AMERGE- naratriptan hydrochloride tablet, film coated
GlaxoSmithKline LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use AMERGE safely and effectively. See full prescribing information for AMERGE.
AMERGE (naratriptan hydrochloride) tablets, for oral use Initial U.S. Approval: 1998 INDICATIONS AND USAGEAMERGE is a serotonin (5-HT1B/1D) receptor agonist (triptan) indicated for the acute treatment of migraine with or without aura in adults. (1) Limitations of Use DOSAGE AND ADMINISTRATIONCONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (≥2% and >placebo) were paresthesias, nausea, dizziness, drowsiness, malaise/fatigue, and throat/neck symptoms. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact GlaxoSmithKline at 1-888-825-5249 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch USE IN SPECIFIC POPULATIONSPregnancy: Based on animal data, may cause fetal harm. (8.1) See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 10/2020 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
AMERGE is indicated for the acute treatment of migraine with or without aura in adults.
Limitations of Use
- •
- Use only if a clear diagnosis of migraine has been established. If a patient has no response to the first migraine attack treated with AMERGE, reconsider the diagnosis of migraine before AMERGE is administered to treat any subsequent attacks.
- •
- AMERGE is not indicated for the prevention of migraine attacks.
- •
- Safety and effectiveness of AMERGE have not been established for cluster headache.
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The recommended dose of AMERGE is 1 mg or 2.5 mg.
If the migraine returns or if the patient has only partial response, the dose may be repeated once after 4 hours, for a maximum dose of 5 mg in a 24-hour period.
The safety of treating an average of more than 4 migraine attacks in a 30‑day period has not been established.
2.2 Dosage Adjustment in Patients with Renal Impairment
AMERGE is contraindicated in patients with severe renal impairment (creatinine clearance: <15 mL/min) because of decreased clearance of the drug [see Contraindications (4), Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
In patients with mild to moderate renal impairment, the maximum daily dose should not exceed 2.5 mg over a 24‑hour period and a 1-mg starting dose is recommended [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
2.3 Dosage Adjustment in Patients with Hepatic Impairment
AMERGE is contraindicated in patients with severe hepatic impairment (Child-Pugh Grade C) because of decreased clearance [see Contraindications (4), Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
In patients with mild or moderate hepatic impairment (Child-Pugh Grade A or B), the maximum daily dose should not exceed 2.5 mg over a 24-hour period and a 1-mg starting dose is recommended [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
1-mg white tablets, D-shaped, film-coated, and debossed with “GX CE3”
2.5-mg green tablets, D-shaped, film-coated, and debossed with “GX CE5”
4 CONTRAINDICATIONS
AMERGE is contraindicated in patients with:
- •
- Ischemic coronary artery disease (CAD) (angina pectoris, history of myocardial infarction, or documented silent ischemia) or coronary artery vasospasm, including Prinzmetal’s angina [see Warnings and Precautions (5.1)]
- •
- Wolff-Parkinson-White syndrome or arrhythmias associated with other cardiac accessory conduction pathway disorders [see Warnings and Precautions (5.2)]
- •
- History of stroke or transient ischemic attack (TIA) or history of hemiplegic or basilar migraine because such patients are at a higher risk of stroke [see Warnings and Precautions (5.4)]
- •
- Peripheral vascular disease [see Warnings and Precautions (5.5)]
- •
- Ischemic bowel disease [see Warnings and Precautions (5.5)]
- •
- Uncontrolled hypertension [see Warnings and Precautions (5.8)]
- •
- Recent use (i.e., within 24 hours) of another 5-HT1 agonist, ergotamine-containing medication, ergot-type medication (such as dihydroergotamine or methysergide) [see Drug Interactions (7.1, 7.2)]
- •
- Hypersensitivity to AMERGE (angioedema and anaphylaxis seen) [see Warnings and Precautions (5.9)]
- •
- Severe renal or hepatic impairment [see Use in Specific Populations (8.6, 8.7), Clinical Pharmacology (12.3)]
5 WARNINGS AND PRECAUTIONS
5.1 Myocardial Ischemia, Myocardial Infarction, and Prinzmetal’s Angina
AMERGE is contraindicated in patients with ischemic or vasospastic CAD. There have been rare reports of serious cardiac adverse reactions, including acute myocardial infarction, occurring within a few hours following administration of AMERGE. Some of these reactions occurred in patients without known CAD. AMERGE may cause coronary artery vasospasm (Prinzmetal’s angina) even in patients without a history of CAD.
Perform a cardiovascular evaluation in triptan-naive patients who have multiple cardiovascular risk factors (e.g., increased age, diabetes, hypertension, smoking, obesity, strong family history of CAD) prior to receiving AMERGE. If there is evidence of CAD or coronary artery vasospasm, AMERGE is contraindicated. For patients with multiple cardiovascular risk factors who have a negative cardiovascular evaluation, consider administering the first dose of AMERGE in a medically supervised setting and performing an electrocardiogram (ECG) immediately following administration of AMERGE. For such patients, consider periodic cardiovascular evaluation in intermittent long-term users of AMERGE.
5.2 Arrhythmias
Life-threatening disturbances of cardiac rhythm, including ventricular tachycardia and ventricular fibrillation leading to death, have been reported within a few hours following the administration of 5-HT1 agonists. Discontinue AMERGE if these disturbances occur. AMERGE is contraindicated in patients with Wolff-Parkinson-White syndrome or arrhythmias associated with other cardiac accessory conduction pathway disorders.
5.3 Chest, Throat, Neck, and/or Jaw Pain/Tightness/Pressure
Sensations of tightness, pain, and pressure in the chest, throat, neck, and jaw commonly occur after treatment with AMERGE and are usually non-cardiac in origin. However, perform a cardiac evaluation if these patients are at high cardiac risk. 5-HT1 agonists, including AMERGE, are contraindicated in patients with CAD and those with Prinzmetal’s variant angina.
