatic decompensation in patients co-infected with HIV-1 and hepatitis C [see Warnings and Precautions (5.5)].
- Exacerbation of anemia in HIV-1/HCV co-infected patients receiving ribavirin and zidovudine [see Warnings and Precautions (5.5)].
- Pancreatitis [see Warnings and Precautions (5.6)].
- Immune reconstitution syndrome [see Warnings and Precautions (5.7)].
- Lipoatrophy [see Warnings and Precautions (5.8)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Lamivudine plus Zidovudine Administered as Separate Formulations
In 4 randomized, controlled trials of EPIVIR 300 mg per day plus RETROVIR 600 mg per day, the following selected adverse reactions and laboratory abnormalities were observed (Tables 1 and 2).
| Adverse Reaction | EPIVIR plus RETROVIR (n = 251) |
|---|---|
| Body as a whole
| |
| Headache | 35% |
| Malaise & fatigue | 27% |
| Fever or chills | 10% |
| Digestive
| |
| Nausea | 33% |
| Diarrhea | 18% |
| Nausea & vomiting | 13% |
| Anorexia and/or decreased appetite | 10% |
| Abdominal pain | 9% |
| Abdominal cramps | 6% |
| Dyspepsia | 5% |
| Nervous system
| |
| Neuropathy | 12% |
| Insomnia & other sleep disorders | 11% |
| Dizziness | 10% |
| Depressive disorders | 9% |
| Respiratory
| |
| Nasal signs & symptoms | 20% |
| Cough | 18% |
| Skin
| |
| Skin rashes | 9% |
| Musculoskeletal
| |
| Musculoskeletal pain | 12% |
| Myalgia | 8% |
| Arthralgia | 5% |
Pancreatitis was observed in 9 of the 2,613 adult subjects (0.3%) who received EPIVIR in controlled clinical trials [see Warnings and Precautions (5.6)].
Selected laboratory abnormalities observed during therapy are listed in Table 2.
| Test (Abnormal Level) | EPIVIR plus RETROVIR % (n) |
|---|---|
| ULN = Upper limit of normal. ANC = Absolute neutrophil count. n = Number of subjects assessed. a Frequencies of these laboratory abnormalities were higher in subjects with mild laboratory abnormalities at baseline. |
|
| Neutropenia (ANC <750/mm3) | 7.2% (237) |
| Anemia (Hgb <8.0 g/dL) | 2.9% (241) |
| Thrombocytopenia (platelets <50,000/mm3) | 0.4% (240) |
| ALT (>5.0 x ULN) | 3.7% (241) |
| AST (>5.0 x ULN) | 1.7% (241) |
| Bilirubin (>2.5 x ULN) | 0.8% (241) |
| Amylase (>2.0 x ULN) | 4.2% (72) |
6.2 Postmarketing Experience
The following adverse reactions have been identified during postmarketing use. Because these reactions are reported voluntarily from a population of unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Body as a Whole
Redistribution/accumulation of body fat [see Warnings and Precautions (5.8)].
Cardiovascular
Cardiomyopathy.
Endocrine and Metabolic
Gynecomastia, hyperglycemia.
Gastrointestinal
Oral mucosal pigmentation, stomatitis.
General
Vasculitis, weakness.
Hemic and Lymphatic
Anemia, (including pure red cell aplasia and anemias progressing on therapy), lymphadenopathy, splenomegaly.
Hepatic and Pancreatic
Lactic acidosis and hepatic steatosis, pancreatitis, posttreatment exacerbations of hepatitis B [see Boxed Warning, Warnings and Precautions (5.3), (5.4), (5.6)].
Hypersensitivity
Sensitization reactions (including anaphylaxis), urticaria.
Musculoskeletal
Muscle weakness, CPK elevation, rhabdomyolysis.
Nervous
Paresthesia, peripheral neuropathy, seizures.
Respiratory
Abnormal breath sounds/wheezing.
Skin
Alopecia, erythema multiforme, Stevens-Johnson syndrome.
7 DRUG INTERACTIONS
7.1 Zidovudine
Agents Antagonistic with Zidovudine
Concomitant use of zidovudine with the following drugs should be avoided since an antagonistic relationship has been demonstrated in vitro:
- Stavudine
- Doxorubicine
- Nucleoside analogues, e.g., ribavirin
Hematologic/Bone Marrow Suppressive/Cytotoxic Agents
Coadministration with the following drugs may increase the hematologic toxicity of zidovudine:
- Ganciclovir
- Interferon alfa
- Ribavirin
- Other bone marrow suppressive or cytotoxic agents
7.2 Lamivudine
Sorbitol
Coadministration of single doses of lamivudine and sorbitol resulted in a sorbitol dose-dependent reduction in lamivudine exposures. When possible, avoid use of sorbitol-containing medicines with lamivudine-containing medicines [see Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to lamivudine and zidovudine during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
Available data from the APR show no difference in the overall risk of birth defects for lamivudine or zidovudine compared with the background rate for birth defects of 2.7% in the Metropolitan Atlanta Congenital Defects Program (MACDP) reference population (see Data). The APR uses the MACDP as the U.S. reference population for birth defects in the general population. The MACDP evaluates women and infants from a limited geographic area and does not include outcomes for births that occurred at less than 20 weeks' gestation. The rate of miscarriage is not reported in the APR. The estimated background rate of miscarriage in clinically recognized pregnancies in the U.S. general population is 15% to 20%. The background risk for major birth defects and miscarriage for the indicated population is unknown.
