ALLEGRA D-12 HOUR- fexofenadine hydrochloride and pseudoephedrine hydrochloride tablet, extended release
sanofi-aventis U.S. LLC
----------
ALLEGRA-D® 12 HOUR
(fexofenadine HCl 60 mg and pseudoephedrine HCl 120 mg)
Extended-Release Tablets
DESCRIPTION
ALLEGRA-D® 12 HOUR (fexofenadine hydrochloride and pseudoephedrine hydrochloride) Extended-Release Tablets for oral administration contain 60 mg fexofenadine hydrochloride for immediate release and 120 mg pseudoephedrine hydrochloride for extended release. Tablets also contain as excipients: microcrystalline cellulose, pregelatinized starch, croscarmellose sodium, magnesium stearate, carnauba wax, stearic acid, silicon dioxide, hypromellose and polyethylene glycol.
Fexofenadine hydrochloride, one of the active ingredients of ALLEGRA-D 12 HOUR, is a histamine H1-receptor antagonist with the chemical name (±)-4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-butyl]-α,α-dimethyl benzeneacetic acid hydrochloride and the following chemical structure:
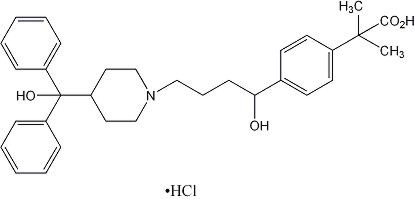
The molecular weight is 538.13 and the empirical formula is C32H39NO4•HCl. Fexofenadine hydrochloride is a white to off-white crystalline powder. It is freely soluble in methanol and ethanol, slightly soluble in chloroform and water, and insoluble in hexane. Fexofenadine hydrochloride is a racemate and exists as a zwitterion in aqueous media at physiological pH.
Pseudoephedrine hydrochloride, the other active ingredient of ALLEGRA-D 12 HOUR, is an adrenergic (vasoconstrictor) agent with the chemical name [S-(R*,R*)]-α-[1-(methylamino)ethyl]-benzenemethanol hydrochloride and the following chemical structure:
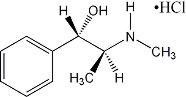
The molecular weight is 201.70. The molecular formula is C10H15NO•HCl. Pseudoephedrine hydrochloride occurs as fine, white to off-white crystals or powder, having a faint characteristic odor. It is very soluble in water, freely soluble in alcohol, and sparingly soluble in chloroform.
CLINICAL PHARMACOLOGY
Mechanism of Action
Fexofenadine hydrochloride, the major active metabolite of terfenadine, is an antihistamine with selective peripheral H1-receptor antagonist activity. Fexofenadine hydrochloride inhibited antigen-induced bronchospasm in sensitized guinea pigs and histamine release from peritoneal mast cells in rats. In laboratory animals, no anticholinergic or alpha1-adrenergic-receptor blocking effects were observed. Moreover, no sedative or other central nervous system effects were observed. Radiolabeled tissue distribution studies in rats indicated that fexofenadine does not cross the blood-brain barrier.
Pseudoephedrine hydrochloride is an orally active sympathomimetic amine and exerts a decongestant action on the nasal mucosa. Pseudoephedrine hydrochloride is recognized as an effective agent for the relief of nasal congestion due to allergic rhinitis. Pseudoephedrine produces peripheral effects similar to those of ephedrine and central effects similar to, but less intense than, amphetamines. It has the potential for excitatory side effects. At the recommended oral dose, it has little or no pressor effect in normotensive adults.
Pharmacokinetics
The pharmacokinetics of fexofenadine hydrochloride in subjects with seasonal allergic rhinitis were similar to those in healthy volunteers.
Absorption
The pharmacokinetics of fexofenadine hydrochloride and pseudoephedrine hydrochloride when administered separately have been well characterized. Fexofenadine pharmacokinetics were linear for oral doses of fexofenadine hydrochloride up to a total daily dose of 240 mg (120 mg twice daily). Peak fexofenadine plasma concentrations were similar between adolescent (12–16 years of age) and adult subjects.