5.4 Cerebrovascular Events
Cerebral hemorrhage, subarachnoid hemorrhage, and stroke have occurred in patients treated with 5-HT1 agonists, and some have resulted in fatalities. In a number of cases, it appears possible that the cerebrovascular events were primary, the 5-HT1 agonist having been administered in the incorrect belief that the symptoms experienced were a consequence of migraine when they were not. Also, patients with migraine may be at increased risk of certain cerebrovascular events (e.g., stroke, hemorrhage, TIA). Discontinue AMERGE if a cerebrovascular event occurs.
Before treating headaches in patients not previously diagnosed as migraineurs, and in migraineurs who present with symptoms atypical for migraine, exclude other potentially serious neurological conditions. AMERGE is contraindicated in patients with a history of stroke or TIA.
5.5 Other Vasospasm Reactions
AMERGE may cause non-coronary vasospastic reactions, such as peripheral vascular ischemia, gastrointestinal vascular ischemia and infarction (presenting with abdominal pain and bloody diarrhea), splenic infarction, and Raynaud’s syndrome. In patients who experience symptoms or signs suggestive of non-coronary vasospasm reaction following the use of any 5-HT1 agonist, rule out a vasospastic reaction before receiving additional doses of AMERGE.
Reports of transient and permanent blindness and significant partial vision loss have been reported with the use of 5-HT1 agonists. Since visual disorders may be part of a migraine attack, a causal relationship between these events and the use of 5-HT1 agonists have not been clearly established.
5.6 Medication Overuse Headache
Overuse of acute migraine drugs (e.g., ergotamine, triptans, opioids, or combination of these drugs for 10 or more days per month) may lead to exacerbation of headache (medication overuse headache). Medication overuse headache may present as migraine-like daily headaches or as a marked increase in frequency of migraine attacks. Detoxification of patients, including withdrawal of the overused drugs, and treatment of withdrawal symptoms (which often includes a transient worsening of headache) may be necessary.
5.7 Serotonin Syndrome
Serotonin syndrome may occur with AMERGE, particularly during coadministration with selective serotonin reuptake inhibitors (SSRIs), serotonin norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants (TCAs), and monoamine oxidase (MAO) inhibitors [see Drug Interactions (7.3)]. Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g., hyperreflexia, incoordination), and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). The onset of symptoms usually occurs within minutes to hours of receiving a new or a greater dose of a serotonergic medication. Discontinue AMERGE if serotonin syndrome is suspected.
5.8 Increase in Blood Pressure
Significant elevation in blood pressure, including hypertensive crisis with acute impairment of organ systems, has been reported on rare occasions in patients treated with 5-HT1 agonists, including patients without a history of hypertension. Monitor blood pressure in patients treated with AMERGE. AMERGE is contraindicated in patients with uncontrolled hypertension.
5.9 Anaphylactic Reactions
There have been reports of anaphylaxis and hypersensitivity reactions, including angioedema, in patients receiving AMERGE. Such reactions can be life threatening or fatal. In general, anaphylactic reactions to drugs are more likely to occur in individuals with a history of sensitivity to multiple allergens. AMERGE is contraindicated in patients with a history of hypersensitivity reaction to AMERGE.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in more detail in other sections of the prescribing information:
- •
- Myocardial ischemia, myocardial infarction, and Prinzmetal’s angina [see Warnings and Precautions (5.1)]
- •
- Arrhythmias [see Warnings and Precautions (5.2)]
- •
- Chest, throat, neck, and/or jaw pain/tightness/pressure [see Warnings and Precautions (5.3)]
- •
- Cerebrovascular events [see Warnings and Precautions (5.4)]
- •
- Other vasospasm reactions [see Warnings and Precautions (5.5)]
- •
- Medication overuse headache [see Warnings and Precautions (5.6)]
- •
- Serotonin syndrome [see Warnings and Precautions (5.7)]
- •
- Increase in blood pressure [see Warnings and Precautions (5.8)]
- •
- Hypersensitivity reactions [see Contraindications (4), Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In a long-term open-label trial where patients were allowed to treat multiple migraine attacks for up to 1 year, 15 patients (3.6%) discontinued treatment due to adverse reactions.
In controlled clinical trials, the most common adverse reactions were paresthesias, dizziness, drowsiness, malaise/fatigue, and throat/neck symptoms, which occurred at a rate of 2% and at least 2 times placebo rate.
Table 1 lists the adverse reactions that occurred in 5 placebo-controlled clinical trials of approximately 1,752 exposures to placebo and AMERGE in adult patients with migraine. Only reactions that occurred at a frequency of 2% or more in groups treated with AMERGE 2.5 mg and that occurred at a frequency greater than the placebo group in the 5 pooled trials are included in Table 1.
Table 1. Adverse Reactions Reported by at Least 2% of Patients Treated with AMERGE and at a Frequency Greater than Placebo
|
Adverse Reaction |
Percent of Patients Reporting |
||
|
AMERGE 1 mg (n = 627) |
AMERGE 2.5 mg (n = 627) |
Placebo (n = 498) |
|
|
Atypical sensation |
2 |
4 |
1 |
|
1 |
2 |
<1 |
|
Gastrointestinal |
6 |
7 |
5 |
|
4 |
5 |
4 |
|
Neurological |
4 |
7 |
3 |
|
1 |
2 |
1 |
|
1 |
2 |
<1 |
|
2 |
2 |
1 |
|
Pain and pressure sensation |
2 |
4 |
2 |
|
1 |
2 |
1 |
The incidence of adverse reactions in controlled clinical trials was not affected by age or weight of the patients, duration of headache prior to treatment, presence of aura, use of prophylactic medications, or tobacco use. There were insufficient data to assess the impact of race on the incidence of adverse reactions.
