POTIGA- ezogabine tablet, film coated
GlaxoSmithKline LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use POTIGA safely and effectively. See full prescribing information for POTIGA.
POTIGA (ezogabine) tablets, for oral use, CV Initial U.S. Approval: 2011 WARNING: RETINAL ABNORMALITIES AND POTENTIAL VISION LOSSSee full prescribing information for complete boxed warning.
RECENT MAJOR CHANGES
INDICATIONS AND USAGEPOTIGA is a potassium channel opener indicated as adjunctive treatment of partial-onset seizures in patients aged 18 years and older who have responded inadequately to several alternative treatments and for whom the benefits outweigh the risk of retinal abnormalities and potential decline in visual acuity. (1) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSTablets: 50 mg, 200 mg, 300 mg, and 400 mg. (3) CONTRAINDICATIONSNone. (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (incidence ≥4% and twice placebo) were dizziness, somnolence, fatigue, confusional state, vertigo, tremor, abnormal coordination, diplopia, disturbance in attention, memory impairment, asthenia, blurred vision, gait disturbance, aphasia, dysarthria, and balance disorder. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact GlaxoSmithKline at 1-888-825-5249 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONSEzogabine plasma levels may be reduced by concomitant administration of phenytoin or carbamazepine. An increase in dosage of POTIGA should be considered when adding phenytoin or carbamazepine. (7.1) USE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 5/2016 |
FULL PRESCRIBING INFORMATION
WARNING: RETINAL ABNORMALITIES AND POTENTIAL VISION LOSS
POTIGA can cause retinal abnormalities with funduscopic features similar to those seen in retinal pigment dystrophies, which are known to result in damage to the photoreceptors and vision loss. In addition, macular abnormalities characterized as vitelliform lesions have been observed. These lesions have been identified most consistently with optical coherence tomography imaging [see Warnings and Precautions (5.1), Adverse Reactions (6.2)].
Some patients with retinal abnormalities have been found to have abnormal visual acuity. It is not possible to determine whether POTIGA caused this decreased visual acuity, as baseline assessments are not available for these patients.
Approximately one third of the patients who had eye examinations performed after approximately 4 years of treatment were found to have retinal pigmentary abnormalities. An earlier onset cannot be ruled out, and it is possible that retinal abnormalities were present earlier in the course of exposure to POTIGA. The rate of progression of retinal abnormalities and their reversibility are unknown. Reversibility of retinal pigmentary abnormalities and partial resolution of vitelliform lesions has been reported after discontinuation of ezogabine in some patients.
POTIGA should only be used in patients who have responded inadequately to several alternative treatments and for whom the benefits outweigh the potential risk of vision loss. Patients who fail to show substantial clinical benefit after adequate titration should be discontinued from POTIGA.
All patients taking POTIGA should have baseline and periodic (every 6 months) systematic visual monitoring by an ophthalmic professional. Testing should include visual acuity, dilated fundus photography, and optical coherence tomography. Additional testing may include fluorescein angiograms, perimetry, and electroretinograms.
If retinal pigmentary abnormalities or vision changes are detected, POTIGA should be discontinued unless no other suitable treatment options are available and the benefits of treatment outweigh the potential risk of vision loss.
1 INDICATIONS AND USAGE
POTIGA® is indicated as adjunctive treatment of partial-onset seizures in patients aged 18 years and older who have responded inadequately to several alternative treatments and for whom the benefits outweigh the risk of retinal abnormalities and potential decline in visual acuity [see Warnings and Precautions (5.1)].
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The initial dosage should be 100 mg 3 times daily (300 mg per day). The dosage should be increased gradually at weekly intervals by no more than 50 mg 3 times daily (increase in the daily dose of no more than 150 mg per day) up to a maintenance dosage of 200 mg to 400 mg 3 times daily (600 mg to 1,200 mg per day), based on individual patient response and tolerability. This information is summarized in Table 1 under Dosing in Specific Populations. In the controlled clinical trials, 400 mg 3 times daily showed limited evidence of additional improvement in seizure reduction, but an increase in adverse events and discontinuations, compared with the 300 mg 3 times daily dosage. The safety and efficacy of dosages greater than 400 mg 3 times daily (1,200 mg per day) have not been examined in controlled trials.