Hyperlactatemia, which may be due to mitochondrial dysfunction, has been reported in infants with in utero exposure to zidovudine-containing products. These events were transient and asymptomatic in most cases. There have been few reports of developmental delay, seizures, and other neurological disease. However, a causal relationship between these events and exposure to zidovudine-containing products in utero or peri-partum has not been established (see Data).
In animal reproduction studies, oral administration of lamivudine to pregnant rabbits during organogenesis resulted in embryolethality at systemic exposure (AUC) similar to the recommended clinical dose; however, no adverse development effects were observed with oral administration of lamivudine to pregnant rats during organogenesis at plasma concentrations (Cmax) 35 times the recommended clinical dose. Administration of oral zidovudine to female rats prior to mating and throughout gestation resulted in embryotoxicity at doses that produced systemic exposure (AUC) approximately 33 times higher than exposure at the recommended clinical dose. However, no embryotoxicity was observed after oral administration of zidovudine to pregnant rats during organogenesis at doses that produced systemic exposure (AUC) approximately 117 times higher than exposures at the recommended clinical dose. Administration of oral zidovudine to pregnant rabbits during organogenesis resulted in embryotoxicity at doses that produced systemic exposure (AUC) approximately 108 times higher than exposure at the recommended clinical dose. However, no embryotoxicity was observed at doses that produced systemic exposure (AUC) approximately 23 times higher than exposures at the recommended clinical dose (see Data).
Data
Human Data: Lamivudine: Based on prospective reports to the APR of over 11,000 exposures to lamivudine during pregnancy resulting in live births (including over 4,500 exposed in the first trimester), there was no difference between the overall risk of birth defects for lamivudine compared with the background birth defect rate of 2.7% in a U.S. reference population of the MACDP. The prevalence of birth defects in live births was 3.1% (95% CI: 2.6% to 3.6%) following first trimester exposure to lamivudine-containing regimens and 2.8% (95% CI: 2.5% to 3.3%) following second/third trimester exposure to lamivudine-containing regimens.
Lamivudine pharmacokinetics were studied in pregnant women during 2 clinical trials conducted in South Africa. The trial assessed pharmacokinetics in 16 women at 36 weeks' gestation using 150 mg lamivudine twice daily with zidovudine, 10 women at 38 weeks' gestation using 150 mg lamivudine twice daily with zidovudine, and 10 women at 38 weeks' gestation using lamivudine 300 mg twice daily without other antiretrovirals. These trials were not designed or powered to provide efficacy information. Lamivudine concentrations were generally similar in maternal, neonatal, and umbilical cord serum samples. In a subset of subjects, amniotic fluid specimens were collected following natural rupture of membranes and confirmed that lamivudine crosses the placenta in humans. Based on limited data at delivery, median (range) amniotic fluid concentrations of lamivudine were 3.9 (1.2 to 12.8)–fold greater compared with paired maternal serum concentration (n = 8).
Zidovudine: Based on prospective reports to the APR of over 13,000 exposures to zidovudine during pregnancy resulting in live births (including over 4,000 exposed in the first trimester), there was no difference between the overall risk of birth defects for zidovudine compared with the background birth defect rate of 2.7% in a U.S. reference population of the MACDP. The prevalence of birth defects in live births was 3.2% (95% CI: 2.7% to 3.8%) following first trimester exposure to zidovudine-containing regimens and 2.8% (95% CI: 2.5% to 3.2%) following second/third trimester exposure to zidovudine-containing regimens.
A randomized, double-blind, placebo-controlled trial was conducted in HIV-1-infected pregnant women to determine the utility of zidovudine for the prevention of maternal-fetal HIV-1 transmission. Zidovudine treatment during pregnancy reduced the rate of maternal-fetal HIV-1 transmission from 24.9% for infants born to placebo-treated mothers to 7.8% for infants born to mothers treated with zidovudine. There were no differences in pregnancy-related adverse events between the treatment groups. Of the 363 neonates that were evaluated, congenital abnormalities occurred with similar frequency between neonates born to mothers who received zidovudine and neonates born to mothers who received placebo. The observed abnormalities included problems in embryogenesis (prior to 14 weeks) or were recognized on ultrasound before or immediately after initiation of trial drug [see Clinical Studies (14.2)].
Zidovudine has been shown to cross the placenta and concentrations in neonatal plasma at birth were essentially equal to those in maternal plasma at delivery [see Clinical Pharmacology (12.3)]. There have been reports of mild, transient elevations in serum lactate levels, which may be due to mitochondrial dysfunction, in neonates and infants exposed in utero or peri-partum to zidovudine-containing products. There have been few reports of developmental delay, seizures, and other neurological disease. However, a causal relationship between these events and exposure to zidovudine-containing products in utero or peri-partum has not been established. The clinical relevance of transient elevations in serum lactate is unknown.