The bioavailability of fexofenadine hydrochloride and pseudoephedrine hydrochloride from ALLEGRA-D 12 HOUR Extended-Release Tablets is similar to that achieved with separate administration of the components. Coadministration of fexofenadine and pseudoephedrine does not significantly affect the bioavailability of either component.
Fexofenadine hydrochloride was rapidly absorbed following single-dose administration of the 60 mg fexofenadine hydrochloride/120 mg pseudoephedrine hydrochloride tablet with median time to mean maximum fexofenadine plasma concentration of 191 ng/mL occurring 2 hours post-dose. Pseudoephedrine hydrochloride produced a mean single-dose pseudoephedrine peak plasma concentration of 206 ng/mL which occurred 6 hours post-dose. Following multiple dosing to steady-state, a fexofenadine peak concentration of 255 ng/mL was observed 2 hours post-dose. Following multiple dosing to steady-state, a pseudoephedrine peak concentration of 411 ng/mL was observed 5 hours post-dose. The administration of ALLEGRA-D 12 HOUR with a high fat meal decreased the bioavailability of fexofenadine by approximately 50% (AUC 42% and Cmax 46%). Time to maximum concentration (Tmax) was delayed by 50%. The rate or extent of pseudoephedrine absorption was not affected by food. Therefore, ALLEGRA-D 12 HOUR should be taken on an empty stomach with water (see DOSAGE AND ADMINISTRATION).
Distribution
Fexofenadine is 60% to 70% bound to plasma proteins, primarily albumin and α1-acid glycoprotein. The protein binding of pseudoephedrine in humans is not known. Pseudoephedrine hydrochloride is extensively distributed into extravascular sites (apparent volume of distribution between 2.6 and 3.5 L/kg).
Metabolism
Approximately 5% of the total dose of fexofenadine hydrochloride and less than 1% of the total oral dose of pseudoephedrine hydrochloride were eliminated by hepatic metabolism.
Elimination
The mean elimination half-life of fexofenadine was 14.4 hours following administration of 60 mg fexofenadine hydrochloride, twice daily, to steady-state in healthy volunteers. Human mass balance studies documented a recovery of approximately 80% and 11% of the [14C] fexofenadine hydrochloride dose in the feces and urine, respectively. Because the absolute bioavailability of fexofenadine hydrochloride has not been established, it is unknown if the fecal component is primarily unabsorbed drug or the result of biliary excretion.
Pseudoephedrine has been shown to have a mean elimination half-life of 4–6 hours which is dependent on urine pH. The elimination half-life is decreased at urine pH lower than 6 and may be increased at urine pH higher than 8.
Special Populations
Pharmacokinetics in special populations (for renal, hepatic impairment, and age), obtained after a single dose of 80 mg fexofenadine hydrochloride, were compared to those from healthy subjects in a separate study of similar design.
Effect of Age
In older subjects (≥65 years old), peak plasma levels of fexofenadine were 99% greater than those observed in younger subjects (<65 years old). Mean fexofenadine elimination half-lives were similar to those observed in younger subjects.
Renally Impaired
In subjects with mild (creatinine clearance 41–80 mL/min) to severe (creatinine clearance 11–40 mL/min) renal impairment, peak plasma levels of fexofenadine were 87% and 111% greater, respectively, and mean elimination half-lives were 59% and 72% longer, respectively, than observed in healthy volunteers. Peak plasma levels in subjects on dialysis (creatinine clearance ≤10 mL/min) were 82% greater and half-life was 31% longer than observed in healthy volunteers.
No data are available on the pharmacokinetics of pseudoephedrine in renally-impaired subjects. However, most of the oral dose of pseudoephedrine hydrochloride (43–96%) is excreted unchanged in the urine. A decrease in renal function is, therefore, likely to decrease the clearance of pseudoephedrine significantly, thus prolonging the half-life and resulting in accumulation.
