EVUSHELD- azd7442
AstraZeneca Pharmaceuticals LP
----------
FACT SHEET FOR HEALTHCARE PROVIDERS: EMERGENCY USE AUTHORIZATION FOR EVUSHELD™ (tixagevimab co-packaged with cilgavimab)
|
HIGHLIGHTS OF EMERGENCY USE AUTHORIZATION (EUA) These highlights of the EUA do not include all the information needed to use EVUSHELD™ under the EUA. See the FULL FACT SHEET FOR HEALTHCARE PROVIDERS for EVUSHELD. EVUSHELD (tixagevimab) injection; (cilgavimab) injection, co-packaged for intramuscular use Original EUA Authorized Date: 12/2021 Revised EUA Authorized Date: 01/2023 -----------RECENT MAJOR CHANGES---------- Limitations of Authorized Use: updated based on variant susceptibility 01/2023 Microbiology (12.4): updated neutralizing data 01/2023 Microbiology (12.4): updated neutralizing data 12/2022 Microbiology (12.4): updated neutralizing data 11/2022 Emergency Use Authorization (1): updated examples 10/2022 Warnings and Precautions (5.3, 17): addition of a new warning 10/2022 Microbiology (12.4): updated neutralizing data 10/2022 Dosage and Administration (2.1, 17): modification of initial dosage and repeat dosing 06/2022 Microbiology (12.4): updated neutralizing data 06/2022 Warnings and Precautions (5.2): addition of new warning 05/2022 Dosage and Administration (2.3) 05/2022 Adverse Reactions (6.1, 12.3): addition of TACKLE data 02/2022 -------------EUA FOR EVUSHELD--------------- The U.S. Food and Drug Administration has issued an EUA for the emergency use of the unapproved product EVUSHELD (tixagevimab co-packaged with cilgavimab), SARS-CoV-2 spike protein-directed attachment inhibitor, for the pre-exposure prophylaxis of coronavirus disease 2019 (COVID-19) in adults and pediatric individuals (12 years of age and older weighing at least 40 kg):
EVUSHELD may only be prescribed for an individual patient by physicians, advanced practice registered nurses, and physician assistants that are licensed or authorized under state law to prescribe drugs in the therapeutic class to which EVUSHELD belongs (i.e., anti-infectives). EVUSHELD has been authorized by FDA for the emergency use described above. EVUSHELD is not FDA-approved for any use, including use for pre-exposure prophylaxis of COVID-19. (1) EVUSHELD is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of EVUSHELD under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner. LIMITATIONS OF AUTHORIZED USE
|
See Full Fact Sheet for Healthcare Providers for examples of medical conditions or treatments that may result in moderate to severe immune compromise and an inadequate immune response to COVID-19 vaccination, the justification for emergency use of drugs during the COVID-19 pandemic, information on available alternatives, and additional information on COVID-19. (1) -------------DOSAGE AND ADMINISTRATION------------ The dosage of EVUSHELD for emergency use is:
See Full Fact Sheet for Healthcare Providers for detail on preparation and administration. (2) --------------DOSAGE FORMS AND STRENGTHS--------- Injection:
--------------CONTRAINDICATIONS---------------------- EVUSHELD is contraindicated in individuals with previous severe hypersensitivity reactions, including anaphylaxis, to EVUSHELD. (4) --------------WARNINGS AND PRECAUTIONS-------------
-------------ADVERSE REACTIONS--------------- Most common adverse events (all grades, incidence ≥3%) are headache, fatigue, and cough. (6.1) You or your designee must report all SERIOUS ADVERSE EVENTS or MEDICATION ERRORS potentially related to EVUSHELD (1) by submitting FDA Form 3500 online, (2) by downloading this form and then submitting by mail or fax, or (3) contacting the FDA at 1-800-FDA-1088 to request this form. Please also provide a copy of this form to AstraZeneca by Fax at 1-866-742-7984 or call 1-800-236-9933. (6.4) See PATIENT AND PARENTS/CAREGIVER FACT SHEET. Revised: 01/2023 |
TABLE OF CONTENTS*
1 EMERGENCY USE AUTHORIZATION
The U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) for the emergency use of the unapproved product EVUSHELD (tixagevimab co-packaged with cilgavimab) for the pre-exposure prophylaxis of coronavirus disease 2019 (COVID-19) in adults and pediatric individuals (12 years of age and older weighing at least 40 kg):
- •
- Who are not currently infected with SARS-CoV-2 and who have not had a known recent exposure to an individual infected with SARS-CoV-2 and
- ∘
- Who have moderate to severe immune compromise due to a medical condition or receipt of immunosuppressive medications or treatments and may not mount an adequate immune response to COVID-19 vaccination or
- ∘
- For whom vaccination with any available COVID-19 vaccine, according to the approved or authorized schedule, is not recommended due to a history of severe adverse reaction to a COVID-19 vaccine(s) and/or COVID-19 vaccine component(s) [see Warnings and Precautions (5.2)].
EVUSHELD may only be prescribed for an individual patient by physicians, advanced practice registered nurses, and physician assistants that are licensed or authorized under state law to prescribe drugs in the therapeutic class to which EVUSHELD belongs (i.e., anti-infectives).
EVUSHELD has been authorized by FDA for the emergency use described above. EVUSHELD is not FDA-approved for any use, including use for pre-exposure prophylaxis of COVID-19.
EVUSHELD is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of EVUSHELD under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
Medical conditions or treatments that may result in moderate to severe immune compromise and an inadequate immune response to COVID-19 vaccination include but are not limited to:
- •
- Active treatment for solid tumor and hematologic malignancies
- •
- Hematologic malignancies associated with poor responses to COVID-19 vaccines regardless of current treatment status (e.g., chronic lymphocytic leukemia, non-Hodgkin lymphoma, multiple myeloma, acute leukemia)
- •
- Receipt of solid-organ transplant or an islet transplant and taking immunosuppressive therapy
- •
- Receipt of chimeric antigen receptor (CAR)-T-cell or hematopoietic stem cell transplant (within 2 years of transplantation or taking immunosuppression therapy)
- •
- Moderate or severe primary immunodeficiency (e.g., common variable immunodeficiency disease, severe combined immunodeficiency, DiGeorge syndrome, Wiskott-Aldrich syndrome)
- •
- Advanced or untreated HIV infection (people with HIV and CD4 cell counts <200/mm3, history of an AIDS-defining illness without immune reconstitution, or clinical manifestations of symptomatic HIV)
- •
- Active treatment with high-dose corticosteroids (i.e., ≥20 mg prednisone or equivalent per day when administered for ≥2 weeks), alkylating agents, antimetabolites, transplant-related immunosuppressive drugs, cancer chemotherapeutic agents classified as severely immunosuppressive, and biologic agents that are immunosuppressive or immunomodulatory (e.g., B-cell depleting agents)
LIMITATIONS OF AUTHORIZED USE
- •
- EVUSHELD is not authorized for use in individuals:
- ∘
- For treatment of COVID-19, or
- ∘
- For post-exposure prophylaxis of COVID-19 in individuals who have been exposed to someone infected with SARS-CoV-2.
- •
- EVUSHELD is authorized for use only when the combined frequency of non-susceptible
- variants nationally is less than or equal to 90%, based on available information including
- variant susceptibility to EVUSHELD and national variant frequencies1.
- •
- Pre-exposure prophylaxis with EVUSHELD is not a substitute for vaccination in individuals for whom COVID-19 vaccination is recommended. Individuals for whom COVID-19 vaccination is recommended, including individuals with moderate to severe immune compromise who may derive benefit from COVID-19 vaccination, should receive COVID-19 vaccination.
- •
- In individuals who have received a COVID-19 vaccine, EVUSHELD should be administered at least two weeks after vaccination.
Justification for Emergency Use of Drugs During the COVID-19 Pandemic
There is currently an outbreak of COVID-19 caused by SARS-CoV-2, a novel coronavirus. The Secretary of HHS has declared that:
- •
- A public health emergency related to COVID-19 has existed since January 27, 2020.
- •
- Circumstances exist justifying the authorization of emergency use of drugs and biological products during the COVID-19 pandemic (March 27, 2020 declaration).
An EUA is a FDA authorization for the emergency use of an unapproved product or unapproved use of an approved product (i.e., drug, biological product, or device) in the United States under certain circumstances including, but not limited to, when the Secretary of HHS declares that there is a public health emergency that affects the national security or the health and security of United States citizens living abroad, and that involves biological agent(s) or a disease or condition that may be attributable to such agent(s). Criteria for issuing an EUA include:
- •
- The biological agent(s) can cause a serious or life-threatening disease or condition;
- •
- Based on the totality of the available scientific evidence (including data from adequate and well-controlled clinical trials, if available), it is reasonable to believe that
- ∘
- The product may be effective in diagnosing, treating, or preventing the serious or life-threatening disease or condition; and
- ∘
- The known and potential benefits of the product - when used to diagnose, prevent, or treat such disease or condition - outweigh the known and potential risks of the product, taking into consideration the material threat posed by the biological agent(s);
- •
- There is no adequate, approved, and available alternative to the product for diagnosing, preventing, or treating the serious or life-threatening disease or condition.
Information Regarding Available Alternatives for the EUA Authorized Use
There are no adequate, approved and available alternatives to EVUSHELD for the pre-exposure prophylaxis of COVID-19 in individuals who may not mount an adequate immune response to COVID-19 vaccination or for whom COVID-19 vaccination is not recommended due to a history of severe adverse reaction to a COVID-19 vaccine or its components.
For information on clinical studies of EVUSHELD and other therapies for the prophylaxis of COVID-19, see www.clinicaltrials.gov.
- 1
- FDA will monitor conditions to determine whether use is consistent with the scope of authorization, referring to available information, including information on variant susceptibility (e.g., Section 12.4 of the authorized Fact Sheet for Healthcare Providers) and CDC variant frequency data available at: https://covid.cdc.gov/covid-data-tracker/#variant-proportions.
2 DOSAGE AND ADMINISTRATION
2.1 Dosage for Emergency Use of EVUSHELD
Initial Dosing
The initial dosage of EVUSHELD in adults and pediatric individuals (12 years of age and older weighing at least 40 kg) is 300 mg of tixagevimab and 300 mg of cilgavimab administered as two separate consecutive intramuscular (IM) injections [see Clinical Pharmacology (12.3)]. Refer to Table 1 below.
Dosing for Individuals Who Initially Received 150 mg of Tixagevimab and 150 mg of Cilgavimab
Individuals who have already received the previously authorized initial dose (150 mg of tixagevimab and 150 mg of cilgavimab) should receive an additional EVUSHELD dose as soon as possible, with the dose based on the following criteria:
- •
- If the patient received their initial dose ≤ 3 months ago, the patient should receive a dose of 150 mg of tixagevimab and 150 mg of cilgavimab, refer to Table 2 below.