Animal Data: Lamivudine: Lamivudine was administered orally to pregnant rats (at 90, 600, and 4,000 mg per kg per day) and rabbits (at 90, 300, and 1,000 mg per kg per day and at 15, 40, and 90 mg per kg per day) during organogenesis (on gestation Days 7 through 16 [rat] and 8 through 20 [rabbit]). No evidence of fetal malformations due to lamivudine was observed in rats and rabbits at doses producing plasma concentrations (Cmax) approximately 35 times higher than human exposure at the recommended daily dose. Evidence of early embryolethality was seen in the rabbit at system exposures (AUC) similar to those observed in humans, but there was no indication of this effect in the rat at plasma concentrations (Cmax) 35 times higher than human exposure at the recommended daily dose. Studies in pregnant rats showed that lamivudine is transferred to the fetus through the placenta. In the fertility/pre-and postnatal development study in rats, lamivudine was administered orally at doses of 180, 900, and 4,000 mg per kg per day (from prior to mating through postnatal Day 20). In the study, development of the offspring, including fertility and reproductive performance, was not affected by maternal administration of lamivudine.
Zidovudine: A study in pregnant rats (at 50, 150, or 450 mg per kg per day starting 26 days prior to mating through gestation to postnatal Day 21) showed increased fetal resorptions at doses that produced systemic exposures (AUC) approximately 33 times higher than exposure at the recommended daily human dose (300 mg twice daily). However, in an oral embryo-fetal development study in rats (at 125, 250, or 500 mg per kg per day on gestation Days 6 through 15), no fetal resorptions were observed at doses that produced systemic exposure (AUC) approximately 117 times higher than exposures at the recommended daily human dose. An oral embryo-fetal development study in rabbits (at 75, 150, or 500 mg per kg per day on gestation Days 6 through 18) showed increased fetal resorptions at the 500 mg per kg per day dose, which produced systemic exposures (AUC) approximately 108 times higher than exposure at the recommended daily human dose; however, no fetal resorptions were noted at doses up to 150 mg per kg per day, which produced systemic exposure (AUC) approximately 23 times higher than exposures at the recommended daily human dose. These oral embryo-fetal development studies in the rat and rabbit revealed no evidence of fetal malformations with zidovudine. In another developmental toxicity study, pregnant rats (dosed at 3,000 mg per kg per day from Days 6 through 15 of gestation) showed marked maternal toxicity and an increased incidence of fetal malformations at exposures greater than 300 times the recommended daily human dose based on AUC. However, there were no signs of fetal malformations at doses up to 600 mg per kg per day.
8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommends that HIV-1-infected mothers in the United States not breastfeed their infants to avoid risking postnatal transmission of HIV-1 infection. Lamivudine and zidovudine are present in human milk. There is no information on the effects of lamivudine or zidovudine on the breastfed infant or the effects of the drugs on milk production. Because of the potential for (1) HIV-1 transmission (in HIV-negative infants), (2) developing viral resistance (in HIV-positive infants), and (3) adverse reactions in a breastfed infant similar to those seen in adults, instruct mothers not to breastfeed if they are receiving lamivudine and zidovudine.
8.4 Pediatric Use
Lamivudine and zidovudine is not recommended for use in pediatric patients who weigh less than 30 kg because it is a fixed-dose combination tablet that cannot be adjusted for this patient population [see Dosage and Administration (2.2)].
8.5 Geriatric Use
Clinical trials of lamivudine and zidovudine did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, caution should be exercised in the administration of lamivudine and zidovudine in elderly patients reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)].
8.6 Patients with Impaired Renal Function
Lamivudine and zidovudine is not recommended for patients with creatinine clearance less than 50 mL per min because lamivudine and zidovudine is a fixed-dose combination and the dosage of the individual components cannot be adjusted. If a dose reduction of the lamivudine or zidovudine components of lamivudine and zidovudine is required for patients with renal impairment then the individual components should be used [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)].
8.7 Patients with Impaired Hepatic Function
Lamivudine and zidovudine is a fixed-dose combination and the dosage of the individual components cannot be adjusted. Zidovudine is primarily eliminated by hepatic metabolism and zidovudine concentrations are increased in patients with impaired hepatic function, which may increase the risk of hematologic toxicity. Frequent monitoring of hematologic toxicities is advised.
10 OVERDOSAGE
There is no known specific treatment for overdose with lamivudine and zidovudine. If overdose occurs, the patient should be monitored and standard supportive treatment applied as required.
Lamivudine
Because a negligible amount of lamivudine was removed via (4-hour) hemodialysis, continuous ambulatory peritoneal dialysis, and automated peritoneal dialysis, it is not known if continuous hemodialysis would provide clinical benefit in a lamivudine overdose event.