Based on increases in bioavailability and half-life of fexofenadine hydrochloride and pseudoephedrine hydrochloride, a dose of one tablet once daily is recommended as the starting dose in patients with decreased renal function (see DOSAGE AND ADMINISTRATION).
Pharmacodynamics
Wheal and Flare
Human histamine skin wheal and flare studies following single and twice daily doses of 20 mg and 40 mg fexofenadine hydrochloride demonstrated that the drug exhibits an antihistamine effect by 1 hour, achieves maximum effect at 2–3 hours, and an effect is still seen at 12 hours. There was no evidence of tolerance to these effects after 28 days of dosing. The clinical significance of these observations is unknown.
Effects on QTc
In dogs (30 mg/kg orally twice daily for 5 days) and rabbits (10 mg/kg intravenously over 1 hour), fexofenadine hydrochloride did not prolong QTc at plasma concentrations that were at least 17 and 38 times, respectively, the therapeutic plasma concentrations in man (based on a 60 mg twice daily fexofenadine hydrochloride dose). No effect was observed on calcium channel current, delayed K+ channel current, or action potential duration in guinea pig myocytes, Na+ current in rat neonatal myocytes, or on the delayed rectifier K+ channel cloned from human heart at concentrations up to 1 × 10-5 M of fexofenadine. This concentration was at least 21 times the therapeutic plasma concentration in man (based on a 60 mg twice daily fexofenadine hydrochloride dose).
No statistically significant increase in mean QTc interval compared to placebo was observed in 714 subjects with seasonal allergic rhinitis given fexofenadine hydrochloride capsules in doses of 60 mg to 240 mg twice daily for 2 weeks or in 40 healthy volunteers given fexofenadine hydrochloride as an oral solution at doses up to 400 mg twice daily for 6 days.
A 1-year study designed to evaluate safety and tolerability of 240 mg of fexofenadine hydrochloride (n=240) compared to placebo (n=237) in healthy volunteers, did not reveal a statistically significant increase in the mean QTc interval for the fexofenadine hydrochloride treated group when evaluated pretreatment and after 1, 2, 3, 6, 9, and 12 months of treatment.
Administration of the 60 mg fexofenadine hydrochloride/120 mg pseudoephedrine hydrochloride combination tablet for approximately 2 weeks to 213 subjects with seasonal allergic rhinitis demonstrated no statistically significant increase in the mean QTc interval compared to fexofenadine hydrochloride administered alone (60 mg twice daily, n=215), or compared to pseudoephedrine hydrochloride (120 mg twice daily, n=215) administered alone.
Clinical Studies
In a 2-week, multicenter, randomized, double-blind, active-controlled trial in subjects 12–65 years of age with seasonal allergic rhinitis due to ragweed allergy (n=651), the 60 mg fexofenadine hydrochloride/120 mg pseudoephedrine hydrochloride combination tablet administered twice daily significantly reduced the intensity of sneezing, rhinorrhea, itchy nose/palate/throat, itchy/watery/red eyes, and nasal congestion.
In three, 2-week, multicenter, randomized, double-blind, placebo-controlled trials in subjects 12–68 years of age with seasonal allergic rhinitis (n=1634), fexofenadine hydrochloride 60 mg twice daily significantly reduced total symptom scores (the sum of the individual scores for sneezing, rhinorrhea, itchy nose/palate/throat, itchy/watery/red eyes) compared to placebo. Statistically significant reductions in symptom scores were observed following the first 60 mg dose, with the effect maintained throughout the 12-hour interval. In general, there was no additional reduction in total symptom scores with higher doses of fexofenadine hydrochloride up to 240 mg twice daily. Although the number of subjects in some of the subgroups was small, there were no significant differences in the effect of fexofenadine hydrochloride across subgroups of subjects defined by gender, age, and race. Onset of action for reduction in total symptom scores, excluding nasal congestion, was observed at 60 minutes compared to placebo following a single 60 mg fexofenadine hydrochloride dose administered to subjects with seasonal allergic rhinitis who were exposed to ragweed pollen in an environmental exposure unit.