- •
- If the patient received their initial dose > 3 months ago, the patient should receive a dose of 300 mg of tixagevimab and 300 mg of cilgavimab, refer to Table 1 below.
Repeat Dosing
The repeat dosage of EVUSHELD in adults and pediatric individuals (12 years of age and older weighing at least 40 kg) is 300 mg of tixagevimab and 300 mg of cilgavimab administered every 6 months, refer to Table 1 below. Repeat dosing should be timed from the date of the most recent EVUSHELD dose.
The recommendations for dosing are based on the totality of the scientific evidence including clinical pharmacology data, antiviral activity data, and clinical trial data [see Clinical Pharmacology (12.3), Microbiology (12.4), and Clinical Studies (14)]. EVUSHELD has only been studied for the prophylaxis of COVID-19 at the EVUSHELD (150 mg of tixagevimab and 150 mg of cilgavimab) dose. There are no data available in a prophylaxis setting for the EVUSHELD (300 mg of tixagevimab and 300 mg of cilgavimab) dose. The clinical safety of the EVUSHELD (300 mg of tixagevimab and 300 mg of cilgavimab) dose is supported by safety data from a treatment study in subjects with mild to moderate COVID-19 [see Adverse Reactions (6.1)]. There are limited safety and no efficacy data available with repeat dosing.
To access the most recent EVUSHELD Fact Sheets, please visit http://www.evusheld.com or scan the QR code:

2.2 Dosage Adjustment in Specific Populations
No dosage adjustment is recommended in pregnant or lactating individuals, in geriatrics, and in individuals with renal impairment [see Use in Specific Populations (8)].
2.3 Dose Preparation and Administration
Each EVUSHELD carton contains two vials; one of each antibody. Each vial contains an overfill to allow the withdrawal of 150 mg (1.5 mL).
|
|||
|
EVUSHELD* (tixagevimab co-packaged with cilgavimab) |
Antibody dose |
Number of vials needed |
Volume to withdraw from vial(s) |
|
tixagevimab 300 mg |
2 vials |
3 mL |
|
|
cilgavimab 300 mg |
2 vials |
3 mL |
|
|
|||
|
EVUSHELD* (tixagevimab co-packaged with cilgavimab) |
Antibody dose |
Number of vials needed |
Volume to withdraw from vial |
|
tixagevimab 150 mg |
1 vial |
1.5 mL |
|
|
cilgavimab 150 mg |
1 vial |
1.5 mL |
|
Preparation
- •
- Tixagevimab and cilgavimab must be prepared by a qualified healthcare provider.
- •
- Tixagevimab and cilgavimab are each supplied in individual single-dose vials. Do not shake the vials.
- •
- Visually inspect the vials for particulate matter and discoloration. Tixagevimab and cilgavimab are clear to opalescent, colorless to slightly yellow solutions. Discard the vials if the solution is cloudy, discolored or visible particles are observed.
- •
- Administer EVUSHELD as TWO separate, consecutive intramuscular (IM) injections, 1 injection of tixagevimab and 1 injection of cilgavimab.
- •
- Withdraw the appropriate amount of tixagevimab solution and the appropriate amount of cilgavimab solution into TWO separate syringes (see Table 1 and Table 2). Discard unused portion in vials.
- •
- This product is preservative-free and therefore, the prepared syringes should be administered immediately. If immediate administration is not possible, and the prepared tixagevimab and cilgavimab syringes need to be stored, the total time from vial puncture to administration must not exceed 4 hours:
- ∘
- in a refrigerator at 2ºC to 8ºC (36ºF to 46ºF), or
- ∘
- at room temperature up to 25ºC (77ºF).
Administration
- •
- Tixagevimab and cilgavimab should be administered by a qualified healthcare provider with appropriate medical support to manage severe hypersensitivity reactions.
- •
- Administer the two components of EVUSHELD consecutively.
- •
- Administer the IM injections at different injection sites, preferably one in each of the gluteal muscles, one after the other.
- ∘
- For the 300 mg tixagevimab and 300 mg cilgavimab dose, ensure that the administration sites are appropriate for the volume (3 mL per injection).
- •
- Clinically monitor individuals after injections and observe for at least 1 hour [see Warnings and Precautions (5.1, 5.2)].
3 DOSAGE FORMS AND STRENGTHS
EVUSHELD is available as an individual single-dose vial of tixagevimab as a clear to opalescent, colorless to slightly yellow solution co-packaged with an individual single-dose vial of cilgavimab as a clear to opalescent, colorless to slightly yellow solution as:
- •
- Injection: 150 mg/1.5 mL (100 mg/mL) of tixagevimab
- •
- Injection: 150 mg/1.5 mL (100 mg/mL) of cilgavimab
4 CONTRAINDICATIONS
EVUSHELD is contraindicated in individuals with previous severe hypersensitivity reactions, including anaphylaxis, to EVUSHELD [see Warnings and Precautions (5.1, 5.2)].
5 WARNINGS AND PRECAUTIONS
There are limited clinical data available for EVUSHELD. Serious and unexpected adverse events may occur that have not been previously reported with EVUSHELD use.
5.1 Hypersensitivity Including Anaphylaxis
Serious hypersensitivity reactions, including anaphylaxis, have been observed with EVUSHELD [see Adverse Reactions (6.1)]. Signs and symptoms of hypersensitivity reactions may include: dyspnea, chills, fatigue/asthenia, tachycardia, chest pain or discomfort, nausea/vomiting, angioedema, dizziness, urticaria, wheezing, pruritus, flushing, hyperhidrosis, myalgia, vaso-vagal reactions (e.g., pre-syncope, syncope), or throat irritation.
Administration of EVUSHELD should be done under the supervision of a healthcare provider with appropriate medical support to manage severe hypersensitivity reactions. If signs and symptoms of a clinically significant hypersensitivity reaction or anaphylaxis occur while taking EVUSHELD, immediately discontinue administration and initiate appropriate medications and/or supportive care. Clinically monitor individuals after injections and observe for at least 1 hour.
5.2 Risk of Cross-Hypersensitivity with COVID-19 Vaccines
EVUSHELD contains polysorbate 80, which is in some COVID-19 vaccines and is structurally similar to polyethylene glycol (PEG), an ingredient in other COVID-19 vaccines [see Description (11)]. For individuals with a history of a severe hypersensitivity reaction to a COVID-19 vaccine, consider consultation with an allergist-immunologist prior to EVUSHELD administration.
Administration of EVUSHELD should be done under the supervision of a healthcare provider with appropriate medical support to manage severe hypersensitivity reactions. If signs and symptoms of a clinically significant hypersensitivity reaction or anaphylaxis occur while taking EVUSHELD, immediately discontinue administration and initiate appropriate medications and/or supportive care. Clinically monitor individuals after injections and observe for at least 1 hour.
5.3 Risk for COVID-19 Due to SARS-CoV-2 Viral Variants Not Neutralized by EVUSHELD
Certain SARS-CoV-2 viral variants may not be neutralized by monoclonal antibodies such as tixagevimab and cilgavimab, the components of EVUSHELD. EVUSHELD may not be effective at preventing COVID-19 caused by these SARS-CoV-2 viral variants. The in-vitro neutralization activity of EVUSHELD against SARS-CoV-2 viral variants is shown in Table 6 [see Microbiology (12.4)].
Inform individuals of the increased risk, compared to other variants, for COVID-19 due to SARS-CoV-2 viral variants not neutralized by EVUSHELD. If signs or symptoms of COVID-19 occur, advise individuals to test for COVID-19 and seek medical attention, including starting treatment for COVID-19 as appropriate. Symptoms of COVID-19 may include: fever or chills, cough, shortness of breath or difficulty breathing, fatigue, muscle or body aches, headache, new loss of taste or smell, sore throat, congestion or runny nose, nausea or vomiting, or diarrhea2 .
- 2
- For additional information on the symptoms of COVID-19, please see https://www.cdc.gov/coronavirus/2019-ncov/symptoms-testing/symptoms.
5.4 Clinically Significant Bleeding Disorders
As with any other intramuscular injection, EVUSHELD should be given with caution to individuals with thrombocytopenia or any coagulation disorder.
5.5 Cardiovascular Events
In PROVENT there was a higher rate of cardiovascular serious adverse events (SAEs), including myocardial infarction (one fatal SAE) and cardiac failure, in subjects who received EVUSHELD compared to placebo [see Adverse Reactions (6.1)]. All subjects who experienced cardiac SAEs had cardiac risk factors and/or a prior history of cardiovascular disease, and there was no clear temporal pattern. A causal relationship between EVUSHELD and these events has not been established. There was no signal for cardiac toxicity or thrombotic events identified in the nonclinical studies.
Consider the risks and benefits prior to initiating EVUSHELD in individuals at high risk for cardiovascular events, and advise individuals to seek immediate medical attention if they experience any signs or symptoms suggestive of a cardiovascular event.
6 ADVERSE REACTIONS
6.1 Adverse Reactions from Clinical Studies
The following adverse events have been observed in the clinical studies of EVUSHELD that supported the EUA. The adverse event rates observed in these clinical studies cannot be directly compared to rates in the clinical studies of other products and may not reflect the rates observed in clinical practice. Additional adverse events associated with EVUSHELD may become apparent with more widespread use.
Approximately 4,220 subjects have been exposed to EVUSHELD (150 mg of tixagevimab and 150 mg of cilgavimab) in two ongoing Phase III trials, PROVENT and STORM CHASER, for the prophylaxis of COVID-19. The primary safety analysis was based on data through to event driven efficacy data cut-offs, such that individual subjects had variable follow-up times [see Clinical Studies (14)], with a median (range) of follow-up of 83 days (3-166 days) for PROVENT and 49 days (5-115 days) for STORM CHASER. An additional data cut-off was conducted to provide updated analyses with a median (range) of follow-up of 6.5 months (3-282 days) for PROVENT and approximately 6 months (5-249 days) for STORM CHASER. The median and range of follow-up times were similar between EVUSHELD and placebo recipients in each trial.
Four hundred and fifty two (452) non-hospitalized subjects (with the exception of those hospitalized for isolation purposes) with mild to moderate COVID-19 have been exposed to EVUSHELD (300 mg of tixagevimab and 300 mg of cilgavimab) in one ongoing Phase III clinical trial, TACKLE. The median (range) duration of follow-up was 84 days (1-183 days). EVUSHELD is not authorized for treatment of COVID-19 [see Limitations of Authorized Use (1)].
In all studies, adults received EVUSHELD administered as two separate, consecutive IM injections of tixagevimab and cilgavimab or placebo [see Clinical Studies (14)].
PROVENT (EVUSHELD [150 mg of tixagevimab and 150 mg of cilgavimab])
PROVENT enrolled adults ≥18 years of age who were either ≥60 years of age, had pre-specified co-morbidities [see Clinical Studies (14)], or were at increased risk of SARS-CoV-2 infection due to their living situation or occupation. Subjects could not have previously received a COVID-19 vaccine or have known prior or current SARS-CoV-2 infection. Subjects received a single dose of EVUSHELD (N= 3,461) or placebo (N= 1,736).