Zidovudine
Acute overdoses of zidovudine have been reported in pediatric patients and adults. These involved exposures up to 50 grams. No specific symptoms or signs have been identified following acute overdosage with zidovudine apart from those listed as adverse events such as fatigue, headache, vomiting, and occasional reports of hematological disturbances. Patients recovered without permanent sequelae. Hemodialysis and peritoneal dialysis appear to have a negligible effect on the removal of zidovudine, while elimination of its primary metabolite, 3'-azido-3'-deoxy-5'-O-β-D-glucopyranuronosylthymidine (GZDV), is enhanced.
11 DESCRIPTION
Lamivudine and Zidovudine tablets USP are combination tablets containing lamivudine and zidovudine. Lamivudine (EPIVIR) and zidovudine (RETROVIR, azidothymidine, AZT, or ZDV) are synthetic nucleoside analogues with activity against HIV-1.
Lamivudine and Zidovudine tablets USP are for oral administration. Each film-coated tablet contains 150 mg of lamivudine, 300 mg of zidovudine, and the inactive ingredients colloidal silicon dioxide, hypromellose, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polysorbate 80, sodium starch glycolate, and titanium dioxide.
Lamivudine
The chemical name of lamivudine is (2R,cis)-4-amino-l-(2-hydroxymethyl-l,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one. Lamivudine is the (-)enantiomer of a dideoxy analogue of cytidine. Lamivudine has also been referred to as (-)2',3'-dideoxy, 3'-thiacytidine. It has a molecular formula of C8H11N3O3S and a molecular weight of 229.3 g per mol. It has the following structural formula:
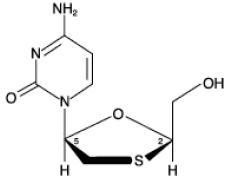
Lamivudine USP is a white to off-white crystalline solid and is soluble in water.
Zidovudine
The chemical name of zidovudine is 3’-azido-3’-deoxythymidine. It has a molecular formula of C10H13N5O4 and a molecular weight of 267.24 g per mol. It has the following structural formula:
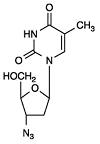
Zidovudine USP is a white to light yellowish powder with a solubility of 20.1 mg per mL in water at 25°C.
Meets USP Dissolution Test 2.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lamivudine and Zidovudine is an antiretroviral agent [see Microbiology (12.4)].
12.3 Pharmacokinetics
Pharmacokinetics in Adults
One lamivudine and zidovudine tablet was bioequivalent to 1 EPIVIR tablet (150 mg) plus 1 RETROVIR tablet (300 mg) following single-dose administration to fasting healthy subjects (n = 24).
Lamivudine: Following oral administration, lamivudine is rapidly absorbed and extensively distributed. Binding to plasma protein is low. Approximately 70% of an intravenous dose of lamivudine is recovered as unchanged drug in the urine. Metabolism of lamivudine is a minor route of elimination (approximately 5% of an oral dose after 12 hours). In humans, the only known metabolite is the trans-sulfoxide metabolite (approximately 5% of an oral dose after 12 hours).
Zidovudine: Following oral administration, zidovudine is rapidly absorbed and extensively distributed. Binding to plasma protein is low. Zidovudine is eliminated primarily by hepatic metabolism. The major metabolite of zidovudine is GZDV. GZDV area under the curve (AUC) is about 3-fold greater than the zidovudine AUC. Urinary recovery of zidovudine and GZDV accounts for 14% and 74% of the dose following oral administration, respectively. A second metabolite, 3’-amino-3’-deoxythymidine (AMT), has been identified in plasma. The AMT AUC was one-fifth of the zidovudine AUC.
In humans, lamivudine and zidovudine are not significantly metabolized by cytochrome P450 enzymes.
The pharmacokinetic properties of lamivudine and zidovudine in fasting subjects are summarized in Table 3.
Table 3. Pharmacokinetic Parametersa for Lamivudine and Zidovudine in Adults
| Parameter
| Lamivudine
| Zidovudine
|
||
| Oral bioavailability (%) | 86 ± 16 | n = 12 | 64 ± 10 | n = 5 |
| Apparent volume of distribution (L/kg) | 1.3 ± 0.4 | n = 20 | 1.6 ± 0.6 | n = 8 |
| Plasma protein binding (%) | <36 | <38 |
||
| CSF:plasma ratiob
| 0.12 [0.04 to 0.47] | n = 38c
| 0.60 [0.04 to 2.62] | n = 39d
|
| Systemic clearance (L/h/kg) | 0.33 ± 0.06 | n = 20 | 1.6 ± 0.6 | n = 6 |
| Renal clearance (L/h/kg) | 0.22 ± 0.06 | n = 20 | 0.34 ± 0.05 | n = 9 |
| Elimination half-life (h)e
| 5 to 7 | 0.5 to 3 |
||
a Data presented as mean ± standard deviation except where noted.
b Median [range].
c Children.
d Adults.
e Approximate range.
Effect of Food on Absorption of Lamivudine and Zidovudine: Lamivudine and Zidovudine may be administered with or without food. The lamivudine and zidovudine AUC following administration of lamivudine and zidovudine with food was similar when compared with fasting healthy subjects (n = 24).
Specific Populations
Patients with Renal Impairment: Lamivudine and Zidovudine: The effect of renal impairment on the combination of lamivudine and zidovudine has not been evaluated (see the U.S. prescribing information for the individual lamivudine and zidovudine components).