INDICATIONS AND USAGE
ALLEGRA-D 12 HOUR Extended-Release Tablets are indicated for the relief of symptoms associated with seasonal allergic rhinitis in adults and children 12 years of age and older. Symptoms treated effectively include sneezing, rhinorrhea, itchy nose/palate/ and/or throat, itchy/watery/red eyes, and nasal congestion.
ALLEGRA-D 12 HOUR should be administered when both the antihistaminic properties of fexofenadine hydrochloride and the nasal decongestant properties of pseudoephedrine hydrochloride are desired (see CLINICAL PHARMACOLOGY).
CONTRAINDICATIONS
ALLEGRA-D 12 HOUR is contraindicated in patients with known hypersensitivity to any of its ingredients.
Due to its pseudoephedrine component, ALLEGRA-D 12 HOUR is contraindicated in patients with narrow-angle glaucoma or urinary retention, and in patients receiving monoamine oxidase (MAO) inhibitor therapy or within fourteen (14) days of stopping such treatment (see Drug Interactions section). It is also contraindicated in patients with severe hypertension, or severe coronary artery disease, and in those who have shown idiosyncrasy to its components, to adrenergic agents, or to other drugs of similar chemical structures. Manifestations of patient idiosyncrasy to adrenergic agents include: insomnia, dizziness, weakness, tremor, or arrhythmias.
WARNINGS
Sympathomimetic amines should be used with caution in patients with hypertension, diabetes mellitus, ischemic heart disease, increased intraocular pressure, hyperthyroidism, renal impairment, or prostatic hypertrophy (see CONTRAINDICATIONS). Sympathomimetic amines may produce central nervous system stimulation with convulsions or cardiovascular collapse with accompanying hypotension.
PRECAUTIONS
General
Patients with decreased renal function should be given a lower initial dose (one tablet per day) because they have reduced elimination of fexofenadine and pseudoephedrine (see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
Information for Patients
Patients taking ALLEGRA-D 12 HOUR tablets should receive the following information: ALLEGRA-D 12 HOUR tablets are prescribed for the relief of symptoms of seasonal allergic rhinitis. Patients should be instructed to take ALLEGRA-D 12 HOUR tablets only as prescribed. Do not exceed the recommended dose. If nervousness, dizziness, or sleeplessness occur, discontinue use and consult the doctor. Patients should also be advised against the concurrent use of ALLEGRA-D 12 HOUR tablets with over-the-counter antihistamines and decongestants.
The product should not be used by patients who are hypersensitive to it or to any of its ingredients. Due to its pseudoephedrine component, this product should not be used by patients with narrow-angle glaucoma, urinary retention, or by patients receiving a monoamine oxidase (MAO) inhibitor or within 14 days of stopping use of MAO inhibitor. It also should not be used by patients with severe hypertension or severe coronary artery disease.
Patients should be told that this product should be used in pregnancy or lactation only if the potential benefit justifies the potential risk to the fetus or nursing infant. Patients should be advised to take the tablet on an empty stomach with water. Patients should be directed to swallow the tablet whole. Patients should be cautioned not to break or chew the tablet. Patients should also be instructed to store the medication in a tightly closed container in a cool, dry place, away from children.
Patients should be told that the inactive ingredients may occasionally be eliminated in the feces in a form that may resemble the original tablet (see DOSAGE AND ADMINISTRATION).
Drug Interactions
Fexofenadine hydrochloride and pseudoephedrine hydrochloride do not influence the pharmacokinetics of each other when administered concomitantly.