Adverse events were reported in 1,221 (35%) subjects receiving EVUSHELD and 593 (34%) receiving placebo. SAEs were reported in 50 (1%) subjects receiving EVUSHELD and 23 (1%) receiving placebo. There was 1 adverse event reported as anaphylaxis among subjects who received EVUSHELD. The event began within minutes of EVUSHELD administration and was treated with epinephrine. The event resolved.
Of the reported adverse events (N= 4,507), the majority were mild (73%) or moderate (24%) in severity. All adverse events, occurring in at least 1% of subjects, were reported at similar incidence rates among subjects receiving EVUSHELD compared to those receiving placebo (difference <1%). The most common treatment-emergent adverse events, occurring in at least 3% of subjects receiving EVUSHELD or placebo are shown in Table 3.
|
EVUSHELD N= 3,461 |
Placebo N= 1,736 |
|
|
Headache |
6% |
5% |
|
Fatigue |
4% |
3% |
|
Cough |
3% |
3% |
At the additional data cut-off (median follow-up 6.5 months), the overall adverse event profile for subjects who received EVUSHELD remained similar to events displayed in Table 3.
Cardiac Serious Adverse Events
Through the additional data cut-off in PROVENT, a higher proportion of subjects who received EVUSHELD versus placebo in PROVENT reported myocardial infarction SAEs, one of which resulted in death, and cardiac failure SAEs (see Table 4 below). All subjects who experienced cardiac SAEs had cardiac risk factors and/or a prior history of cardiovascular disease at baseline. There was no clear temporal pattern, with events reported from several hours after EVUSHELD receipt through the end of the follow-up period.
| EVUSHELD
N= 3,461 | Placebo
N= 1,736 |
|
|---|---|---|
|
||
|
Subjects with any cardiac SAE* |
22 (0.6%) |
3 (0.2%) |
|
10 (0.3%) |
2 (0.1%) |
|
8 (0.2%) |
1 (0.1%) |
|
6 (0.2%) |
1 (0.1%) |
|
4 (0.1%) |
1 (0.1%) |
|
3 (0.1%) |
0 |
STORM CHASER (EVUSHELD [150 mg tixagevimab and 150 mg cilgavimab])
STORM CHASER enrolled adults ≥18 years of age following potential exposure (within 8 days) to an identified individual with a laboratory-confirmed SARS-CoV-2 infection (symptomatic or asymptomatic). Subjects could not have previously received a COVID-19 vaccine, have symptoms consistent with COVID-19, or have a known prior SARS-CoV-2 infection. Subjects received a single dose of EVUSHELD (N= 749) or placebo (N= 372).
Adverse events were reported in 162 (22%) subjects receiving EVUSHELD and 111 (30%) receiving placebo. SAEs were reported in 5 (<1%) subjects receiving EVUSHELD and 3 (<1%) receiving placebo. Of the reported adverse events (N= 777), the majority were mild (75%) or moderate (23%) in severity.
At the additional data cut-off (median follow-up approximately 6 months), the overall adverse event profile for subjects who received EVUSHELD remained similar to earlier results. EVUSHELD is not authorized for post-exposure prophylaxis of COVID-19 in individuals who have been exposed to someone infected with SARS-CoV-2 [see Emergency Use Authorization (1)].
Cardiac Serious Adverse Events
In STORM CHASER (N= 1,121) no cardiac SAEs were reported (median follow-up approximately 6 months). Compared to PROVENT, the subjects in STORM CHASER were younger (median age 48 versus 57 years) and had fewer baseline cardiac risk factors (24% versus 36% with hypertension, 11% versus 14% with diabetes, and 3% versus 8% with cardiovascular disease in STORM CHASER versus PROVENT, respectively).
TACKLE (EVUSHELD [300 mg tixagevimab and 300 mg cilgavimab])
TACKLE enrolled adults ≥18 years of age with mild to moderate COVID-19 who were within ≤7 days of symptom onset. Approximately 90% of study subjects had risk factors that put them at high risk for progression to severe COVID-19. Subjects received a single dose of EVUSHELD (N= 452) or placebo (N= 451).
Adverse events were reported in 132 (29%) subjects receiving EVUSHELD and 163 (36%) receiving placebo. Serious adverse events were reported in 33 (7%) subjects receiving EVUSHELD and 54 (12%) receiving placebo. Of the reported adverse events (N= 520), the majority were mild (56%) or moderate (27%) in severity. There were no reports of anaphylaxis or serious hypersensitivity reactions.
Adverse events of insomnia (1% vs. <1%) and dizziness (1% vs. none) were reported at a higher rate with EVUSHELD compared to placebo. No other treatment-emergent adverse events, occurring in at least 1% of subjects, were reported at higher incidence rates (difference ≥1%) among subjects receiving EVUSHELD compared to those receiving placebo.
Cardiac Serious Adverse Events
In TACKLE (N= 903) four subjects reported cardiac SAEs. Acute myocardial infarction was reported for two subjects who received EVUSHELD (one of whom also experienced cardiac failure leading to death) and sudden cardiac death was reported for one subject who received EVUSHELD. One subject who received placebo reported arrhythmia. All subjects who experienced cardiac SAEs had cardiac risk factors and/or a prior history of cardiovascular disease at baseline.
6.4 Required Reporting for Serious Adverse Events and Medication Errors
The prescribing healthcare provider and/or the provider’s designee is/are responsible for mandatory reporting of all serious adverse events* and medication errors potentially related to EVUSHELD within 7 calendar days from the healthcare provider’s awareness of the event, using FDA Form 3500 (for information on how to access this form, see below). The FDA requires that such reports, using FDA Form 3500, include the following:
- •
- Patient demographics and baseline characteristics (e.g., patient identifier, age or date of birth, gender, weight, ethnicity, and race)
- •
- A statement "EVUSHELD use for COVID-19 under Emergency Use Authorization (EUA)” under the “Describe Event, Problem, or Product Use/Medication Error” heading
- •
- Information about the serious adverse event or medication error (e.g., signs and symptoms, test/laboratory data, complications, timing of drug initiation in relation to the occurrence of the event, duration of the event, treatments required to mitigate the event, evidence of event improvement/disappearance after stopping or reducing the dosage, evidence of event reappearance after reintroduction, clinical outcomes)
- •
- Patient’s preexisting medical conditions and use of concomitant products
- •
- Information about the product (e.g., dosage, route of administration, NDC #)
Submit adverse event and medication error reports, using Form 3500, to FDA MedWatch using one of the following methods:
- •
- Complete and submit the report online: www.fda.gov/medwatch/report.htm
- •
- Complete and submit a postage-paid FDA Form 3500 (https://www.fda.gov/media/76299/download) and return by:
- ∘
- Mail to MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787, or
- ∘
- Fax to 1-800-FDA-0178, or
- •
- Call 1-800-FDA-1088 to request a reporting form
In addition, please provide a copy of all FDA MedWatch forms to AstraZeneca:
- •
- Fax 1-866-742-7984
and to report adverse events please:
- •
- Visit https://contactazmedical.astrazeneca.com, or
- •
- Call AstraZeneca at 1-800-236-9933.
The prescribing healthcare provider and/or the provider’s designee is/are responsible for mandatory responses to requests from FDA for information about adverse events and medication errors following receipt of EVUSHELD.
*Serious adverse events are defined as:
- •
- Death
- •
- A life-threatening adverse event;
- •
- A persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions;
- •
- A congenital anomaly/birth defect;
- •
- Other important medical event, which may require a medical or surgical intervention to prevent death, a life-threatening event, hospitalization, disability, or congenital anomaly;
- •
- Inpatient hospitalization or prolongation of existing hospitalization.
7 DRUG INTERACTIONS
Drug-drug interaction studies have not been performed.
Tixagevimab and cilgavimab are not renally excreted or metabolized by cytochrome P450 (CYP) enzymes; therefore, interactions with concomitant medications that are renally excreted or that are substrates, inducers, or inhibitors of CYP enzymes are unlikely [see Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are insufficient data to evaluate a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. EVUSHELD should only be used during pregnancy if the potential benefit outweighs the potential risk for the mother and the fetus.
Nonclinical reproductive toxicity studies have not been conducted with tixagevimab and cilgavimab. In a tissue cross-reactivity study assessing off-target binding of tixagevimab and cilgavimab to human fetal tissues no binding of clinical concern was observed. Human immunoglobulin G1 (IgG1) antibodies are known to cross the placental barrier; therefore, tixagevimab and cilgavimab have the potential to be transferred from the mother to the developing fetus. It is unknown whether the potential transfer of tixagevimab and cilgavimab provides any treatment benefit or risk to the developing fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
8.2 Lactation
Risk Summary
There are no available data on the presence of tixagevimab or cilgavimab in human milk or animal milk, the effects on the breastfed infant, or the effects of the drug on milk production. Maternal IgG is known to be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for EVUSHELD and any potential adverse effects on the breastfed infant from EVUSHELD.
8.4 Pediatric Use
EVUSHELD is not authorized for use in pediatric individuals under 12 years of age or weighing less than 40 kg. The safety and effectiveness of EVUSHELD have not been established in pediatric individuals. The dosing regimen is expected to result in comparable serum exposures of tixagevimab and cilgavimab in individuals 12 years of age and older and weighing at least 40 kg as observed in adults, since adults with similar body weight have been included in the trials PROVENT, STORM CHASER and TACKLE [see Adverse Reactions (6.1) and Clinical Studies (14)].
8.5 Geriatric Use
Of the 2,555 subjects in the pooled pharmacokinetics (PK) analysis (Phase I and Phase III studies), 21% (N= 533) were 65 years of age or older and 3% (N= 81) were 75 years of age or older. There is no clinically meaningful difference in the PK of tixagevimab and cilgavimab in geriatric subjects (≥65 years) compared to younger subjects.
8.6 Renal Impairment
Tixagevimab and cilgavimab are not eliminated intact in the urine, renal impairment is not expected to affect the exposure of tixagevimab and cilgavimab. Similarly, dialysis is not expected to impact the PK of tixagevimab and cilgavimab.
8.7 Hepatic Impairment
The effect of hepatic impairment on the PK of tixagevimab and cilgavimab is unknown.
8.8 Other Specific Populations
Based on a population PK analysis, the PK profile of tixagevimab and cilgavimab was not affected by sex, age, race, or ethnicity. Population PK model-based simulations suggest that body weight had no clinically relevant effect on the PK of tixagevimab and cilgavimab in healthy adults over the range of 36 kg to 177 kg.
10 OVERDOSAGE
Treatment of overdose with EVUSHELD should consist of general supportive measures including the monitoring of the clinical status of the individual. There is no specific treatment for overdose with EVUSHELD.