Patients with Hepatic Impairment: Lamivudine and Zidovudine: The effect of hepatic impairment on the combination of lamivudine, and zidovudine has not been evaluated (see the U.S. prescribing information for the individual lamivudine and zidovudine components).
Pregnant Women: Lamivudine: Lamivudine pharmacokinetics were studied in 36 pregnant women during 2 clinical trials conducted in South Africa. Lamivudine pharmacokinetics in pregnant women were similar to those seen in non-pregnant adults and in postpartum women. Lamivudine concentrations were generally similar in maternal, neonatal, and umbilical cord serum samples.
Zidovudine: Zidovudine pharmacokinetics have been studied in a Phase 1 trial of 8 women during the last trimester of pregnancy. Zidovudine pharmacokinetics were similar to those of non-pregnant adults. Consistent with passive transmission of the drug across the placenta, zidovudine concentrations in neonatal plasma at birth were essentially equal to those in maternal plasma at delivery.
Although data are limited, methadone maintenance therapy in 5 pregnant women did not appear to alter zidovudine pharmacokinetics.
Geriatric Patients: The pharmacokinetics of lamivudine and zidovudine have not been studied in subjects over 65 years of age.
Male and Female Patients: There are no significant or clinically relevant gender differences in the pharmacokinetics of the individual components (lamivudine or zidovudine) based on the available information that was analyzed for each of the individual components.
Racial Groups: Lamivudine: There are no significant or clinically relevant racial differences in lamivudine pharmacokinetics based on the available information that was analyzed for the individual lamivudine component.
Zidovudine: The pharmacokinetics of zidovudine with respect to race have not been determined.
Drug Interaction Studies
No drug interaction trials have been conducted using lamivudine and zidovudine tablets.
Lamivudine and Zidovudine: No clinically significant alterations in lamivudine or zidovudine pharmacokinetics were observed in 12 asymptomatic HIV-1-infected adult subjects given a single dose of zidovudine (200 mg) in combination with multiple doses of lamivudine (300 mg every 12 hours).
Interferon Alfa: There was no significant pharmacokinetic interaction between lamivudine and interferon alfa in a trial of 19 healthy male subjects.
Ribavirin: In vitro data indicate ribavirin reduces phosphorylation of lamivudine, stavudine, and zidovudine. However, no pharmacokinetic (e.g., plasma concentrations or intracellular triphosphorylated active metabolite concentrations) or pharmacodynamic (e.g., loss of HIV-1/HCV virologic suppression) interaction was observed when ribavirin and lamivudine (n = 18), stavudine (n = 10), or zidovudine (n = 6) were coadministered as part of a multi-drug regimen to HIV-1/HCV co-infected subjects.
Sorbitol (Excipient): Lamivudine and sorbitol solutions were coadministered to 16 healthy adult subjects in an open-label, randomized-sequence, 4-period, crossover trial. Each subject received a single 300 mg dose of lamivudine oral solution alone or coadministered with a single dose of 3.2 grams, 10.2 grams, or 13.4 grams of sorbitol in solution. Coadministration of lamivudine with sorbitol resulted in dose-dependent decreases of 20%, 39%, and 44% in the AUC(0-24); 14%, 32%, and 36% in the AUC(∞); and 28%, 52%, and 55% in the Cmax: of lamivudine, respectively.
Table 4 presents drug interaction information for the individual components of lamivudine and zidovudine.
Table 4. Effect of Coadministered Drugs on Lamivudine and Zidovudine AUCa
| Coadministered Drug and Dose | Drug and Dose | n
| Concentrations of Lamivudine or Zidovudine
| Concentration of
Coadministered Drug |
|
| AUC
| Variability
|
||||
| Nelfinavir 750 mg every 8 h x 7 to 10 days | Lamivudine single 150 mg | 11 | ↑10% | 95% CI: 1% to 20% | ↔ |
| Trimethoprim 160 mg/ Sulfamethoxazole 800 mg daily x 5 days | Lamivudine single 300 mg | 14 | ↑43% | 90% CI: 32% to 55% | ↔ |
| Atovaquone 750 mg every 12 h with food | Zidovudine 200 mg every 8 h | 14 | ↑31% | Range: 23% to 78%b | ↔ |
| Clarithromycin 500 mg twice daily | Zidovudine 100 mg every 4 h x 7 days | 4 | ↓12% | Range: ↓34% to ↑14% | Not Reported |
| Fluconazole 400 mg daily | Zidovudine 200 mg every 8 h | 12 | ↑74% | 95% CI: 54% to 98% | Not Reported |
| Methadone 30 to 90 mg daily | Zidovudine 200 mg every 4 h | 9 | ↑43% | Range: 16% to 64% b | ↔ |
| Nelfinavir 750 mg every 8 h x 7 to 10 days | Zidovudine single 200 mg | 11 | ↓35% | Range: 28% to 41% | ↔ |
| Probenecid 500 mg every 6 h x 2 days | Zidovudine 2 mg/kg every 8 h x 3 days | 3 | ↑106% | Range: 100% to 170% b | Not Assessed |
| Rifampin 600 mg daily x 14 days | Zidovudine 200 mg every 8 h x 14 days | 8 | ↓47% | 90% CI: 41% to 53% | Not Assessed |
| Ritonavir 300 mg every 6 h x 4 days | Zidovudine 200 mg every 8 h x 4 days | 9 | ↓25 % | 95% CI: 15% to 34% | ↔ |
| Valproic acid 250 mg or 500 mg every 8 h x 4 days | Zidovudine 100 mg every 8 h x 4 days | 6 | ↑80% | Range: 64% to 130% b | Not Assessed |
↑ = Increase; ↓ = Decrease; ↔ = No significant change; AUC = Area under the concentration versus time curve; CI = Confidence interval.