7 DRUG INTERACTIONS
7.1 Ergot-Containing Drugs
Ergot-containing drugs have been reported to cause prolonged vasospastic reactions. Because these effects may be additive, use of ergotamine-containing or ergot-type medications (like dihydroergotamine or methysergide) and AMERGE within 24 hours of each other is contraindicated.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with use of AMERGE in pregnant women. Data from a prospective pregnancy exposure registry and epidemiological studies of pregnant women have documented outcomes in women exposed to naratriptan during pregnancy; however, due to small sample sizes, no definitive conclusions can be drawn regarding the risk of birth defects following exposure to naratriptan [see Data]. In animal studies, naratriptan produced developmental toxicity (including embryolethality and fetal abnormalities) when administered to pregnant rats and rabbits. The lowest doses producing evidence of developmental toxicity in animals were associated with plasma exposures 2.5 (rabbit) to 11 (rat) times that in humans at the maximum recommended daily dose (MRDD) [see Data].
In the U.S. general population, the estimated background risk of major birth defects and of miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The reported rate of major birth defects among deliveries to women with migraine ranged from 2.2% to 2.9% and of miscarriage was 17%, which were similar to rates reported in women without migraine.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Several studies have suggested that women with migraine may be at increased risk of preeclampsia during pregnancy.
Data
Human Data: The numbers of exposed pregnancy outcomes accumulated during the Sumatriptan/Naratriptan/Treximet (sumatriptan and naproxen sodium) Pregnancy Registry, a population-based international prospective study that collected data from October 1997 to September 2012, and smaller observational studies, were insufficient to define a level of risk for naratriptan in pregnant women. The Registry documented outcomes of 57 infants and fetuses exposed to naratriptan during pregnancy (52 exposed during the first trimester and 5 exposed during the second trimester). The occurrence of major birth defects (excluding fetal deaths and induced abortions without reported defects and all spontaneous pregnancy losses) during first-trimester exposure to naratriptan was 2.2% (1/46 [95% CI: 0.1% to 13.0%]) and during any trimester of exposure was 2.0% (1/51 [95% CI: 0.1% to 11.8%]). Seven infants were exposed to both naratriptan and sumatriptan in utero, and one of these infants with first-trimester exposure was born with a major birth defect (ventricular septal defect). The sample size in this study had 80% power to detect at least a 3.8- to 4.6-fold increase in the rate of major malformations.
In a study using data from the Swedish Medical Birth Register, women who used triptans or ergots during pregnancy were compared with women who did not. Of the 22 births with first-trimester exposure to naratriptan, one infant was born with a malformation (congenital deformity of the hand).
Animal Data: When naratriptan was administered to pregnant rats during the period of organogenesis at doses of 10, 60, or 340 mg/kg/day, there was a dose-related increase in embryonic death; incidences of fetal structural variations (incomplete/irregular ossification of skull bones, sternebrae, ribs) were increased at all doses. The maternal plasma exposures (AUC) at these doses were approximately 11, 70, and 470 times the exposure in humans at the MRDD. The high dose was maternally toxic, as evidenced by decreased maternal body weight gain during gestation. A no-effect dose for developmental toxicity in rats exposed during organogenesis was not established.
When naratriptan was administered orally (1, 5, or 30 mg/kg/day) to pregnant Dutch rabbits throughout organogenesis, the incidence of a specific fetal skeletal malformation (fused sternebrae) was increased at the high dose, the incidence of fetal variations (major blood vessel variations, supernumerary ribs, incomplete skeletal ossification) was increased at the mid and high doses, and embryonic death was increased at all doses (4, 20, and 120 times, respectively, the MRDD on a body surface area basis). Maternal toxicity (decreased body weight gain) was evident at the high dose. In a similar study in New Zealand White rabbits (1, 5, or 30 mg/kg/day throughout organogenesis), decreased fetal weights and increased incidences of fetal skeletal variations were observed at all doses (maternal exposures equivalent to 2.5, 19, and 140 times exposure in humans receiving the MRDD), while maternal body weight gain was reduced at 5 mg/kg or greater. A no-effect dose for developmental toxicity in rabbits exposed during organogenesis was not established.
When female rats were treated orally with naratriptan (10, 60, or 340 mg/kg/day) during late gestation and lactation, offspring behavioral impairment (tremors) and decreased offspring viability and growth were observed at doses of 60 mg/kg or greater, while maternal toxicity occurred only at the highest dose. Maternal exposures at the no-effect dose for developmental effects in this study were approximately 11 times the exposure in humans receiving the MRDD.
8.2 Lactation
Risk Summary
There are no data on the presence of naratriptan in human milk, the effects of naratriptan on the breastfed infant, or the effects of naratriptan on milk production. Naratriptan is present in rat milk.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for AMERGE and any potential adverse effects on the breastfed infant from naratriptan or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established. Therefore, AMERGE is not recommended for use in patients younger than 18 years of age.
One controlled clinical trial evaluated AMERGE (0.25 to 2.5 mg) in 300 adolescent migraineurs aged 12 to 17 years who received at least 1 dose of AMERGE for an acute migraine. In this study, 54% of the patients were female and 89% were Caucasian. There were no statistically significant differences between any of the treatment groups. The headache response rates at 4 hours (n) were 65% (n = 74), 67% (n = 78), and 64% (n = 70) for placebo, 1-mg, and 2.5-mg groups, respectively. This trial did not establish the efficacy of AMERGE compared with placebo in the treatment of migraine in adolescents. Adverse reactions observed in this clinical trial were similar in nature to those reported in clinical trials in adults.
8.5 Geriatric Use
Clinical trials of AMERGE did not include sufficient numbers of patients aged 65 and older to determine whether they respond differently from younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy.
Naratriptan is known to be substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in elderly patients who have reduced renal function. In addition, elderly patients are more likely to have decreased hepatic function, they are at higher risk for CAD, and blood pressure increases may be more pronounced in the elderly.