11 DESCRIPTION
Tixagevimab, a SARS-CoV-2 spike protein-directed attachment inhibitor, is a human immunoglobulin G1 (IgG1κ) monoclonal antibody produced in Chinese hamster ovary (CHO) cells by recombinant DNA technology. The molecular weight is approximately 149 kDa.
Tixagevimab injection is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow solution supplied in a single-dose vial for intramuscular use. The vial stoppers are not made with natural rubber latex. Each 1.5 mL contains 150 mg tixagevimab, L- histidine (2.4 mg), L- histidine hydrochloride monohydrate (3.0 mg), polysorbate 80 (0.6 mg), sucrose (123.2 mg), and Water for Injection, USP. The pH is 6.0.
Cilgavimab, a SARS-CoV-2 spike protein-directed attachment inhibitor, is a human IgG1κ monoclonal antibody produced in CHO cells by recombinant DNA technology. The molecular weight is approximately 152 kDa.
Cilgavimab injection is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow solution supplied in a single-dose vial for intramuscular use. The vial stoppers are not made with natural rubber latex. Each 1.5 mL contains 150 mg cilgavimab, L- histidine (2.4 mg), L- histidine hydrochloride monohydrate (3.0 mg), polysorbate 80 (0.6 mg), sucrose (123.2 mg), and Water for Injection, USP. The pH is 6.0.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tixagevimab and cilgavimab are two recombinant human IgG1 monoclonal antibodies with amino acid substitutions to extend antibody half-life (YTE), reduce antibody effector function, and minimize the potential risk of antibody-dependent enhancement of disease (TM). Tixagevimab and cilgavimab can simultaneously bind to non-overlapping regions of the receptor binding domain (RBD) of SARS-CoV-2 spike protein. Tixagevimab, cilgavimab, and their combination bind to spike protein with equilibrium dissociation constants of KD = 2.76 pM, 13.0 pM and 13.7 pM, respectively, blocking its interaction with human ACE2, the SARS-CoV-2 receptor, which is required for virus attachment. Tixagevimab, cilgavimab, and their combination blocked RBD binding to human ACE2 with IC50 values of 0.32 nM (48 ng/mL), 0.53 nM (80 ng/mL), and 0.43 nM (65 ng/mL), respectively.
12.3 Pharmacokinetics
A summary of PK parameters and properties of tixagevimab and cilgavimab following administration of a single EVUSHELD (300 mg of tixagevimab and 300 mg of cilgavimab) intramuscular dose is provided in Table 5.
| PK Parameters | Tixagevimab | Cilgavimab |
|---|---|---|
|
21.9 (61.7) |
20.3 (63.6) |
|
14.9 (1.1 – 86) |
15.0 (1.1 – 85) |
|
9.5 (77) |
9.1 (80) |
|
15 (48) |
14 (51) |
|
1408 (54) |
1307 (58) |
|
Absorption |
||
|
68.5 |
65.8 |
|
|
Distribution |
||
|
7.7 (1.97) |
8.7 (2.73) |
|
Elimination |
||
|
87.9 (13.9) |
82.9 (12.3) |
|
|
0.062 (0.019) |
0.074 (0.028) |
|
Metabolism |
Catabolic pathways; Same manner as endogenous IgG |
|
|
Excretion |
Not likely to undergo renal excretion |
|
In the PROVENT repeat dose sub-study, following a second IM dose of EVUSHELD (150 mg of tixagevimab and 150 mg of cilgavimab) administered 10 to 14 months after the initial IM dose of EVUSHELD (150 mg of tixagevimab and 150 mg of cilgavimab) (N= 53), the geometric mean serum concentration was 26.4 µg/mL on post-administration Day 29. This serum concentration was similar to the geometric mean drug concentration on post-administration Day 29 (23.3 μg/mL) following the initial IM EVUSHELD dose (150 mg of tixagevimab and 150 mg of cilgavimab) in the PROVENT parent study.
The primary analysis in the clinical efficacy study PROVENT was conducted prior to the emergence of the Omicron variant; the dominant variants in circulation at that time were Alpha, Beta, Gamma, and Delta. Pharmacokinetic and pharmacodynamic modeling using cell-based EC50 values of EVUSHELD against the currently circulating variants in the U.S. suggest in vivo activity against these variants may be retained at drug concentrations achieved following a single EVUSHELD initial dose of 300 mg tixagevimab and 300 mg cilgavimab for 6 months [see Dosage and Administration (2.1)].
Specific Populations
The PK profile of tixagevimab and cilgavimab were not affected by sex, age, race or ethnicity. Body weight had no clinically relevant effect on the PK of tixagevimab and cilgavimab in adults over the range of 36 kg to 177 kg.
Pediatric Population
The PK of tixagevimab and cilgavimab in pediatric individuals have not been evaluated.
The dosing regimen is expected to result in comparable plasma exposures of tixagevimab and cilgavimab in pediatric individuals ages 12 years of age or older who weigh at least 40 kg as observed in adult individuals [see Use in Specific Populations (8.4)].
Renal impairment
Tixagevimab and cilgavimab are not eliminated intact in the urine.
Renal impairment is not expected to impact the PK of tixagevimab and cilgavimab, since monoclonal antibodies with molecular weight >69 kDa are known not to undergo renal elimination. Similarly, dialysis is not expected to impact the PK of tixagevimab and cilgavimab.
There is no difference in the clearance of tixagevimab and cilgavimab in individuals with mild or moderate renal impairment compared to individuals with normal renal function. There were insufficient subjects with severe renal impairment to draw conclusions [see Use in Specific Populations (8.6)].
Hepatic impairment
No specific studies have been conducted to examine the effects of hepatic impairment on the PK of tixagevimab and cilgavimab. The impact of hepatic impairment on the PK of tixagevimab and cilgavimab is unknown [see Use in Specific Populations (8.7)].
Drug Interaction Studies
Drug-drug interaction studies have not been performed. Based on key elimination pathways, tixagevimab and cilgavimab interactions with concomitant medications that are renally excreted or that are substrates, inducers, or inhibitors of CYP enzymes are unlikely [see Drug Interactions (7)].
12.4 Microbiology
Antiviral Activity
In a neutralization assay on Vero E6 cells, tixagevimab, cilgavimab, and their combination neutralized SARS-CoV-2 (USA-WA1/2020 isolate) with EC50 values of 60.7 pM (9 ng/mL), 211.5 pM (32 ng/mL), and 65.9 pM (10 ng/mL), respectively.
Tixagevimab, cilgavimab, and their combination showed reduced or no antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), or antibody-dependent natural killer cell activation (ADNKA) in cell culture studies. Tixagevimab, cilgavimab, and their combination did not mediate antibody-dependent complement deposition (ADCD) activity with guinea pig complement proteins.
Antibody Dependent Enhancement (ADE) of Infection
The potential of tixagevimab and cilgavimab to mediate antibody-dependent viral entry was assessed in FcγRII-expressing Raji cells co-incubated with recombinant virus-like particles (VLPs) pseudotyped with SARS-CoV-2 spike protein, with antibody concentrations at a range of 6.6 nM (1 µg/mL) to 824 pM (125 ng/mL). Tixagevimab, cilgavimab, and their combination did not mediate entry of VLPs into these cells under the tested conditions.
The potential for ADE was also evaluated in a non-human primate model of SARS-CoV-2 using EVUSHELD. Intravascular administration prior to virus inoculation resulted in a dose-dependent improvement in all measured outcomes (total viral RNA in the lungs or nasal mucosae, infectious virus levels in the lungs based on TCID50 measurements, or lung injury and pathology based on histology measurements). No evidence of enhancement of viral replication or disease was observed at any dose evaluated, including sub-neutralizing doses down to 0.04 mg/kg.
Antiviral Resistance
There is a potential risk of treatment failure due to the development of viral variants that are resistant to tixagevimab and cilgavimab. Prescribing healthcare providers should consider the prevalence of SARS-CoV-2 variants in their area, where data are available, when considering prophylactic treatment options.
Escape variants were identified following serial passage in cell culture of SARS-CoV-2 or replication competent recombinant vesicular stomatitis virus (VSV) expressing SARS-CoV-2 spike protein in the presence of tixagevimab or cilgavimab individually or in combination. Tixagevimab selected a variant expressing F486S in the spike protein with a >800-fold reduction in susceptibility to tixagevimab. Cilgavimab selected variants that expressed spike protein amino acid substitutions R346G, R346I, K444E, K444N, K444Q, K444R, K444T or N450D were each associated with a >200-fold reduction in susceptibility to cilgavimab. No escape variants to the tixagevimab and cilgavimab combination were selected.
In neutralization assays using recombinant VLPs pseudotyped with SARS-CoV-2 spike and harboring
individual spike amino acid substitutions identified in circulating SARS-CoV-2, variants with reduced
susceptibility to cilgavimab alone included those with R346I (>200-fold), K444E (>200-fold), K444Q
(>200-fold), K444R (>200-fold), V445A (21- to 51-fold), G446V (4.2-fold), N450K (9.1-fold), or L452R
(5.8-fold) substitutions. Variants with reduced susceptibility to tixagevimab alone included those with
Q414R (4.6-fold), L455F (2.5- to 4.7-fold), G476S (3.3-fold), E484D (7.1-fold), E484K (6.2- to 12-
fold), E484Q (3.0-fold), F486S (>600-fold), F486V (121- to 149-fold), Q493K (2.4- to 3.2-fold), Q493R (7.9-fold), E990A (6.1-fold), or T1009I (8.2-fold) substitutions. Variants harboring an E484K (2.4- to
5.4-fold), Q493R (3.4-fold), E990A (5.7-fold), or T1009I (4.5-fold) substitution exhibited low level
reduced susceptibility to tixagevimab and cilgavimab in combination.
VLPs pseudotyped with the SARS-CoV-2 spike of variant strains with reduced susceptibility to cilgavimab included those with R346K+E484K+N501Y (Mu, 21-fold), and those with reduced susceptibility to tixagevimab included those harboring E484K (Alpha, 18.5-fold; Beta, 3.5- to 15-fold; Zeta, 7.3-fold). Similar results were observed, where data were available, in neutralization assays using authentic SARS-CoV-2 variant strains.