Fexofenadine has been shown to exhibit minimal (ca. 5%) metabolism. However, co-administration of fexofenadine hydrochloride with either ketoconazole or erythromycin led to increased plasma concentrations of fexofenadine. Fexofenadine had no effect on the pharmacokinetics of either erythromycin or ketoconazole. In 2 separate studies, fexofenadine hydrochloride 120 mg twice daily (twice the recommended dose) was co-administered with erythromycin 500 mg every 8 hours or ketoconazole 400 mg once daily under steady-state conditions to healthy volunteers (n=24, each study). No differences in adverse events or QTc interval were observed when subjects were administered fexofenadine hydrochloride alone or in combination with either erythromycin or ketoconazole. The findings of these studies are summarized in the following table.
| Concomitant Drug | Cmax SS
(Peak plasma concentration) | AUCSS(0–12h) (Extent of systemic exposure) |
|---|---|---|
| Erythromycin (500 mg every 8 hrs) | +82% | +109% |
| Ketoconazole (400 mg once daily) | +135% | +164% |
The changes in plasma levels were within the range of plasma levels achieved in adequate and well-controlled clinical trials.
The mechanism of these interactions has been evaluated in in vitro, in situ, and in vivo animal models. These studies indicate that ketoconazole or erythromycin co-administration enhances fexofenadine gastrointestinal absorption. This observed increase in the bioavailability of fexofenadine may be due to transport-related effects, such as p-glycoprotein. In vivo animal studies also suggest that in addition to enhancing absorption, ketoconazole decreases fexofenadine gastrointestinal secretion, while erythromycin may also decrease biliary excretion.
Due to the pseudoephedrine component, ALLEGRA-D 12 HOUR is contraindicated in patients taking monoamine oxidase inhibitors and for 14 days after stopping use of an MAO inhibitor. Concomitant use with antihypertensive drugs which interfere with sympathetic activity (e.g., methyldopa, mecamylamine, and reserpine) may reduce their antihypertensive effects. Increased ectopic pacemaker activity can occur when pseudoephedrine is used concomitantly with digitalis. Care should be taken in the administration of ALLEGRA-D 12 HOUR concomitantly with other sympathomimetic amines because combined effects on the cardiovascular system may be harmful to the patient (see WARNINGS).
Drug Interactions with Antacids
Administration of 120 mg of fexofenadine hydrochloride (2 × 60 mg capsule) within 15 minutes of an aluminum and magnesium containing antacid (Maalox®) decreased fexofenadine AUC by 41% and Cmax by 43%. ALLEGRA-D 12 HOUR should not be taken closely in time with aluminum and magnesium containing antacids.
Interactions with Fruit Juices
Fruit juices such as grapefruit, orange and apple may reduce the bioavailability and exposure of fexofenadine. This is based on the results from 3 clinical studies using histamine induced skin wheals and flares coupled with population pharmacokinetic analysis. The size of wheal and flare were significantly larger when fexofenadine hydrochloride was administered with either grapefruit or orange juices compared to water. Based on the literature reports, the same effects may be extrapolated to other fruit juices such as apple juice. The clinical significance of these observations is unknown. In addition, based on the population pharmacokinetics analysis of the combined data from grapefruit and orange juices studies with the data from a bioequivalence study, the bioavailability of fexofenadine was reduced by 36%. Therefore, to maximize the effects of fexofenadine, it is recommended that ALLEGRA-D 12 HOUR should be taken with water (see DOSAGE AND ADMINISTRATION).
Carcinogenesis, Mutagenesis, Impairment of Fertility
There are no animal or in vitro studies on the combination product fexofenadine hydrochloride and pseudoephedrine hydrochloride to evaluate carcinogenesis, mutagenesis, or impairment of fertility.
The carcinogenic potential and reproductive toxicity of fexofenadine hydrochloride were assessed using terfenadine studies with adequate fexofenadine exposure (area-under-the plasma concentration versus time curve [AUC]). No evidence of carcinogenicity was observed when mice and rats were given daily oral doses up to 150 mg/kg of terfenadine for 18 and 24 months, respectively. In both species, 150 mg/kg of terfenadine produced AUC values of fexofenadine that were approximately 3 times the human AUC at the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR.
Two-year feeding studies in rats and mice conducted under the auspices of the National Toxicology Program (NTP) demonstrated no evidence of carcinogenic potential with ephedrine sulfate, a structurally related drug with pharmacological properties similar to pseudoephedrine, at doses up to 10 and 27 mg/kg, respectively (less than the maximum recommended human daily oral dose of pseudoephedrine hydrochloride on a mg/m2 basis).