a This table is not all inclusive.
b Estimated range of percent difference.
12.4 Microbiology
Mechanism of Action
Lamivudine: Lamivudine is a synthetic nucleoside analogue. Intracellularly, lamivudine is phosphorylated to its active 5’-triphosphate metabolite, lamivudine triphosphate (3TC-TP). The principal mode of action of 3TC-TP is inhibition of reverse transcriptase (RT) via DNA chain termination after incorporation of the nucleotide analogue.
Zidovudine: Zidovudine is a synthetic nucleoside analogue. Intracellularly, zidovudine is phosphorylated to its active 5’-triphosphate metabolite, zidovudine triphosphate (ZDV-TP). The principal mode of action of ZDV-TP is inhibition of RT via DNA chain termination after incorporation of the nucleotide analogue.
Antiviral Activity
Lamivudine plus Zidovudine: In HIV-1-infected MT-4 cells, lamivudine in combination with zidovudine at various ratios was not antagonistic.
Lamivudine: The antiviral activity of lamivudine against HIV-1 was assessed in a number of cell lines including monocytes and fresh human peripheral blood lymphocytes (PBMCs) using standard susceptibility assays. EC50 values were in the range of 0.003 to 15 microM (1 microM = 0.23 mcg per mL). The median EC50 values of lamivudine were 60 nM (range: 20 to 70 nM), 35 nM (range: 30 to 40 nM), 30 nM (range: 20 to 90 nM), 20 nM (range: 3 to 40 nM), 30 nM (range: 1 to 60 nM), 30 nM (range: 20 to 70 nM), 30 nM (range: 3 to 70 nM), and 30 nM (range: 20 to 90 nM) against HIV-1 clades A-G and group O viruses (n = 3 except n = 2 for clade B) respectively. The EC50 values against HIV-2 isolates (n = 4) ranged from 0.003 to 0.120 microM in PBMCs. Ribavirin (50 microM) used in the treatment of chronic HCV infection decreased the anti-HIV-1 activity of lamivudine by 3.5-fold in MT-4 cells.
Zidovudine: The antiviral activity of zidovudine against HIV-1 was assessed in a number of cell lines including monocytes and fresh human peripheral blood lymphocytes. The EC50 and EC90 values for zidovudine were 0.01 to 0.49 microM (1 microM = 0.27 mcg per mL) and 0.1 to 9 microM, respectively. HIV-1 from therapy-naive subjects with no amino acid substitutions associated with resistance gave median EC50 values of 0.011 microM (range: 0.005 to 0.110 microM) from Virco (n = 92 baseline samples) and 0.0017 microM (range: 0.006 to 0.0340 microM) from Monogram Biosciences (n = 135 baseline samples). The EC50 values of zidovudine against different HIV-1 clades (A-G) ranged from 0.00018 to 0.02 microM, and against HIV-2 isolates from 0.00049 to 0.004 microM. Ribavirin has been found to inhibit the phosphorylation of zidovudine in cell culture.
Neither lamivudine nor zidovudine was antagonistic to tested anti-HIV agents, with the exception of stavudine where an antagonistic relationship with zidovudine has been demonstrated in cell culture. See full prescribing information for EPIVIR (lamivudine) and RETROVIR (zidovudine).
Resistance
In subjects receiving lamivudine monotherapy or combination therapy with lamivudine plus zidovudine, HIV-1 isolates from most subjects became phenotypically and genotypically resistant to lamivudine within 12 weeks.
HIV-1 strains resistant to both lamivudine and zidovudine have been isolated from subjects after prolonged lamivudine/zidovudine therapy. Dual resistance required the presence of multiple amino acid substitutions, the most essential of which may be G333E. The incidence of dual resistance and the duration of combination therapy required before dual resistance occurs are unknown.
Lamivudine: Lamivudine-resistant isolates of HIV-1 have been selected in cell culture and have also been recovered from subjects treated with lamivudine or lamivudine plus zidovudine. Genotypic analysis of isolates selected in cell culture and recovered from lamivudine-treated subjects showed that the resistance was due to a specific amino acid substitution in the HIV-1 reverse transcriptase at codon 184 changing the methionine to either valine or isoleucine (M184V/I).