A cardiovascular evaluation is recommended for geriatric patients who have other cardiovascular risk factors (e.g., diabetes, hypertension, smoking, obesity, strong family history of CAD) prior to receiving AMERGE [see Warnings and Precautions (5.1)].
8.6 Renal Impairment
The use of AMERGE is contraindicated in patients with severe renal impairment (creatinine clearance: <15 mL/min) because of decreased clearance of the drug. In patients with mild to moderate renal impairment, the recommended starting dose is 1 mg, and the maximum daily dose should not exceed 2.5 mg over a 24-hour period [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
The use of AMERGE is contraindicated in patients with severe hepatic impairment (Child-Pugh Grade C) because of decreased clearance. In patients with mild or moderate hepatic impairment (Child-Pugh Grade A or B), the recommended starting dose is 1 mg, and the maximum daily dose should not exceed 2.5 mg over a 24-hour period [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)].
10 OVERDOSAGE
Adverse reactions observed after overdoses of up to 25 mg included increases in blood pressure resulting in lightheadedness, neck tension, tiredness, and loss of coordination. Also, ischemic ECG changes likely due to coronary artery vasospasm have been reported.
The elimination half-life of naratriptan is about 6 hours [see Clinical Pharmacology (12.3)]; therefore monitoring of patients after overdose with AMERGE should continue for at least 24 hours or while symptoms or signs persist. There is no specific antidote to naratriptan. It is unknown what effect hemodialysis or peritoneal dialysis has on the serum concentrations of naratriptan.
11 DESCRIPTION
AMERGE contains naratriptan hydrochloride, a selective 5-HT1B/1D receptor agonist. Naratriptan hydrochloride is chemically designated as N-methyl-3-(1-methyl-4-piperidinyl)-1H-indole-5-ethanesulfonamide monohydrochloride, and it has the following structure:
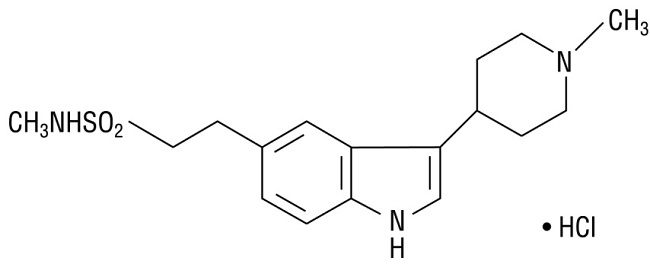
The empirical formula is C17H25N3O2S•HCl, representing a molecular weight of 371.93. Naratriptan hydrochloride is a white to pale yellow powder that is readily soluble in water.
Each AMERGE tablet for oral administration contains 1.11 or 2.78 mg of naratriptan hydrochloride, equivalent to 1 or 2.5 mg of naratriptan, respectively. Each tablet also contains the inactive ingredients croscarmellose sodium; hypromellose; lactose; magnesium stearate; microcrystalline cellulose; triacetin; and titanium dioxide, iron oxide yellow (2.5-mg tablet only), and indigo carmine aluminum lake (FD&C Blue No. 2) (2.5-mg tablet only) for coloring.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Naratriptan binds with high affinity to human cloned 5-HT1B/1D receptors. Migraines are likely due to local cranial vasodilatation and/or to the release of sensory neuropeptides (including substance P and calcitonin gene-related peptide) through nerve endings in the trigeminal system. The therapeutic activity of AMERGE for the treatment of migraine headache is thought to be due to the agonist effects at the 5-HT1B/1D receptors on intracranial blood vessels (including the arterio-venous anastomoses) and sensory nerves of the trigeminal system, which result in cranial vessel constriction and inhibition of pro-inflammatory neuropeptide release.
12.2 Pharmacodynamics
In the anesthetized dog, naratriptan has been shown to reduce the carotid arterial blood flow with little or no effect on arterial blood pressure or total peripheral resistance. While the effect on blood flow was selective for the carotid arterial bed, increases in vascular resistance of up to 30% were seen in the coronary arterial bed. Naratriptan has also been shown to inhibit trigeminal nerve activity in rat and cat.
In 10 subjects with suspected CAD undergoing coronary artery catheterization, there was a 1% to 10% reduction in coronary artery diameter following subcutaneous injection of 1.5 mg of naratriptan [see Contraindications (4)].
12.3 Pharmacokinetics
Absorption
Naratriptan is well absorbed, with about 70% oral bioavailability. Following administration of a 2.5-mg tablet, the peak concentrations are obtained in 2 to 3 hours. After administration of 1- or 2.5-mg tablets, the Cmax is somewhat (about 50%) higher in women (not corrected for milligram-per-kilogram dose) than in men. During a migraine attack, absorption is slower, with a Tmax of 3 to 4 hours. Food does not affect the pharmacokinetics of naratriptan. Naratriptan displays linear kinetics over the therapeutic dose range.
Distribution
The steady-state volume of distribution of naratriptan is 170 L. Plasma protein binding is 28% to 31% over the concentration range of 50 to 1,000 ng/mL.
Metabolism
In vitro, naratriptan is metabolized by a wide range of cytochrome P450 isoenzymes into a number of inactive metabolites.
Elimination
Naratriptan is predominantly eliminated in urine, with 50% of the dose recovered unchanged and 30% as metabolites in urine. The mean elimination half-life of naratriptan is 6 hours. The systemic clearance of naratriptan is 6.6 mL/min/kg. The renal clearance (220 mL/min) exceeds glomerular filtration rate, indicating active tubular secretion. Repeat administration of naratriptan tablets does not result in drug accumulation.
Special Populations
Age: A small decrease in clearance (approximately 26%) was observed in healthy elderly subjects (65 to 77 years) compared with younger subjects, resulting in slightly higher exposure [see Use in Specific Populations (8.5)].