VLPs pseudotyped with the SARS-CoV-2 spike of Omicron BA.1 or BA.1.1 (BA.1+R346K) showed
reduced susceptibility to tixagevimab (>600- to >1,000-fold or 460-fold, respectively) and to
cilgavimab (>700- to >1,000-fold or >500-fold, respectively). VLPs pseudotyped with the SARS-CoV-2
spike of Omicron BA.2 or BA.2.12.1 showed reduced susceptibility to tixagevimab (>1,000-fold or
>500-fold, respectively) but not to cilgavimab (1.9-fold or 2-fold, respectively). VLPs pseudotyped with
the SARS-CoV-2 spike of Omicron BA.2.75 or BA.2.75.2 showed reduced susceptibility to
tixagevimab (7- to 53-fold or >3,333- to >10,000-fold, respectively) and to cilgavimab (6- to 40-fold or
>769- to >5,000-fold, respectively). VLPs pseudotyped with the SARS-CoV-2 spike of Omicron BA.3
showed reduced susceptibility to tixagevimab (>5,000-fold) but not to cilgavimab (4-fold). VLPs
pseudotyped with the SARS-CoV-2 spike of Omicron BA.4/BA.5 showed reduced susceptibility to
tixagevimab (>10,000-fold) and cilgavimab (7.5- to 9-fold). VLPs pseudotyped with the SARS-CoV-2
spike of Omicron BA.4.6 showed reduced susceptibility to tixagevimab (>1,000-fold) and to
cilgavimab (>1,000-fold). VLPs pseudotyped with the SARS-CoV-2 spike of Omicron BF.7 or BJ.1
showed reduced susceptibility to tixagevimab (>3,333- to >10,000-fold or 85- to 172-fold,
respectively) and to cilgavimab (>769- to >5,000-fold or >769- to >5,000-fold, respectively). VLPs
pseudotyped with the SARS-CoV-2 spike of Omicron BQ.1 or BQ.1.1 showed reduced susceptibility
to tixagevimab (>1,250- to >10,000-fold) and to cilgavimab (>667- to >5,000-fold). VLPs pseudotyped
with the SARS-CoV-2 spike of Omicron BA.5.2.6 or BF.11 showed reduced susceptibility to
tixagevimab (>333-fold) and to cilgavimab (>77-fold). VLPs pseudotyped with the SARS-CoV-2 spike
of Omicron BN.1 or XBB showed reduced susceptibility to tixagevimab (24- to 44-fold or >2,600- to
>10,000-fold, respectively) and to cilgavimab (>3,700- to >5,000-fold or >565- to >5,000-fold,
respectively). VLPs pseudotyped with the SARS-CoV-2 spike of Omicron XBB.1.5 showed reduced
susceptibility to tixagevimab (>10,000-fold) and to cilgavimab (>2,900-fold). The effects of the
individual substitutions in Omicron spike glycoproteins on neutralization susceptibility are being
investigated.
The neutralizing activity of tixagevimab and cilgavimab in combination was tested against
pseudotyped VLPs and/or authentic SARS-CoV-2 variant strains harboring all spike substitutions
identified in Alpha (B.1.1.7, 0.5- to 5.2-fold), Beta (B.1.351, 1.0- to 3.8-fold), Gamma (P.1, 0.4- to 2.0-
fold), Delta (B.1.617.2, 0.6- to 1.2-fold), and Delta [+K417N] (AY.1/ AY.2, 1.0-fold) variants of
concern, and Eta (B.1.525, 3.1-fold), Iota (B.1.526, 0.3- to 3.4-fold), Kappa (B.1.617.1, 0.5- to 3.4-
fold) Lambda (C.37, 0.7-fold), and Mu (B.1.621, 7.5-fold) variants of interest. Tixagevimab and
cilgavimab in combination was also tested against Epsilon (B.1.427 / B.1.429, 0.8- to 3.5-fold), R.1
(3.5-fold), B.1.1.519 (1.4-fold), C.36.3 (2.3-fold), B.1.214.2 (0.8-fold), and B.1.619.1 (3.3-fold) variant
alerts for further monitoring and B.1.616 (0.5-fold), A.23.1 (0.4-fold), A.27 (0.8-fold), and AV.1 (5.9-
fold) variants de-escalated from further monitoring (Table 6).
Preliminary data for the neutralizing activities of tixagevimab and cilgavimab in combination against circulating Omicron subvariants are available. VLPs pseudotyped with the SARS-CoV-2 spike of Omicron BA.1 or BA.1.1 (BA.1+R346K) showed reduced neutralizing activity (132- to 183-fold or 424-
fold, respectively), Omicron BA.2 showed no change in neutralizing activity (3.2-fold). VLPs
pseudotyped with the spike of Omicron BA.2.12.1, BA.2.75, BA.2.75.2, BA.3, BA.4/BA.5, or BA.4.6
showed 5-fold, 2.4- to 15-fold, >5,000- to >10,000-fold, 16-fold, 33- to 65-fold, or >1,000-fold
reductions in neutralizing activity, respectively. VLPs pseudotyped with the spike of Omicron BF.7,
BJ.1, BQ.1 or BQ.1.1 showed >5,000- to >10,000-fold, 228- to 424-fold, >2,000- to >10,000-fold or
>2,000- to >10,000-fold reductions in neutralizing activity, respectively. VLPs pseudotyped with the
spike of Omicron BA.5.2.6, BF.11, BN.1, XBB, or XBB.1.5 showed >500-fold, >500-fold, 68-fold,
>1,400- to >10,000-fold, or >5,000-fold reductions in neutralizing activity, respectively. Authentic
Omicron BA.1, BA.1.1, BA.2, or BA.5 viruses showed 12- to 30-fold, 176-fold, 5.4-fold, or 2.8- to 16-
fold reductions in susceptibility, respectively.
Data collection is ongoing to better understand how the reductions in activity seen in pseudotyped VLP assays or authentic SARS-CoV-2 assays may correlate with clinical outcomes. Emerging Omicron subvariants that are resistant to neutralization by cilgavimab harbor the spike substitution R346T or K444T, while those resistant to neutralization by tixagevimab harbor the spike substitution F486S or F486V. EVUSHELD is unlikely to neutralize SARS-CoV-2 Omicron subvariants harboring R346T or K444T in combination with F486S or F486V.
| Lineage with Spike Protein Substitution | Country First Identified | WHO Nomenclature | Key Substitutions Tested | Fold Reduction in
Susceptibility*(Pseudotyped VLPs†) | Fold Reduction in
Susceptibility* (Authentic virus‡) |
|---|---|---|---|---|---|
|
|||||
|
B.1.1.7 |
UK |
Alpha |
N501Y |
0.5- to 5.2‑fold |
No Change§ |
|
B.1.351 |
South Africa |
Beta |
K417N+E484K+N501Y |
No Change§ |
No Change§ |
|
|
|
K417T+E484K+N501Y |
No Change§ |
No Change§ |
|
|
|
L452R+T478K |
No Change§ |
No Change§ |
|
|
|
K417N+L452R+T478K |
No Change§ |
No Change§ |
|
|
|
G339D+S371L+S373P+ S375F+K417N+N440K+ G446S+S477N+T478K+ E484A+Q493R+G496S+ Q489R+N501Y+Y505H |
132- to 183-fold¶ |
12- to 30-fold |
|
|
|
BA.1+R346K |
424-fold |
176-fold |
|
|
|
G339D+S371F+S373P+ S375F+T376A+D405N+ R408S+K417N+N440K+ S477N+T478K+E484A+ Q493R+Q498R+N501Y+ Y505H |
No Change§ |
5.4-fold |
|
|
|
BA.2+L452Q |
5-fold |
ND |
|
|
|
G339H+S371F+S373P+ S375F+T376A+D405N+ R408S+K417N+N440K+ G446S+N460K+S477N+ T478K+E484A+Q498R+ N501Y+ Y505H |
2.4- to 15-fold |
ND |
|
|
|
BA.2.75+R346T+F486S |
>5000-fold# |
ND |
|
|
|
G339D+S371F+ S373P+S375F+D405N+ K417N+N440K+G446S+ S477N+T478K+E484A+ Q493R+Q498R+N501Y+ Y505H |
16-fold |
ND |
|
|
|
G339D+S371F+S373P+ S375F+T376A+D405N+ R408S+K417N+N440K+ L452R+S477N+T478K+ E484A+F486V+Q498R+ N501Y+Y505H |
33- to 65-fold |
ND |
|
|
|
BA.4+R346T |
>1000-fold# |
ND |
|
|
|
+G339D+S371F+S373P+ S375F+T376A+D405N+ R408S+K417N+N440K+ L452R+S477N+T478K+ E484A+F486V+Q498R+ N501Y+Y505H |
33- to 65-fold |
2.8- to 16-fold |
|
|
|
G339D+R346T+S371F+ S373P+S375F+T376A+ D405N+R408S+K417N+ N440K+L452R+S477N+ T478K+E484A+F486V+ Q498R+N501Y+Y505H |
>500-fold |
ND |
|
|
|
BA.4+R346T |
>5000-fold# |
ND |
|
|
|
G339D+R346T+S371F+ S373P+ S375F+T376A+ D405N+R408S+K417N+ N440K+L452R+S477N+ T478K+E484A+F486V+ Q498R+N501Y+Y505H |
>500-fold |
ND |
|
|
|
G339H+R346T+L368I+ S371F+S373P+S375F+ T376A+D405N+R408S+ K417N+N440K+V445P+ G446S+S477N+T478K+ V483A+E484A+F490V+ Q493R+Q498R+N501Y+ Y505H |
228- to 424-fold |
ND |
|
|
|
G339D+R346T+K356T+ S371F+S373P+S375F+ D405N+ R408S+ K417N+N440K+G446S+ N460K+S477N+T478K+ E484A+F490S+ Q493R+Q498R+Y505H |
68-fold |
ND |
|
|
|
BA.5+K444T+N460K |
>2000-fold# |
ND |
|
|
|
BA.5+R346T+K444T+ N460K |
>2000-fold# |
ND |
|
|
|
G339H+R346T+L368I+ S371F+S373P+S375F+ T376A+D405N+R408S+ K417N+N440K+V445P+ G446S+N460K+S477N+ T478K+ E484A+F486S+ F490S+Q498R+N501Y+ Y505H |
>1400-fold# |
ND |
|
|
|
G339H+R346T+L368I+ S371F+S373P+S375F+ T376A+D405N+R408S+ K417N+N440K+V445P+ G446S+N460K+S477N+ T478K+E484A+F486P+ F490S+Q498R+N501Y +Y505H |
>5000-fold# |
ND |
|
|
|
E484K |
No Change§ |
ND |
|
|
|
E484K |
No Change§ |
No Change§ |
|
|
|
L452R+E484Q |
No Change§ |
No Change§ |
|
|
|
L452Q+F490S |
No Change§ |
ND |
|
|
|
R346K+E484K +N501Y |
7.5-fold |
ND |
|
|
|
L452R |
No Change§ |
No Change§ |
|
|
|
E484K |
No Change§ |
ND |
|
|
|
T478K |
No Change§ |
ND |
|
|
|
R346S:L452R |
No Change§ |
ND |
|
|
|
Q414K:N450K |
No Change§ |
ND |
|
|
|
N440K:E484K |
No Change§ |
ND |
|
|
|
E484K |
No Change§ |
ND |
|
|
|
V483A |
No Change§ |
ND |
|
|
|
V367F |
No Change§ |
ND |
|
|
|
L452R+N501Y |
No Change§ |
ND |
|
|
|
N439K+E484K |
5.9-fold |
ND |
|
ND, not determined; RBD, receptor binding domain |
|||||
It is not known how pseudotyped VLPs or authentic SARS-CoV-2 neutralization susceptibility data correlate with clinical outcome.