In in vitro (Bacterial Reverse Mutation, CHO/HGPRT Forward Mutation, and Rat Lymphocyte Chromosomal Aberration assays) and in vivo (Mouse Bone Marrow Micronucleus assay) tests, fexofenadine hydrochloride revealed no evidence of mutagenicity.
Reproduction and fertility studies with terfenadine in rats produced no effect on male or female fertility at oral doses up to 300 mg/kg/day. However, reduced implants and post implantation losses were reported at 300 mg/kg. A reduction in implants was also observed at an oral dose of 150 mg/kg/day. Oral doses of 150 and 300 mg/kg of terfenadine produced AUC values of fexofenadine that were approximately 4 times the AUC at the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR. In mice, fexofenadine produced no effect on male or female fertility at average dietary doses up to 4438 mg/kg (approximately 15 times the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR based on comparison of the AUCs).
Pregnancy
Teratogenic Effects
Category C. Terfenadine alone was not teratogenic in rats and rabbits at oral doses up to 300 mg/kg; 300 mg/kg of terfenadine produced fexofenadine AUC values that were approximately 4 and 30 times, respectively, the AUC at the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR.
In mice, no adverse effects and no teratogenic effects during gestation were observed with fexofenadine at dietary doses up to 3730 mg/kg (approximately 15 times the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR based on comparison of the AUCs).
The combination of terfenadine and pseudoephedrine hydrochloride in a ratio of 1:2 by weight was studied in rats and rabbits. In rats, an oral combination dose of 150/300 mg/kg produced reduced fetal weight and delayed ossification with a finding of wavy ribs. The dose of 150 mg/kg of terfenadine in rats produced an AUC value of fexofenadine that was approximately 4 times the AUC at the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR. The dose of 300 mg/kg of pseudoephedrine hydrochloride in rats was approximately 10 times the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR on a mg/m2 basis. In rabbits, an oral combination dose of 100/200 mg/kg produced decreased fetal weight. By extrapolation, the AUC of fexofenadine for 100 mg/kg orally of terfenadine was approximately 10 times the AUC at the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR. The dose of 200 mg/kg of pseudoephedrine hydrochloride was approximately 15 times the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR on a mg/m2 basis.
There are no adequate and well-controlled studies in pregnant women. ALLEGRA-D 12 HOUR should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nonteratogenic Effects
Dose-related decreases in pup weight gain and survival were observed in rats exposed to an oral dose of 150 mg/kg of terfenadine; this dose produced an AUC of fexofenadine that was approximately 4 times the AUC at the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR.
Nursing Mothers
It is not known if fexofenadine is excreted in human milk. Because many drugs are excreted in human milk, caution should be used when fexofenadine hydrochloride is administered to a nursing woman. Pseudoephedrine hydrochloride administered alone distributes into breast milk of lactating human females. Pseudoephedrine concentrations in milk are consistently higher than those in plasma. The total amount of drug in milk as judged by AUC is 2 to 3 times greater than the plasma AUC. The fraction of a pseudoephedrine dose excreted in milk is estimated to be 0.4% to 0.7%. A decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. Caution should be exercised when ALLEGRA-D 12 HOUR is administered to nursing women.
Pediatric Use
Safety and effectiveness of ALLEGRA-D 12 HOUR in children below the age of 12 years have not been established. In addition, the doses of the individual components in ALLEGRA-D 12 HOUR exceed the recommended individual doses for pediatric patients under 12 years of age. ALLEGRA-D 12 HOUR is not recommended for pediatric patients under 12 years of age.
Geriatric Use
Clinical studies of ALLEGRA-D 12 HOUR did not include sufficient numbers of subjects aged 65 and older to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger subjects, although the elderly are more likely to have adverse reactions to sympathomimetic amines.