Race: The effect of race on the pharmacokinetics of naratriptan has not been examined.
Renal Impairment: Clearance of naratriptan was reduced by 50% in subjects with moderate renal impairment (creatinine clearance: 18 to 39 mL/min) compared with the normal group. Decrease in clearances resulted in an increase of mean half-life from 6 hours (healthy) to 11 hours (range: 7 to 20 hours). The mean Cmax increased by approximately 40%. The effects of severe renal impairment (creatinine clearance: ≤15 mL/min) on the pharmacokinetics of naratriptan have not been assessed [see Contraindications (4)].
Hepatic Impairment: Clearance of naratriptan was decreased by 30% in subjects with moderate hepatic impairment (Child-Pugh Grade A or B). This resulted in an approximately 40% increase in the half-life (range: 8 to 16 hours). The effects of severe hepatic impairment (Child-Pugh Grade C) on the pharmacokinetics of naratriptan have not been assessed [see Contraindications (4)].
Drug Interaction Studies
From population pharmacokinetic analyses, coadministration of naratriptan and fluoxetine, beta-blockers, or tricyclic antidepressants did not affect the clearance of naratriptan.
Oral Contraceptives: Oral contraceptives reduced clearance by 32% and volume of distribution by 22%, resulting in slightly higher concentrations of naratriptan. Hormone replacement therapy had no effect on pharmacokinetics in older female patients.
Monoamine Oxidase and P450 Inhibitors: Naratriptan does not inhibit monoamine oxidase (MAO) enzymes and is a poor inhibitor of P450; metabolic interactions between naratriptan and drugs metabolized by P450 or MAO are therefore unlikely.
Smoking: Smoking increased the clearance of naratriptan by 30%.
Alcohol: In normal volunteers, co-administration of single doses of naratriptan tablets and alcohol did not result in substantial modification of naratriptan pharmacokinetic parameters.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In carcinogenicity studies, mice and rats were given naratriptan by oral gavage for 104 weeks. There was no evidence of an increase in tumors related to naratriptan administration in mice receiving up to 200 mg/kg/day. That dose was associated with a plasma (AUC) exposure that was 110 times the exposure in humans receiving the MRDD of 5 mg. Two rat studies were conducted, one using a standard diet and the other a nitrite-supplemented diet (naratriptan can be nitrosated in vitro to form a mutagenic product that has been detected in the stomachs of rats fed a high-nitrite diet). Doses of 5, 20, and 90 mg/kg were associated with AUC exposures that in the standard-diet study were 7, 40, and 236 times, respectively, and in the nitrite-supplemented–diet study were 7, 29, and 180 times, respectively, the exposure in humans at the MRDD. In both studies, there was an increase in the incidence of thyroid follicular hyperplasia in high-dose males and females and in thyroid follicular adenomas in high-dose males. In the standard-diet study only, there was also an increase in the incidence of benign c-cell adenomas in the thyroid of high-dose males and females. The exposures achieved at the no-effect dose for thyroid tumors were 40 (standard diet) and 29 (nitrite-supplemented diet) times the exposure achieved in humans at the MRDD. In the nitrite-supplemented–diet study only, the incidence of benign lymphocytic thymoma was increased in all treated groups of females. It was not determined if the nitrosated product is systemically absorbed. However, no changes were seen in the stomachs of rats in that study.
Mutagenesis
Naratriptan was not mutagenic when tested in in vitro gene mutation (Ames and mouse lymphoma tk) assays. Naratriptan was also negative in the in vitro human lymphocyte assay and the in vivo mouse micronucleus assay. Naratriptan can be nitrosated in vitro to form a mutagenic product (WHO nitrosation assay) that has been detected in the stomachs of rats fed a nitrite-supplemented diet.
Impairment of Fertility
In a reproductive toxicity study in which male and female rats were administered naratriptan orally prior to and throughout the mating period (10, 60, 170, or 340 mg/kg/day; plasma exposures [AUC] approximately 11, 70, 230, and 470 times, respectively, the human exposure at the MRDD), there was a drug-related decrease in the number of females exhibiting normal estrous cycles at doses of 170 mg/kg/day or greater and an increase in pre-implantation loss at 60 mg/kg/day or greater. In high-dose males, testicular/epididymal atrophy accompanied by spermatozoa depletion reduced mating success and may have contributed to the observed pre-implantation loss. The exposures achieved at the no-effect doses for pre-implantation loss, anestrus, and testicular effects were approximately 11, 70, and 230 times, respectively, the exposures in humans at the MRDD.
In a study in which rats were dosed orally with naratriptan (10, 60, or 340 mg/kg/day) for 6 months, changes in the female reproductive tract including atrophic or cystic ovaries and anestrus were seen at the high dose. The exposure at the no-effect dose of 60 mg/kg was approximately 85 times that in humans at the MRDD.
14 CLINICAL STUDIES
The efficacy of AMERGE in the acute treatment of migraine headaches was evaluated in 3 randomized, double-blind, placebo-controlled trials in adult patients (Trials 1, 2, 3). These trials enrolled adult patients who were predominantly female (86%) and Caucasian (96%) with a mean age of 41 years (range: 18 to 65 years). In all studies, patients were instructed to treat at least 1 moderate to severe headache. Headache response, defined as a reduction in headache severity from moderate or severe pain to mild or no pain, was assessed up to 4 hours after dosing. Associated symptoms such as nausea, vomiting, photophobia, and phonophobia were also assessed. Maintenance of response was assessed for up to 24 hours postdose. A second dose of AMERGE or other rescue medication to treat migraines was allowed 4 to 24 hours after the initial treatment for recurrent headache.