In PROVENT, illness visit sequencing data were available for 21 of 33 subjects with SARS-CoV-2 infection (6 who received tixagevimab and cilgavimab and 15 placebo). Fourteen subjects were infected with variants of concern or variants of interest, including 8 subjects with Alpha (B.1.1.7) (8 who received placebo), 1 subject with Beta (B.1.351) (1 who received tixagevimab and cilgavimab), 3 subjects with Delta (B.1.617.2) (3 who received placebo), and 2 subjects with Epsilon (B.1.429) (2 who received tixagevimab and cilgavimab). Seven additional subjects were infected with B.1.375 (1 who received tixagevimab and cilgavimab) or the A_1 set of lineages containing a constellation of spike protein substitutions including D614G and P681H or Q677P (3 who received tixagevimab and cilgavimab and 3 placebo). Additional spike protein RBD substitutions detected at low frequency (between 3% and 24%) included V503F in the tixagevimab and cilgavimab group.
In STORM CHASER, illness visit sequencing data was available for 19 of 19 subjects with SARS-CoV-2 infections (12 of 12 who received tixagevimab and cilgavimab and 7 of 7 placebo). At an allele fraction ≥25%, 12 of 19 subjects were infected with variants of concern or variants of interest, including 9 subjects with Alpha (B.1.1.7) (5 who received tixagevimab and cilgavimab and 4 placebo) and 3 subjects with Epsilon (B.1.427 / B.1.429) (2 who received tixagevimab and cilgavimab and 1 placebo). Seven additional subjects were infected with B.1.1.519 (1 who received tixagevimab and cilgavimab) or the A_1 set of lineages containing a constellation of spike protein substitutions including D614G and D138H, Q675H, Q677H, or V1176F (4 who received tixagevimab and cilgavimab and 2 placebo). Additional spike protein RBD substitutions detected at an allele fraction ≥3% included S325P, Del342, C361W, Del428, F429V, and F515C in the tixagevimab and cilgavimab group.
Evaluation of neutralization susceptibility of variants identified through global surveillance and in subjects who received tixagevimab and cilgavimab is ongoing.
It is possible that variants resistant to tixagevimab and cilgavimab could have cross-resistance to other monoclonal antibodies targeting the RBD of SARS-CoV-2. The combination of tixagevimab and cilgavimab retained activity against pseudotyped VLPs harboring individual SARS-CoV-2 spike substitutions (K417E/N, D420N, K444Q, V445A, Y453F, L455F, N460K/S/T, E484D/K/Q, F486V, F490S, Q493K/R, and S494P) identified in neutralization escape variants of other monoclonal antibodies targeting the RBD of SARS-CoV-2 spike protein.
12.6 Immunogenicity
There are no immunogenicity data available for the currently authorized dosing regimen (EVUSHELD [300 mg of tixagevimab and 300 mg cilgavimab] administered every 6 months).
There was no apparent clinically significant effect of anti-EVUSHELD antibodies (ADA) on the safety or effectiveness of EVUSHELD in PROVENT (EVUSHELD [150 mg of tixagevimab and 150 mg cilgavimab]), but data are limited at this time. There is up to a 26% decrease, on average, in serum concentrations of EVUSHELD over time through 183 days post-administration in subjects with positive ADA after the initial dose compared to subjects who tested negative for ADA after the initial dose; the clinical significance of this decrease is unknown.
In PROVENT, following a single IM dose of EVUSHELD (150 mg of tixagevimab and 150 mg cilgavimab) (baseline: study Day 1) through study Day 183, treatment-emergent anti-tixagevimab, anti-cilgavimab and anti-EVUSHELD antibodies were detected in 3% (101/3152), 4% (113/3068) and 5% (156/3158) ADA-evaluable participants, respectively, who received EVUSHELD (150 mg of tixagevimab and 150 mg of cilgavimab). The average Day 8, 29, and 183 serum concentrations of EVUSHELD were approximately 0%, 12%, and 26% lower, respectively, in subjects who tested positive for ADA after the initial dose versus subjects who tested negative for ADA after the initial dose.
In the PROVENT repeat dose sub-study, following a subsequent single IM dose of EVUSHELD (150 mg of tixagevimab and 150 mg cilgavimab) (baseline: sub-study Day 1) through sub-study Day 29, treatment-emergent anti-tixagevimab, anti-cilgavimab and anti-EVUSHELD antibodies were detected in 0% (0/49), 10% (5/49) and 10% (5/49) ADA-evaluable subjects, respectively. The average Day 29 concentration of EVUSHELD was approximately 14% lower in subjects who tested positive for ADA after the second dose versus subjects who tested negative for ADA after the second dose. The time between repeat doses was 10 to 14 months (first IM dose administered in the original PROVENT study to second IM dose administered in the PROVENT sub-study).
The observed incidence of ADA is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described above with the incidence of ADA in other studies.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity, genotoxicity, and reproductive toxicology studies have not been conducted with tixagevimab and cilgavimab.
13.2 Animal Toxicology and Pharmacology
In a toxicology study in cynomolgus monkeys, tixagevimab and cilgavimab had no adverse effects when administered via IM injection.
In tissue cross-reactivity studies with tixagevimab and cilgavimab using human adult and fetal tissues no binding of clinical concern was detected.
Tixagevimab and cilgavimab have been assessed in rhesus macaque and cynomolgus macaque models of SARS-CoV-2 infection. Prophylactic administration of tixagevimab and cilgavimab (N= 4 rhesus macaque; N= 3 cynomolgus macaque) three days prior to infection prevented SARS-CoV-2 infection of the upper and lower respiratory tracts in dose-dependent manner. Prophylactic administration of 4 mg/kg tixagevimab and cilgavimab resulted in a 7-log10 reduction in viral sub-genomic messenger RNA (sgmRNA) in nasopharyngeal swabs and 5 to 6-log10 reduction in sgmRNA or infectious virus titer in bronchoalveolar lavage samples at Day 2 post-challenge in all animals relative to placebo-treated animals. Compared to placebo, prophylactic administration of tixagevimab and cilgavimab (N= 3 cynomolgus macaque) reduced lung injury associated with SARS-CoV-2 infection.
The applicability of these findings to a clinical setting is not known.
14 CLINICAL STUDIES
The data supporting this EUA are based on analyses from the Phase III trials PROVENT (NCT04625725) and STORM CHASER (NCT04625972). Both trials are evaluating the safety and efficacy of EVUSHELD (150 mg of tixagevimab and 150 mg of cilgavimab) for the prophylaxis SARS-CoV-2 symptomatic illness (COVID-19).
Efficacy Data from PROVENT
PROVENT is an ongoing Phase III, randomized (2:1), double-blind, placebo-controlled clinical trial studying EVUSHELD for the pre-exposure prophylaxis of COVID-19 in adults ≥18 years of age. All subjects were either ≥60 years of age, had a pre-specified co-morbidity (obesity, congestive heart failure, chronic obstructive pulmonary disease, chronic kidney disease, chronic liver disease, immunocompromised state, or previous history of severe or serious adverse event after receiving any approved vaccine), or were at increased risk of SARS-CoV-2 infection due to their living situation or occupation. Subjects could not have previously received a COVID-19 vaccine. Subjects received a single dose (administered as two IM injections) of EVUSHELD or placebo. The study excluded subjects with a history of laboratory-confirmed SARS-CoV-2 infection or SARS-CoV-2 antibody positivity at screening. Once COVID-19 vaccines were locally available, subjects were permitted on request to unblind to make an informed decision on vaccine timing and to receive COVID-19 vaccination.
The baseline demographics were balanced across the EVUSHELD and placebo arms. The median age was 57 years (with 43% of subjects aged 60 years or older), 46% of subjects were female, 73% were White, 3% were Asian 17% were Black/African American, and 15% were Hispanic/Latino. Of the 5,197 subjects, 78% had baseline co-morbidities or characteristics associated with an increased risk for severe COVID-19, including obesity (42%), diabetes (14%), cardiovascular disease (8%), cancer, including a history of cancer (7%), chronic obstructive pulmonary disease (5%), chronic kidney disease (5%), chronic liver disease (5%), immunosuppressive medications (3%) and immunosuppressive disease (<1%).
For the primary endpoint, a subject was defined as a COVID-19 case if their first case of SARS-CoV-2 RT-PCR-positive symptomatic illness occurred after administration and prior to Day 183. The primary analysis included 5,172 subjects who were SARS-CoV-2 RT-PCR negative at baseline, of which 3,441 received EVUSHELD and 1,731 received placebo. Only events that occurred prior to unblinding or vaccine receipt were included. EVUSHELD receipt resulted in a statistically significant (p-value <0.001) 77% reduction in incidence of SARS-CoV-2 RT-PCR-positive symptomatic illness (COVID-19) when compared to placebo (Table 7). At the time of analysis the median follow-up time post-administration was 83 days (range 3 to 166 days).
Similar results were observed for EVUSHELD recipients compared to placebo recipients in the reduction in incidence of SARS-CoV-2 RT-PCR-positive symptomatic illness or death from any cause (12/3,441 versus 19/1,731, respectively) with relative risk reduction of 69% (95% CI: 36, 85; p value= 0.002), and in the reduction in incidence of SARS-CoV-2 RT-PCR-positive symptomatic illness regardless of unblinding or vaccine receipt (10/3,441 versus 22/1,731, respectively) with relative risk reduction of 77% (95% CI: 52, 89 ; p-value <0.001).
|
N* |
Number of events, n (%) |
Relative Risk Reduction, % (95% CI) |
|
|
EVUSHELD† |
3,441 |
8 (0.2%) |
77% (46, 90) |
|
Placebo |
1,731 |
17 (1.0%) |
|
|
N = number of subjects in analysis; CI = Confidence Interval |
|||
Among subjects who received EVUSHELD, there were no severe/critical COVID 19 events (defined as SARS-CoV-2 RT-PCR-positive symptomatic illness characterized by a minimum of either pneumonia [fever, cough, tachypnoea or dyspnea, and lung infiltrates] or hypoxemia [SpO2 <90% in room air and/or severe respiratory distress] and a WHO Clinical Progression Scale score of 5 or higher) compared to one event (0.1%) among subjects who received placebo.
An additional data cut was conducted to provide post-hoc updated efficacy and safety analysis, the median follow-up was 6.5 months for subjects in both EVUSHELD and placebo arms. The relative risk reduction of SARS-CoV-2 RT-PCR-positive symptomatic illness was 83% (95% CI: 66, 91) with 11/3,441 (0.3%) events in the EVUSHELD arm and 31/1,731 (1.8%) events in the placebo arm, see Figure 1. These results are consistent with the duration of protection predicted by population PK modelling. Among subjects who received EVUSHELD there were no severe/critical COVID-19 events compared to five events among subjects who received placebo.