Zidovudine: HIV-1 isolates with reduced susceptibility to zidovudine have been selected in cell culture and were also recovered from subjects treated with zidovudine. Genotypic analyses of the isolates selected in cell culture and recovered from zidovudine-treated subjects showed thymidine analogue mutation (TAM) substitutions in HIV-1 RT (M41L, D67N, K70R, L210W, T215Y or F, and K219E/R/H/Q/N) that confer zidovudine resistance. In general, higher levels of resistance were associated with greater number of substitutions.
In some subjects harboring zidovudine-resistant virus at baseline, phenotypic sensitivity to zidovudine was restored by 12 weeks of treatment with lamivudine and zidovudine.
Cross-Resistance
Cross-resistance has been observed among NRTIs. Cross-resistance between lamivudine and zidovudine has not been reported. In some subjects treated with lamivudine alone or in combination with zidovudine, isolates have emerged with a substitution at codon 184, which confers resistance to lamivudine.
TAM substitutions are selected by zidovudine and confer cross-resistance to abacavir, didanosine, stavudine, and tenofovir.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
Lamivudine: Long-term carcinogenicity studies with lamivudine in mice and rats showed no evidence of carcinogenic potential at exposures up to 10 times (mice) and 58 times (rats) the human exposures at the recommended dose of 300 mg.
Zidovudine: Zidovudine was administered orally at 3 dosage levels to separate groups of mice and rats (60 females and 60 males in each group). Initial single daily doses were 30, 60, and 120 mg per kg per day in mice and 80, 220, and 600 mg per kg per day in rats. The doses in mice were reduced to 20, 30, and 40 mg per kg per day after Day 90 because of treatment-related anemia, whereas in rats only the high dose was reduced to 450 mg per kg per day on Day 91 and then to 300 mg per kg per day on Day 279.
In mice, 7 late-appearing (after 19 months) vaginal neoplasms (5 non-metastasizing squamous cell carcinomas, 1 squamous cell papilloma, and 1 squamous polyp) occurred in animals given the highest dose. One late-appearing squamous cell papilloma occurred in the vagina of a middle-dose animal. No vaginal tumors were found at the lowest dose.
In rats, 2 late-appearing (after 20 months), non-metastasizing vaginal squamous cell carcinomas occurred in animals given the highest dose. No vaginal tumors occurred at the low or middle dose in rats. No other drug-related tumors were observed in either sex of either species.
At doses that produced tumors in mice and rats, the estimated drug exposure (as measured by AUC) was approximately 3 times (mouse) and 24 times (rat) the estimated human exposure at the recommended therapeutic dose of 100 mg every 4 hours.
It is not known how predictive the results of rodent carcinogenicity studies may be for humans.
Mutagenicity
Lamivudine: Lamivudine was mutagenic in an L5178Y mouse lymphoma assay and clastogenic in a cytogenetic assay using cultured human lymphocytes. Lamivudine was not mutagenic in a microbial mutagenicity assay, in an in vitro cell transformation assay, in a rat micronucleus test, in a rat bone marrow cytogenetic assay, and in an assay for unscheduled DNA synthesis in rat liver.
Zidovudine: Zidovudine was mutagenic in an L5178Y mouse lymphoma assay, positive in an in vitro cell transformation assay, clastogenic in a cytogenetic assay using cultured human lymphocytes, and positive in mouse and rat micronucleus tests after repeated doses. It was negative in a cytogenetic study in rats given a single dose.
Impairment of Fertility
Lamivudine: Lamivudine did not affect male or female fertility in rats at doses up to 4,000 mg per kg per day, associated with concentrations approximately 42 times (male) or 63 times (female) higher than the concentrations (Cmax) in humans at the dose of 300 mg.
Zidovudine: Zidovudine, administered to male and female rats at doses up to 450 mg per kg per day, which is 7 times the recommended adult dose (300 mg twice daily) based on body surface area, had no effect on fertility based on conception rates.
14 CLINICAL STUDIES
One lamivudine and zidovudine tablet given twice daily is an alternative regimen to EPIVIR tablets 150 mg twice daily plus RETROVIR 600 mg per day in divided doses.
14.1 Adults
The NUCB3007 (CAESAR) trial was conducted using EPIVIR 150 mg tablets (150 mg twice daily) and RETROVIR 100 mg capsules (2 x 100 mg 3 times daily). CAESAR was a multi-center, double-blind, placebo-controlled trial comparing continued current therapy (zidovudine alone [62% of subjects] or zidovudine with didanosine or zalcitabine [38% of subjects]) to the addition of EPIVIR or EPIVIR plus an investigational non-nucleoside reverse transcriptase inhibitor, randomized 1:2:1. A total of 1,816 HIV-1-infected adults with 25 to 250 (median 122) CD4 cells per mm3 at baseline were enrolled: median age was 36 years, 87% were male, 84% were nucleoside-experienced, and 16% were therapy-naive. The median duration on trial was 12 months. Results are summarized in Table 5.