In all 3 trials, the percentage of patients achieving headache response 4 hours after treatment, the primary outcome measure, was significantly greater among patients receiving AMERGE compared with those who received placebo. In all trials, response to 2.5 mg was numerically greater than response to 1 mg and in the largest of the 3 trials, there was a statistically significant greater percentage of patients with headache response at 4 hours in the 2.5-mg group compared with the 1-mg group. The results are summarized in Table 2.
Table 2. Percentage of Adult Patients with Headache Response (Mild or No Headache) 4 Hours following Treatment
|
AMERGE 1 mg (n = 491) |
AMERGE 2.5 mg (n = 493) |
Placebo (n = 395) |
|
|
Trial 1 Trial 2 Trial 3 |
50%a 52%a 54%a |
60%a 66%ab 65%a |
34% 27% 32% |
- a P<0.05 compared with placebo.
- b P<0.05 compared with 1 mg.
The estimated probability of achieving an initial headache response in adults over the 4 hours following treatment in pooled Trials 1, 2, and 3 is depicted in Figure 1.
Figure 1. Estimated Probability of Achieving Initial Headache Response within 4 Hours in Pooled Trials 1, 2, and 3a
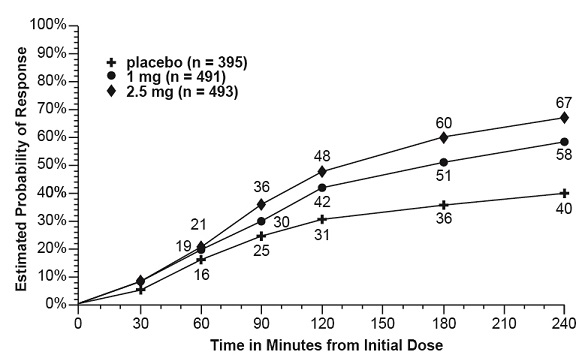
- a The figure shows the probability over time of obtaining headache response (reduction in headache severity from moderate or severe pain to no or mild pain) following treatment with AMERGE. In this Kaplan-Meier plot, patients not achieving response within 240 minutes were censored at 240 minutes.
For patients with migraine-associated nausea, photophobia, and phonophobia at baseline, there was a lower incidence of these symptoms 4 hours following administration of 1-mg and 2.5-mg AMERGE compared with placebo.
Four to 24 hours following the initial dose of study treatment, patients were allowed to use additional treatment for pain relief in the form of a second dose of study treatment or other rescue medication. The estimated probability of patients taking a second dose or other rescue medication to treat migraine over the 24 hours following the initial dose of study treatment is summarized in Figure 2.
Figure 2. Estimated Probability of Patients Taking a Second Dose of AMERGE Tablets or Other Medication to Treat Migraine over the 24 Hours following the Initial Dose of Study Treatment in Pooled Trials 1, 2, and 3a
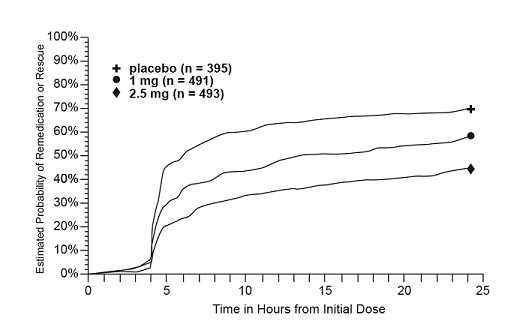
- a Kaplan-Meier plot based on data obtained in the 3 controlled clinical trials (Trials 1, 2, and 3) providing evidence of efficacy with patients not using additional treatments censored at 24 hours. The plot also includes patients who had no response to the initial dose. Remedication was discouraged prior to 4 hours postdose.
There is no evidence that doses of 5 mg provided a greater effect than 2.5 mg. There was no evidence to suggest that treatment with AMERGE was associated with an increase in the severity or frequency of migraine attacks. The efficacy of AMERGE was unaffected by presence of aura; gender, age, or weight of the subject; oral contraceptive use; or concomitant use of common migraine prophylactic drugs (e.g., beta-blockers, calcium channel blockers, tricyclic antidepressants). There was insufficient data to assess the impact of race on efficacy.
16 HOW SUPPLIED/STORAGE AND HANDLING
AMERGE tablets containing 1 mg and 2.5 mg of naratriptan (base) as the hydrochloride salt.
AMERGE tablets, 1 mg, are white, D-shaped, film-coated tablets debossed with “GX CE3” on one side in blister packs of 9 tablets (NDC 0173-0561-00).
AMERGE tablets, 2.5 mg, are green, D-shaped, film-coated tablets debossed with “GX CE5” on one side in blister packs of 9 tablets (NDC 0173-0562-00).
Store at controlled room temperature, 20° to 25°C (68° to 77°F) [see USP].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Risk of Myocardial Ischemia and/or Infarction, Prinzmetal’s Angina, Other Vasospasm-Related Events, Arrhythmias, and Cerebrovascular Events
Inform patients that AMERGE may cause serious cardiovascular side effects such as myocardial infarction or stroke. Although serious cardiovascular events can occur without warning symptoms, patients should be alert for the signs and symptoms of chest pain, shortness of breath, irregular heartbeat, significant rise in blood pressure, weakness, and slurring of speech and should ask for medical advice if any indicative sign or symptoms are observed. Apprise patients of the importance of this follow-up [see Warnings and Precautions (5.1, 5.2, 5.4, 5.5, 5.8)].
Anaphylactic Reactions
Inform patients that anaphylactic reactions have occurred in patients receiving AMERGE. Such reactions can be life-threatening or fatal. In general, anaphylactic reactions to drugs are more likely to occur in individuals with a history of sensitivity to multiple allergens [see Contraindications (4), Warnings and Precautions (5.9)].
Concomitant Use with Other Triptans or Ergot Medications
Inform patients that use of AMERGE within 24 hours of another triptan or an ergot-type medication (including dihydroergotamine or methysergide) is contraindicated [see Contraindications (4), Drug Interactions (7.1, 7.2)].