The pseudoephedrine component of ALLEGRA-D 12 HOUR is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
ADVERSE REACTIONS
ALLEGRA-D 12 HOUR
In one clinical trial (n=651) in which 215 subjects with seasonal allergic rhinitis received the 60 mg fexofenadine hydrochloride/120 mg pseudoephedrine hydrochloride combination tablet twice daily for up to 2 weeks, adverse events were similar to those reported either in subjects receiving fexofenadine hydrochloride 60 mg alone (n=218 subjects) or in subjects receiving pseudoephedrine hydrochloride 120 mg alone (n=218). A placebo group was not included in this study.
The percent of subjects who withdrew prematurely because of adverse events was 3.7% for the fexofenadine hydrochloride/pseudoephedrine hydrochloride combination group, 0.5% for the fexofenadine hydrochloride group, and 4.1% for the pseudoephedrine hydrochloride group. All adverse events that were reported by greater than 1% of subjects who received the recommended daily dose of the fexofenadine hydrochloride/pseudoephedrine hydrochloride combination are listed in the following table.
| Adverse Experience | 60 mg Fexofenadine Hydrochloride/120 mg Pseudoephedrine Hydrochloride Combination Tablet Twice Daily (n=215) | Fexofenadine Hydrochloride 60 mg Twice Daily (n=218) | Pseudoephedrine Hydrochloride 120 mg Twice Daily (n=218) |
|---|---|---|---|
| Headache | 13.0% | 11.5% | 17.4% |
| Insomnia | 12.6% | 3.2% | 13.3% |
| Nausea | 7.4% | 0.5% | 5.0% |
| Dry Mouth | 2.8% | 0.5% | 5.5% |
| Dyspepsia | 2.8% | 0.5% | 0.9% |
| Throat Irritation | 2.3% | 1.8% | 0.5% |
| Dizziness | 1.9% | 0.0% | 3.2% |
| Agitation | 1.9% | 0.0% | 1.4% |
| Back Pain | 1.9% | 0.5% | 0.5% |
| Palpitation | 1.9% | 0.0% | 0.9% |
| Nervousness | 1.4% | 0.5% | 1.8% |
| Anxiety | 1.4% | 0.0% | 1.4% |
| Upper Respiratory Infection | 1.4% | 0.9% | 0.9% |
| Abdominal Pain | 1.4% | 0.5% | 0.5% |
Many of the adverse events occurring in the fexofenadine hydrochloride/pseudoephedrine hydrochloride combination group were adverse events also reported predominately in the pseudoephedrine hydrochloride group, such as insomnia, headache, nausea, dry mouth, dizziness, agitation, nervousness, anxiety, and palpitation.
Fexofenadine Hydrochloride
In placebo-controlled clinical trials, which included 2461 subjects receiving fexofenadine hydrochloride at doses of 20 mg to 240 mg twice daily, adverse events were similar in fexofenadine hydrochloride and placebo-treated subjects. The incidence of adverse events, including drowsiness, was not dose related and was similar across subgroups defined by age, gender, and race. The percent of subjects who withdrew prematurely because of adverse events was 2.2% with fexofenadine hydrochloride vs 3.3% with placebo.
Events that have been reported during controlled clinical trials involving subjects with seasonal allergic rhinitis and chronic idiopathic urticaria at incidences less than 1% and similar to placebo and have been rarely reported during postmarketing surveillance include: insomnia, nervousness, and sleep disorders or paroniria. In rare cases, rash, urticaria, pruritus and hypersensitivity reactions with manifestations such as angioedema, chest tightness, dyspnea, flushing and systemic anaphylaxis have been reported.
Pseudoephedrine Hydrochloride
Pseudoephedrine hydrochloride may cause mild CNS stimulation in hypersensitive patients. Nervousness, excitability, restlessness, dizziness, weakness, or insomnia may occur. Headache, drowsiness, tachycardia, palpitation, pressor activity, cardiac arrhythmias and ischemic colitis have been reported. Sympathomimetic drugs have also been associated with other untoward effects such as fear, anxiety, tenseness, tremor, hallucinations, seizures, pallor, respiratory difficulty, dysuria, and cardiovascular collapse.