Figure 1 Kaplan Meier: Cumulative Incidence of Symptomatic COVID-19* (PROVENT)
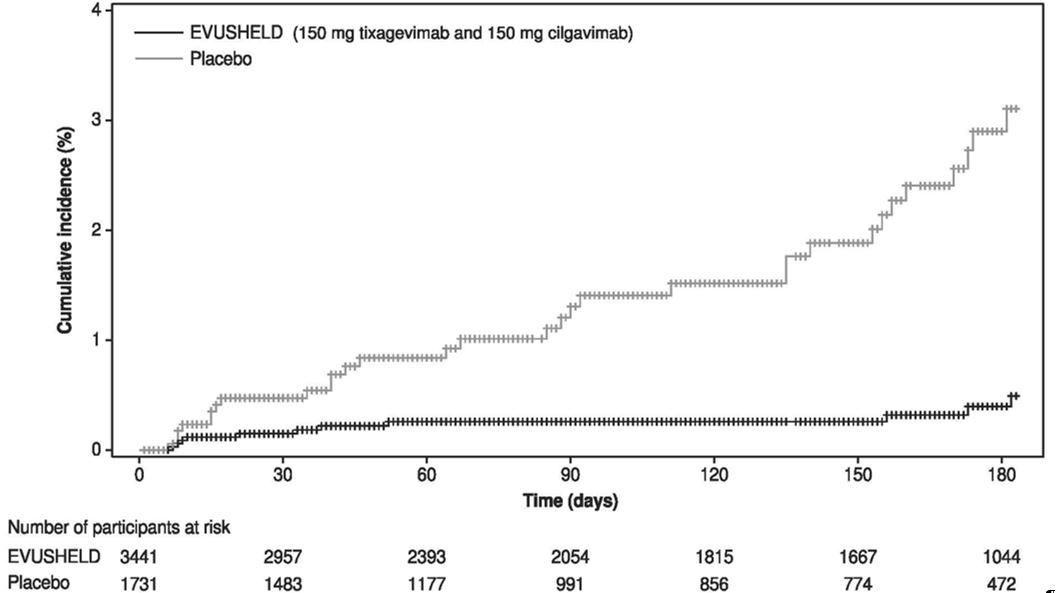
* Subjects who do not experience a primary endpoint event (and had not discontinued) are censored at Day 183. Subjects who were unblinded/vaccinated prior to an event are also censored at the earlier time of unblinding/vaccination.
Efficacy Data from STORM CHASER
STORM CHASER is an ongoing Phase III randomized (2:1), double-blind, placebo-controlled clinical trial of EVUSHELD for the post-exposure prophylaxis of COVID-19 in adults ≥18 years of age. Subjects who had not previously received a COVID-19 vaccine were enrolled following potential exposure (within 8 days) to an identified individual with a laboratory-confirmed SARS-CoV-2 infection (symptomatic or asymptomatic). Subjects received a single dose (administered as two IM injections) of EVUSHELD or placebo. The study excluded subjects with a history of laboratory-confirmed SARS-CoV-2 infection or SARS-CoV-2 antibody positivity at screening. Once COVID-19 vaccines were locally available, subjects were permitted on request to unblind to make an informed decision on vaccine timing and to receive COVID-19 vaccination.
Of the 1,121 subjects who were randomized and received EVUSHELD (N= 749) or placebo (N= 372), 48 subjects were positive for SARS-CoV-2 (RT-PCR analysis of nasopharyngeal swabs) at baseline.
The primary efficacy analysis, comparison of the incidence of a subject’s first case of SARS-CoV-2 RT PCR-positive symptomatic illness occurring post-dose and before Day 183, did not demonstrate a statistically significant effect for EVUSHELD versus placebo with 23 cases of symptomatic COVID-19 in the EVUSHELD arm (3.1%) and 17 cases in the placebo arm (4.6%) (relative risk reduction of 33%, 95% CI: -26, 65). At the time of analysis the median follow-up time post-administration was 49 days (range 5 to 115 days).
The study did not demonstrate benefit for EVUSHELD in preventing symptomatic COVID-19 in the first 30 days after randomization, leading to the limitation of use for post-exposure prophylaxis [see Emergency Use Authorization (1)]. However, there was a higher proportion of symptomatic COVID-19 cases among placebo recipients after Day 29 (see Figure 2 below, data from the post-hoc updated efficacy analysis with a median follow-up time of 6.5 months). EVUSHELD is not authorized for post-exposure prophylaxis of COVID-19 in individuals who have been exposed to someone infected with SARS-CoV-2.
Figure 2 Kaplan Meier: Cumulative Incidence of Symptomatic COVID-19* (STORM CHASER)
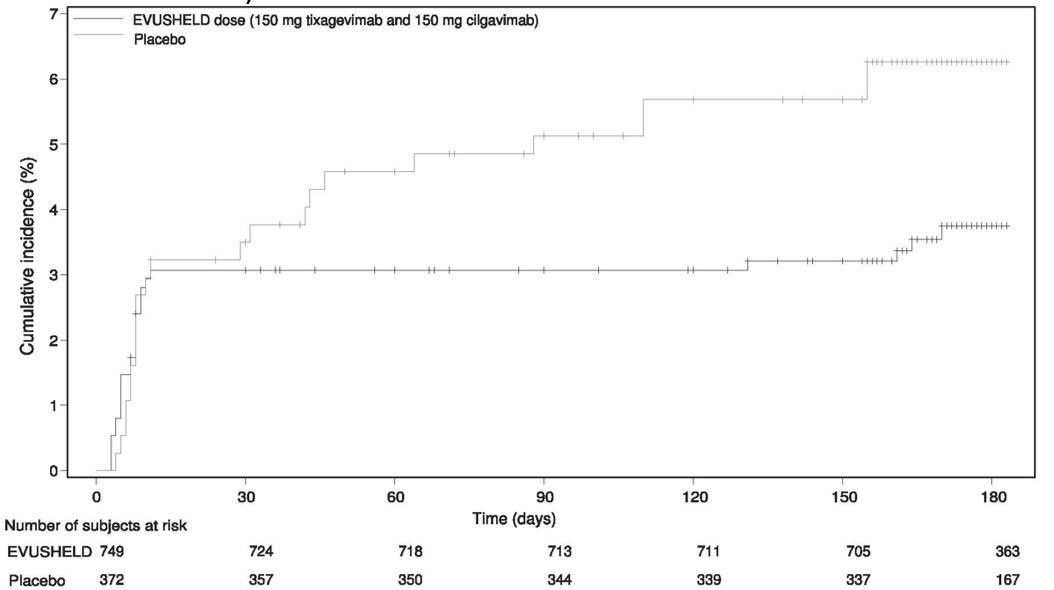
* Subjects who do not experience a primary endpoint event (and had not discontinued) are censored at Day 183.
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Each EVUSHELD co-packaged carton contains two vials (Table 8):
- •
- 1 single-dose vial of tixagevimab injection as a sterile, preservative-free, clear to opalescent and colorless to slightly yellow solution.
- •
- 1 single-dose vial of cilgavimab injection as a sterile, preservative-free, clear to opalescent and colorless to slightly yellow solution.
|
||
|
Carton (2 vials per pack) |
Components |
|
|
1 vial of Tixagevimab 150 mg/1.5 mL (100 mg/mL) (dark grey cap) |
1 vial of Cilgavimab 150 mg/1.5 mL (100 mg/mL) (white cap) |
|
|
NDC 0310-7442-02 |
NDC 0310-8895-01 |
NDC 0310-1061-01 |
|
NDC 0310-8861-02 |
||
Storage and Handling
Store unopened vials in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light. Discard any unused portion.
DO NOT FREEZE. DO NOT SHAKE.
17 PATIENT COUNSELING INFORMATION
As a prescribing healthcare practitioner, you must communicate to the patient, parent and caregiver information consistent with the “FACT SHEET FOR PATIENTS, PARENTS OR CAREGIVERS” and provide them with a copy of this Fact Sheet prior to administration of EVUSHELD.
Dosing
Inform individuals that they will need to receive additional doses of EVUSHELD every 6 months if ongoing protection is needed [see Dosage and Administration (2.1), and Clinical Pharmacology (12.3)].
Risk for COVID-19 Due to SARS-CoV-2 Viral Variants Not Neutralized by EVUSHELD
Certain SARS-CoV-2 viral variants may not be neutralized by monoclonal antibodies such as tixagevimab and cilgavimab, the components of EVUSHELD. EVUSHELD may not be effective at preventing COVID-19 caused by these SARS-CoV-2 viral variants. Inform individuals of the increased risk, compared to other variants, for COVID-19 due to SARS-CoV-2 viral variants not neutralized by EVUSHELD. If signs and symptoms of COVID-19 occur, advise individuals to test for COVID-19 and seek medical attention, including starting treatment for COVID-19 as appropriate [see Warnings and Precautions (5.3)].
Cardiovascular Events
Inform individuals that a higher proportion of subjects who received EVUSHELD versus placebo reported cardiovascular serious adverse events (myocardial infarctions and heart failure). Advise individuals to seek immediate medical attention if they experience any signs or symptoms suggestive of a cardiovascular event [see Warnings and Precautions (5.5)].
For additional information, please visit the website or call the telephone number provided below.
To access the most recent EVUSHELD Fact Sheets, please scan the QR code provided below.
|
Website |
Telephone number |
 |
1-800-236-9933 |
18 MANUFACTURER INFORMATION
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
Manufactured for: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
©AstraZeneca 2023. All rights reserved.
Fact Sheet for Patients, Parents And Caregivers
Emergency Use Authorization (EUA) of
EVUSHELD™ (tixagevimab co-packaged with
cilgavimab) for Coronavirus Disease 2019
(COVID-19)
You are being given this Fact Sheet because your healthcare provider believes it is
necessary to provide you with EVUSHELD (tixagevimab co-packaged with cilgavimab)
for pre-exposure prophylaxis for prevention of coronavirus disease 2019 (COVID-19)
caused by the SARS-CoV-2 virus.
This Fact Sheet contains information to help you understand the potential risks and
potential benefits of taking EVUSHELD, which you have received or may receive.
The U.S. Food and Drug Administration (FDA) has issued an Emergency Use
Authorization (EUA) to make EVUSHELD available during the COVID-19 pandemic
(for more details about an EUA please see “What is an Emergency Use
Authorization?” at the end of this document). EVUSHELD is not an FDA-approved
medicine in the United States.
Read this Fact Sheet for information about EVUSHELD. Talk to your healthcare
provider if you have any questions. It is your choice to receive or not receive
EVUSHELD.
What is COVID-19?
COVID-19 is caused by a virus called a coronavirus. You can get COVID-19 through close contact with another person who has the virus.