Serotonin Syndrome
Caution patients about the risk of serotonin syndrome with the use of AMERGE or other triptans, particularly during combined use with SSRIs, SNRIs, TCAs, and MAO inhibitors [see Warnings and Precautions (5.7), Drug Interactions (7.3)].
Medication Overuse Headache
Inform patients that use of acute migraine drugs for 10 or more days per month may lead to an exacerbation of headache and encourage patients to record headache frequency and drug use (e.g., by keeping a headache diary) [see Warnings and Precautions (5.6)].
Pregnancy
Advise patients to notify their healthcare provider if they become pregnant during treatment or intend to become pregnant [see Use in Specific Populations (8.1)].
Lactation
Advise patients to notify their healthcare provider if they are breastfeeding or plan to breastfeed [see Use in Specific Populations (8.2)].
Ability to Perform Complex Tasks
Treatment with AMERGE may cause somnolence and dizziness; instruct patients to evaluate their ability to perform complex tasks after administration of AMERGE.
AMERGE is a trademark owned by or licensed to the GSK group of companies. The other brand listed is a trademark owned by or licensed to its owner and is not owned by or licensed to the GSK group of companies. The maker of this brand is not affiliated with and does not endorse the GSK group of companies or its products.
Distributed by:
GlaxoSmithKline
Research Triangle Park, NC 27709
©2020 GSK group of companies or its licensor.
AMG:7PI
Patient Information
AMERGE (a-MERJ)
(naratriptan hydrochloride)
tablets
Read this Patient Information before you start taking AMERGE and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or treatment.
What is the most important information I should know about AMERGE?
AMERGE can cause serious side effects, including:
Heart attack and other heart problems. Heart problems may lead to death.
Stop taking AMERGE and get emergency medical help right away if you have any of the following symptoms of a heart attack:
- •
- discomfort in the center of your chest that lasts for more than a few minutes, or that goes away and comes back
- •
- severe tightness, pain, pressure, or heaviness in your chest, throat, neck, or jaw
- •
- pain or discomfort in your arms, back, neck, jaw, or stomach
- •
- shortness of breath with or without chest discomfort
- •
- breaking out in a cold sweat
- •
- nausea or vomiting
- •
- feeling lightheaded
AMERGE is not for people with risk factors for heart disease unless a heart exam is done and shows no problem. You have a higher risk for heart disease if you:
- •
- have high blood pressure
- •
- have high cholesterol levels
- •
- smoke
- •
- are overweight
- •
- have diabetes
- •
- have a family history of heart disease
What is AMERGE?
AMERGE is a prescription medicine used to treat acute migraine headaches with or without aura in adults who have been diagnosed with migraine headaches.
AMERGE is not used to prevent or decrease the number of migraine headaches you have.
AMERGE is not used to treat other types of headaches such as hemiplegic migraines (that make you unable to move on one side of your body) or basilar migraines (rare form of migraine with aura).
It is not known if AMERGE is safe and effective to treat cluster headaches.
It is not known if AMERGE is safe and effective in children younger than 18 years of age.
Who should not take AMERGE?
Do not take AMERGE if you have:
- •
- heart problems or a history of heart problems
- •
- narrowing of blood vessels to your legs, arms, stomach, or kidney (peripheral vascular disease)
- •
- uncontrolled high blood pressure
- •
- severe kidney problems
- •
- severe liver problems
- •
- hemiplegic migraines or basilar migraines. If you are not sure if you have these types of migraines, ask your healthcare provider.
- •
- had a stroke, transient ischemic attacks (TIAs), or problems with your blood circulation
- •
- taken any of the following medicines in the last 24 hours:
- •
- almotriptan (AXERT)
- •
- eletriptan (RELPAX)
- •
- frovatriptan (FROVA)
- •
- rizatriptan (MAXALT, MAXALT-MLT)
- •
- sumatriptan (IMITREX, SUMAVEL DosePro, ALSUMA)
- •
- sumatriptan and naproxen (TREXIMET)
- •
- ergotamines (CAFERGOT, ERGOMAR, MIGERGOT)
- •
- dihydroergotamine (D.H.E. 45, MIGRANAL)
- Ask your healthcare provider if you are not sure if your medicine is listed above.
- •
- an allergy to naratriptan or any of the ingredients in AMERGE. See the end of this leaflet for a complete list of ingredients in AMERGE.
What should I tell my healthcare provider before taking AMERGE?
Before you take AMERGE, tell your healthcare provider about all of your medical conditions, including if you:
- •
- have high blood pressure
- •
- have high cholesterol
- •
- have diabetes
- •
- smoke
- •
- are overweight
- •
- have heart problems or family history of heart problems or stroke
- •
- have kidney problems
- •
- have liver problems
- •
- are not using effective birth control
- •
- are pregnant or plan to become pregnant
- •
- are breastfeeding or plan to breastfeed. It is not known if AMERGE passes into your breast milk. Talk with your healthcare provider about the best way to feed your baby if you take AMERGE.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Using AMERGE with certain other medicines can affect each other, causing serious side effects.
Especially tell your healthcare provider if you take antidepressant medicines called:
- •
- selective serotonin reuptake inhibitors (SSRIs)
- •
- serotonin norepinephrine reuptake inhibitors (SNRIs)
- •
- tricyclic antidepressants (TCAs)
- •
- monoamine oxidase inhibitors (MAOIs)
Ask your healthcare provider or pharmacist for a list of these medicines if you are not sure.
Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine.
How should I take AMERGE?
- •
- Certain people should take their first dose of AMERGE in their healthcare provider’s office or in another medical setting. Ask your healthcare provider if you should take your first dose in a medical setting.
- •
- Take AMERGE exactly as your healthcare provider tells you to take it.