| Endpoint | Current Therapy (n = 460) | EPIVIR plus Current Therapy (n = 896) | EPIVIR plus a NNRTIa plus Current Therapy (n = 460) |
|---|---|---|---|
| a An investigational non-nucleoside reverse transcriptase inhibitor not approved in the United States. |
|||
| HIV-1 progression or death | 90 (19.6%) | 86 (9.6%) | 41 (8.9%) |
| Death | 27 (5.9%) | 23 (2.6%) | 14 (3.0%) |
14.2 Prevention of Maternal-Fetal HIV-1 Transmission
The utility of zidovudine alone for the prevention of maternal-fetal HIV-1 transmission was demonstrated in a randomized, double-blind, placebo-controlled trial conducted in HIV-1-infected pregnant women with CD4+ cell counts of 200 to 1,818 cells per mm3 (median in the treated group: 560 cells per mm3) who had little or no previous exposure to zidovudine. Oral zidovudine was initiated between 14 and 34 weeks of gestation (median 11 weeks of therapy) followed by IV administration of zidovudine during labor and delivery. Following birth, neonates received oral zidovudine syrup for 6 weeks. The trial showed a statistically significant difference in the incidence of HIV-1 infection in the neonates (based on viral culture from peripheral blood) between the group receiving zidovudine and the group receiving placebo. Of 363 neonates evaluated in the trial, the estimated risk of HIV-1 infection was 7.8% in the group receiving zidovudine and 24.9% in the placebo group, a relative reduction in transmission risk of 68.7%. Zidovudine was well tolerated by mothers and infants. There was no difference in pregnancy-related adverse events between the treatment groups.
16 HOW SUPPLIED/STORAGE AND HANDLING
Lamivudine and Zidovudine Tablets USP, containing 150 mg lamivudine and 300 mg zidovudine, are white to off-white, modified capsule shaped, biconvex, film-coated tablets with deep breakline in between ‘J’ and ‘58’ on one side and deep breakline on the other side. They are available as follows:
Bottles of 60 NDC 65862-597-60
Store at 20° to 25°C (68° to 77°F). [See USP Controlled Room Temperature.]
17 PATIENT COUNSELING INFORMATION
Neutropenia and Anemia
Inform patients that the important toxicities associated with zidovudine are neutropenia and/or anemia. Inform them of the extreme importance of having their blood counts followed closely while on therapy, especially for patients with advanced HIV-1 disease [see Boxed Warning, Warnings and Precautions (5.1)].
Myopathy
Inform patients that myopathy and myositis with pathological changes, similar to that produced by HIV-1 disease, have been associated with prolonged use of zidovudine [see Warnings and Precautions (5.2)].
Lactic Acidosis/Hepatomegaly with Steatosis
Advise patients that lactic acidosis and severe hepatomegaly with steatosis have been reported with use of nucleoside analogues and other antiretrovirals. Advise patients to stop taking lamivudine and zidovudine if they develop clinical symptoms suggestive of lactic acidosis or pronounced hepatotoxicity [see Warnings and Precautions (5.3)].
Patients with Hepatitis B or C Co-infection
Advise patients co-infected with HIV-1 and HBV that worsening of liver disease has occurred in some cases when treatment with lamivudine was discontinued. Advise patients to discuss any changes in regimen with their healthcare provider [see Warnings and Precautions (5.4)].
Inform patients with HIV-1/HCV co-infection that hepatic decompensation (some fatal) has occurred in HIV-1/HCV co-infected patients receiving combination antiretroviral therapy for HIV-1 and interferon alfa with or without ribavirin [see Warnings and Precautions (5.5)].
Drug Interactions
Advise patients that other medications may interact with lamivudine and zidovudine and certain medications, including ganciclovir, interferon alfa, and ribavirin, may exacerbate the toxicity of zidovudine, a component of lamivudine and zidovudine [see Drug Interactions (7.1)].
Immune Reconstitution Syndrome
Advise patients to inform their healthcare provider immediately of any signs and symptoms of infection as inflammation from previous infection may occur soon after combination antiretroviral therapy, including when lamivudine and zidovudine is started [see Warnings and Precautions (5.7)].
Lipoatrophy
Advise patients that loss of subcutaneous fat may occur in patients receiving lamivudine and zidovudine and that they will be regularly assessed during therapy [see Warnings and Precautions (5.8)].
Pregnancy Registry
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to lamivudine and zidovudine during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Instruct women with HIV-1 infection not to breastfeed because HIV-1 can be passed to the baby in the breast milk [see Use in Specific Populations (8.2)].
Missed Dose
Instruct patients that if they miss a dose of lamivudine and zidovudine, to take it as soon as they remember. Advise patients not to double their next dose or take more than the prescribed dose [see Dosage and Administration (2)].
Trademarks are owned by or licensed to the ViiV Healthcare group of companies.
Distributed by:
Aurobindo Pharma USA, Inc.
279 Princeton-Hightstown Road
East Windsor, NJ 08520
Manufactured by:
Aurobindo Pharma Limited
Hyderabad-500 038, India
Revised: 06/2019

| LAMIVUDINE AND ZIDOVUDINE
lamivudine and zidovudine tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Aurobindo Pharma Limited (650082092) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Aurobindo Pharma Limited | 650381903 | ANALYSIS(65862-597) , MANUFACTURE(65862-597) | |