OVERDOSAGE
Most reports of fexofenadine hydrochloride overdose contain limited information. However, dizziness, drowsiness, and dry mouth have been reported. For the pseudoephedrine hydrochloride component of ALLEGRA-D 12 HOUR, information on acute overdose is limited to the marketing history of pseudoephedrine hydrochloride. Single doses of fexofenadine hydrochloride up to 800 mg (6 healthy volunteers at this dose level), and doses up to 690 mg twice daily for one month (3 healthy volunteers at this dose level), were administered without the development of clinically significant adverse events.
In large doses, sympathomimetics may give rise to giddiness, headache, nausea, vomiting, sweating, thirst, tachycardia, precordial pain, palpitations, difficulty in micturition, muscular weakness and tenseness, anxiety, restlessness, and insomnia. Many patients can present a toxic psychosis with delusions and hallucinations. Some may develop cardiac arrhythmias, circulatory collapse, convulsions, coma, and respiratory failure.
In the event of overdose, consider standard measures to remove any unabsorbed drug. Symptomatic and supportive treatment is recommended. Following administration of terfenadine, hemodialysis did not effectively remove fexofenadine, the major active metabolite of terfenadine, from blood (up to 1.7% removed). The effect of hemodialysis on the removal of pseudoephedrine is unknown.
No deaths occurred in mature mice and rats at oral doses of fexofenadine hydrochloride up to 5000 mg/kg (approximately 170 and 340 times, respectively, the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR on a mg/m2 basis.) The median oral lethal dose in newborn rats was 438 mg/kg (approximately 30 times the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR on a mg/m2 basis). In dogs, no evidence of toxicity was observed at oral doses up to 2000 mg/kg (approximately 450 times the maximum recommended human daily oral dose on a mg/m2 basis). The oral median lethal dose of pseudoephedrine hydrochloride in rats was 1674 mg/kg (approximately 55 times the maximum recommended human daily oral dose of ALLEGRA-D 12 HOUR on a mg/m2 basis).
DOSAGE AND ADMINISTRATION
The recommended dose of ALLEGRA-D 12 HOUR Extended-Release Tablets is one tablet twice daily administered on an empty stomach with water for adults and children 12 years of age and older. It is recommended that the administration of ALLEGRA-D 12 HOUR with food should be avoided. A dose of one tablet once daily is recommended as the starting dose in patients with decreased renal function. (See CLINICAL PHARMACOLOGY and PRECAUTIONS.)
ALLEGRA-D 12 HOUR must be swallowed whole and never crushed or chewed. Occasionally, the inactive ingredients of ALLEGRA-D 12 HOUR may be eliminated in the feces in a form that may resemble the original tablet. (See PRECAUTIONS, Information for Patients.)
HOW SUPPLIED
ALLEGRA-D 12 HOUR Extended-Release Tablets contain 60 mg fexofenadine hydrochloride for immediate release and 120 mg pseudoephedrine hydrochloride for extended release. ALLEGRA-D 12 HOUR Extended-Release Tablets are available in high-density polyethylene (HDPE) bottles of 100 (NDC 0088-1090-47) with a polypropylene screw cap containing a pulp/wax liner with heat-sealed foil inner seal; HDPE bottles of 500 (NDC 0088-1090-55) with a polypropylene screw cap containing a pulp/wax liner with heat-sealed foil inner seal; and aluminum foil-backed clear blister packs of 100 (NDC 0088-1090-49).
ALLEGRA-D 12 HOUR is a two-layer tablet, one white layer and one tan layer with a clear film coating on the tablet. The tablets are engraved with "06/012D" on the white layer.
Store ALLEGRA-D 12 HOUR Extended-Release Tablets at 20–25°C (68–77°F). (See USP Controlled Room Temperature.)

| ALLEGRA D-12 HOUR
fexofenadine hydrochloride and pseudoephedrine hydrochloride tablet, extended release |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - sanofi-aventis U.S. LLC (783243835) |