- •
- Your healthcare provider may change your dose. Do not change your dose without first talking with your healthcare provider.
- •
- Take AMERGE with water or other liquids.
- •
- If you do not get any relief after your first AMERGE tablet, do not take a second tablet without first talking with your healthcare provider.
- •
- If your headache comes back or you only get some relief from your headache, you can take a second tablet 4 hours after the first tablet.
- •
- Do not take more than a total of 5 mg of AMERGE in a 24-hour period.
- •
- Some people who take too many AMERGE tablets may have worse headaches (medication overuse headache). If your headaches get worse, your healthcare provider may decide to stop your treatment with AMERGE.
- •
- If you take too much AMERGE, call your healthcare provider or go to the nearest hospital emergency room right away.
- •
- You should write down when you have headaches and when you take AMERGE so you can talk with your healthcare provider about how AMERGE is working for you.
What should I avoid while taking AMERGE?
AMERGE can cause dizziness, weakness, or drowsiness. If you have these symptoms, do not drive a car, use machinery, or do anything where you need to be alert.
What are the possible side effects of AMERGE?
AMERGE may cause serious side effects. See “What is the most important information I should know about AMERGE?”
These serious side effects include:
- •
- changes in color or sensation in your fingers and toes (Raynaud’s syndrome)
- •
- stomach and intestinal problems (gastrointestinal and colonic ischemic events). Symptoms of gastrointestinal and colonic ischemic events include:
- •
- sudden or severe stomach pain
- •
- stomach pain after meals
- •
- weight loss
- •
- nausea or vomiting
- •
- constipation or diarrhea
- •
- bloody diarrhea
- •
- fever
- •
- problems with blood circulation to your legs and feet (peripheral vascular ischemia). Symptoms of peripheral vascular ischemia include:
- •
- cramping and pain in your legs or hips
- •
- feeling of heaviness or tightness in your leg muscles
- •
- burning or aching pain in your feet or toes while resting
- •
- numbness, tingling, or weakness in your legs
- •
- cold feeling or color changes in 1 or both legs or feet
- •
- medication overuse headaches. Some people who use too many AMERGE tablets may have worse headaches (medication overuse headache). If your headaches get worse, your healthcare provider may decide to stop your treatment with AMERGE.
- •
- serotonin syndrome. Serotonin syndrome is a rare but serious problem that can happen in people using AMERGE, especially if AMERGE is used with antidepressant medicines called SSRIs, SNRIs, TCAs, or MAOIs. Call your healthcare provider right away if you have any of the following symptoms of serotonin syndrome:
- •
- mental changes such as seeing things that are not there (hallucinations), agitation, or coma
- •
- fast heartbeat
- •
- changes in blood pressure
- •
- high body temperature
- •
- tight muscles
- •
- trouble walking
The most common side effects of AMERGE include:
- •
- tingling or numbness in your fingers or toes
- •
- dizziness
- •
- warm, hot, burning feeling to your face (flushing)
- •
- discomfort or stiffness in your neck
- •
- feeling weak, drowsy, or tired
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of AMERGE. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store AMERGE?
Store AMERGE between 68°F and 77°F (20°C and 25°C).
Keep AMERGE and all medicines out of the reach of children.
General information about the safe and effective use of AMERGE.
Medicines are sometimes prescribed for purposes other than those listed in Patient Information leaflets. Do not use AMERGE for a condition for which it was not prescribed. Do not give AMERGE to other people, even if they have the same symptoms you have. It may harm them.
This Patient Information leaflet summarizes the most important information about AMERGE. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about AMERGE that is written for healthcare professionals.
For more information, go to www.gsk.com or call 1-888-825-5249.
What are the ingredients in AMERGE?
Active ingredient: naratriptan hydrochloride
Inactive ingredients: croscarmellose sodium, hypromellose, lactose, magnesium stearate, microcrystalline cellulose, triacetin, titanium dioxide
2.5-mg tablets also contain iron oxide yellow and indigo carmine aluminum lake (FD&C Blue No. 2) for coloring.
This Patient Information has been approved by the U.S. Food and Drug Administration.
AMERGE and IMITREX are trademarks owned by or licensed to the GSK group of companies. The other brands listed are trademarks owned by or licensed to their respective owners and are not owned by or licensed to the GSK group of companies. The makers of these brands are not affiliated with and do not endorse the GSK group of companies or its products.
Distributed by:
GlaxoSmithKline
Research Triangle Park, NC 27709
©2020 GSK group of companies or its licensor.
October 2020
AMG:7PIL
PRINCIPAL DISPLAY PANEL
NDC 0173-0561-00
Amerge
(NARATRIPTAN HYDROCHLORIDE)
Tablets
1 mg naratriptan
9 Tablets
Rx only
Each tablet contains 1 mg of naratriptan as the hydrochloride.
Store at controlled room temperature, 20o to 25oC (68o to 77oF) (see USP).
Do not use if blisters are torn, broken, or missing.
1 mg
Made in Canada
©2020 GSK group of companies or its licensor.
- 62000000059585 Rev.12/20

PRINCIPAL DISPLAY PANEL
NDC 0173-0562-00
Amerge
(NARATRIPTAN HYDROCHLORIDE)
Tablets
2.5 mg naratriptan
9 Tablets
Rx only
Each tablet contains 2.5 mg of naratriptan as the hydrochloride.
Store at controlled room temperature, 20 o to 25 o C (68 o to 77 o F) (see USP).
Do not use if blisters are torn, broken, or missing.
2.5 mg
Made in Canada
©2020 GSK group of companies or its licensor.
- 62000000059587 Rev. 12/20

| AMERGE
naratriptan hydrochloride tablet, film coated |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| AMERGE
naratriptan hydrochloride tablet, film coated |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Labeler - GlaxoSmithKline LLC (167380711) |