COVID-19 illnesses have ranged from very mild (including some with no reported
symptoms) to severe, including illness resulting in death. While information so far
suggests that most COVID-19 illness is mild, serious illness can happen and may
cause some of your other medical conditions to become worse. Older people and
people of all ages with severe, long-lasting (chronic) medical conditions like heart
disease, lung disease, and diabetes, for example, seem to be at higher risk of
being hospitalized for COVID-19.
What is EVUSHELD (tixagevimab co-packaged with cilgavimab)?
EVUSHELD is an investigational medicine used in adults and adolescents (12 years of age and older who weigh at least 88 pounds [40 kg]) for pre-exposure prophylaxis for prevention of COVID-19 in persons who are:
- •
- not currently infected with SARS-CoV-2 and who have not had recent known close contact with someone who is infected with SARS-CoV-2 and
- ∘
- Who have moderate to severe immune compromise due to a medical condition or have received immunosuppressive medicines or treatments and may not mount an adequate immune response to COVID-19 vaccination or
- ∘
- For whom vaccination with any available COVID-19 vaccine, according to the approved or authorized schedule, is not recommended due to a history of severe adverse reaction to a COVID-19 vaccine(s) or COVID-19 vaccine ingredient(s).
EVUSHELD is investigational because it is still being studied. There is limited information known about the safety and effectiveness of using EVUSHELD for pre-exposure prophylaxis for prevention of COVID 19.
For more information about an EUA, see the “What is an Emergency Use Authorization (EUA)?” section at the end of this Fact Sheet.
EVUSHELD is not authorized for:
- •
- treatment of COVID-19, or
- •
- post-exposure prophylaxis of COVID-19 (use to prevent COVID-19 after
- being around someone infected with SARS-CoV-2), or
- •
- use when EVUSHELD is not expected to work (be active) against more
- than 90% of the circulating SARS-CoV-2 variants in the US. Viruses can
- change over time (mutate) and develop into a slightly different form of the
- virus, called a variant.
Pre-exposure prophylaxis for prevention of COVID-19 with EVUSHELD does not
take the place of vaccination in people for whom COVID-19 vaccination is
recommended. People who may benefit from COVID-19 vaccination should
receive COVID-19 vaccination. For people who have received a COVID-19
vaccine, wait two weeks after vaccination to receive EVUSHELD.
The FDA has authorized the emergency use of EVUSHELD for pre-exposure prophylaxis for prevention of COVID-19 under an Emergency Use Authorization (EUA).
What should I tell my healthcare provider before I receive EVUSHELD?
Tell your healthcare provider if you:
- •
- Have any allergies, including if you have had a severe allergic reaction to a COVID-19 vaccine
- •
- Have low numbers of blood platelets (which help blood clotting), a bleeding disorder, or are taking anticoagulants (to prevent blood clots)
- •
- Have had a heart attack or stroke, have other heart problems, or are at high-risk of cardiac (heart) events
- •
- Are pregnant or plan to become pregnant
- •
- Are breastfeeding a child
- •
- Have any serious illness
- •
- Are taking any medications (prescription, over-the-counter, vitamins, or herbal products)
How will I receive EVUSHELD?
- •
- EVUSHELD consists of two investigational medicines, tixagevimab and cilgavimab.
- •
- You will receive 1 dose of EVUSHELD, consisting of 2 separate injections (tixagevimab and cilgavimab).
- •
- EVUSHELD will be given to you by your healthcare provider as 2 intramuscular injections, given one after the other.
Viruses can change over time (mutate) and develop into a slightly different form of the virus, called a variant. Based on what we know about current SARS-CoV-2 variants, you will need to receive additional doses of EVUSHELD every 6 months if ongoing protection is needed. Talk to your healthcare provider for more information.
Who should generally not take EVUSHELD?
Do not take EVUSHELD if you have had a severe allergic reaction to EVUSHELD.
What are the important possible side effects of EVUSHELD?
Possible side effects of EVUSHELD are:
- •
- Allergic reactions. Allergic reactions can happen during and after injection of EVUSHELD and can sometimes be serious or life-threatening. You may have an increased risk of allergic reaction with EVUSHELD if you have had a severe allergic reaction to a COVID-19 vaccine. EVUSHELD contains polysorbate 80, an ingredient in some COVID-19 vaccines. Also, polysorbate 80 is similar to polyethylene glycol (PEG), an ingredient in other COVID-19 vaccines. Your healthcare provider may consult with a healthcare provider who specializes in allergy and immunology before giving you EVUSHELD if you have had a serious allergic reaction to a COVID-19 vaccine.
- Your healthcare provider will monitor you for allergic reactions during and for at least 1 hour after you receive EVUSHELD. Tell your healthcare provider right away if you get any of the following signs and symptoms of an allergic reaction during or after you receive EVUSHELD:
|
|
|
|
|
|
|
|
|
|
|
|
The side effects of getting any medicine by intramuscular injection may include pain, bruising of the skin, soreness, swelling, and possible bleeding or infection at the injection site.
These are not all the possible side effects of EVUSHELD. Not a lot of people have been given EVUSHELD. Serious and unexpected side effects may happen. EVUSHELD is still being studied so it is possible that all of the risks are not known at this time.
It is possible that EVUSHELD may reduce your body’s immune response to a COVID-19 vaccine. If you have received a COVID-19 vaccine, you should wait to receive EVUSHELD until at least 2 weeks after COVID-19 vaccination.
What other important information do I need to know when receiving EVUSHELD?
Risk of COVID-19 caused by certain SARS-CoV-2 variants: Viruses can change over time (mutate) and develop into a slightly different form of the virus, called a variant. EVUSHELD may not be effective at preventing COVID-19 caused by certain SARS-CoV-2 variants. If you are exposed to these variants, your chance of developing COVID-19 is higher than from other variants. Tell your healthcare provider right away, and test for COVID-19, if you develop any symptoms of COVID-19, including:
|
|
|
|
|
|
|
|
|
|
If you develop COVID-19, your healthcare provider may recommend one of the available COVID-19 treatments.
For more information about the symptoms of COVID-19, go to https://www.cdc.gov/coronavirus/2019-ncov/symptoms-testing/symptoms.
What other prevention choices are there?
Vaccines to prevent COVID-19 are approved or available under Emergency Use Authorization. Use of EVUSHELD does not replace vaccination against COVID-19. For more information about other medicines authorized for treatment or prevention of COVID-19 go to https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization for more information.
It is your choice to receive or not receive EVUSHELD. Should you decide not to receive EVUSHELD, it will not change your standard medical care.
EVUSHELD is not authorized for post-exposure prophylaxis of COVID-19.
What if I am pregnant or breastfeeding?
If you are pregnant or breastfeeding, discuss your options and specific situation with your healthcare provider.
How do I report side effects with EVUSHELD?
Contact your healthcare provider if you have any side effects that bother you or do not go away. Report side effects to FDA MedWatch at www.fda.gov/medwatch or call 1-800-FDA-1088 or call AstraZeneca at 1-800-236-9933.
Additional Information
If you have questions, visit the website or call the telephone number provided below.
To access the most recent EVUSHELD Fact Sheets, please scan the QR code provided below.
|
Website |
Telephone number |
 |
1-800-236-9933 |
How can I learn more about COVID-19?
- •
- Ask your healthcare provider.
- •
- Visit https://www.cdc.gov/COVID19
- •
- Contact your local or state public health department.
What is an Emergency Use Authorization?
The United States FDA has made EVUSHELD (tixagevimab co-packaged with cilgavimab) available under an emergency access mechanism called an Emergency Use Authorization EUA. The EUA is supported by a Secretary of Health and Human Services (HHS) declaration that circumstances exist to justify the emergency use of drugs and biological products during the COVID-19 pandemic.
EVUSHELD for pre-exposure prophylaxis for prevention of coronavirus disease 2019 (COVID-19) caused by the SARS-CoV-2 virus has not undergone the same type of review as an FDA-approved product. In issuing an EUA under the COVID-19 public health emergency, the FDA has determined, among other things, that based on the total amount of scientific evidence available including data from adequate and well-controlled clinical trials, if available, it is reasonable to believe that the product may be effective for diagnosing, treating, or preventing COVID-19, or a serious or life-threatening disease or condition caused by COVID-19; that the known and potential benefits of the product, when used to diagnose, treat, or prevent such disease or condition, outweigh the known and potential risks of such product; and that there are no adequate, approved and available alternatives.
All of these criteria must be met to allow for the product to be used in the treatment of patients during the COVID-19 pandemic. The EUA for EVUSHELD is in effect for the duration of the COVID-19 declaration justifying emergency use of EVUSHELD, unless terminated or revoked (after which EVUSHELD may no longer be used under the EUA).
What are the ingredients in EVUSHELD?
Each EVUSHELD co-packaged carton contains 2 vials.
Active ingredient: Each of the vials contains either: tixagevimab or cilgavimab
Inactive ingredients: Each vial contains L- histidine, L- histidine hydrochloride monohydrate, polysorbate 80, sucrose, water.
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
Manufactured for: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
©AstraZeneca 2023. All rights reserved.
Package/Label Display Panel
tixagevimab
Injection
150 mg/1.5 mL
(100mg/mL)
For Intramuscular Use.
1.5 mL single-dose vial.
MUST ADMINISTER
WITH CILGAVIMAB
Rx only
NDC 0310-8895-01
Discard unused portion.
No preservative. Store at
2° to 8°C (36° to 46°F).
Do not freeze or shake.
Protect from light.
For use under Emergency
Use Authorization (EUA)
AstraZeneca

Package/Label Display Panel
cilgavimab
Injection
150 mg/1.5 mL
(100mg/mL)
For Intramuscular Use.
1.5 mL single-dose vial.
MUST ADMINISTER
WITH TIXAGEVIMAB
Rx only
NDC 0310-1061-01
Discard unused portion.
No preservative. Store at
2° to 8°C (36° to 46°F).
Do not freeze or shake.
Protect from light.
For use under Emergency
Use Authorization (EUA)
AstraZeneca

Package/Label Display Panel
NDC 0310-7442-02
EVUSHELD™
(tixagevimab) Injection
150 mg/1.5 mL
(100mg/mL)
Co-packaged with
(cilgavimab) injection
150 mg/1.5 mL
(100mg/mL)
For use under Emergency Use Authorization (EUA)
For Intramuscular Use
Keep vials in original carton to protect from light.
Discard unused portion.
Each carton contains two (2) vials:
One 1.5 mL single-dose vial of tixagevimab
One 1.5 mL single-dose vial of cilgavimab
ATTENTION HEALTHCARE PROVIDER: EACH
VIAL MUST BE ADMINISTERED SEPARATELY
BY INTRAMUSCULAR INJECTION
Refer to FDA-Authorized Fact Sheets
for Dosage and Administration.
Rx only AstraZeneca

| EVUSHELD
azd7442 kit |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| Labeler - AstraZeneca Pharmaceuticals LP (054743190) |
| Registrant - AstraZeneca PLC (230790719) |