POTIGA should be given orally in 3 equally divided doses daily, with or without food.
POTIGA tablets should be swallowed whole.
If POTIGA is discontinued, the dosage should be gradually reduced over a period of at least 3 weeks, unless safety concerns require abrupt withdrawal.
2.2. Dosing Considerations to Mitigate the Risk of Visual Adverse Reactions
Because POTIGA may cause retinal abnormalities with long-term use, patients who fail to show substantial clinical benefit after adequate titration should be discontinued from POTIGA. Testing of visual function should be done at baseline and every 6 months during therapy with POTIGA. Patients who cannot be monitored should usually not be treated with POTIGA. If retinal pigmentary abnormalities or vision changes are detected, POTIGA should be discontinued unless no other suitable treatment options are available and the benefits of treatment outweigh the potential risk of vision loss [see Warnings and Precautions (5.1)].
2.3 Dosing in Specific Populations
No adjustment in dosage is recommended in patients with mild renal or hepatic impairment (see Table 1). Dosage adjustment is recommended in geriatric and patients with moderate or severe renal or hepatic impairment (see Table 1).
Table 1. Dosing in Specific Populations
|
Specific Population |
Initial Dose |
Titration |
Maximum Dosage |
|
General Dosing |
|||
|
General population (including patients with mild renal or hepatic impairment) |
100 mg 3 times daily (300 mg per day) |
Increase by no more than 50 mg 3 times daily, at weekly intervals |
400 mg 3 times daily (1,200 mg per day) |
|
Dosing in Specific Populations |
|||
|
Geriatrics (patients ≥65 years) |
50 mg 3 times daily (150 mg per day) |
Increase by no more than 50 mg 3 times daily, at weekly intervals |
250 mg 3 times daily (750 mg per day) |
|
Hepatic impairment (patients with Child-Pugh 7-9) |
250 mg 3 times daily (750 mg per day) |
||
|
Hepatic impairment (patients with Child-Pugh >9) |
200 mg 3 times daily (600 mg per day) |
||
|
Renal impairment (patients with CrCL <50 mL per min or end-stage renal disease on hemodialysisa) |
200 mg 3 times daily (600 mg per day) |
||
aImmediately following hemodialysis a single supplemental dose is recommended. If breakthrough seizures occur toward the end of hemodialysis, an additional supplemental dose may be considered at the start of subsequent dialysis sessions [see Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
50 mg, purple, round, film-coated tablets debossed with “RTG 50” on one side.
200 mg, yellow, oblong, film-coated tablets debossed with “RTG-200” on one side.
300 mg, green, oblong, film-coated tablets debossed with “RTG-300” on one side.
400 mg, purple, oblong, film-coated tablets debossed with “RTG-400” on one side.
5 WARNINGS AND PRECAUTIONS
5.1 Retinal Abnormalities and Potential Vision Loss
POTIGA can cause abnormalities of the retina. The abnormalities seen in patients treated with POTIGA have funduscopic features similar to those seen in retinal pigment dystrophies that are known to result in damage to photoreceptors and vision loss. In addition, macular abnormalities characterized as vitelliform lesions have been observed; such lesions may also be seen in macular degenerative and dystrophic disorders. These lesions have been identified most consistently with optical coherence tomography imaging [see Adverse Reactions (6.2)].
The retinal abnormalities observed with POTIGA have been reported in patients who were originally enrolled in clinical trials with POTIGA and who have generally taken the drug for a long period of time in 2 ongoing extension trials. Approximately one third of the patients who had eye examinations performed after approximately 4 years of treatment were found to have retinal pigmentary abnormalities. However, an earlier onset cannot be ruled out, and it is possible that retinal abnormalities were present earlier in the course of exposure to POTIGA. POTIGA causes skin, scleral, nail, and mucous membrane discoloration and it is not clear whether this discoloration is related to retinal abnormalities [see Warnings and Precautions (5.3)]. Approximately 15% of patients with retinal pigmentary abnormalities had no such discoloration.
Funduscopic abnormalities have most commonly been described as perivascular pigmentation (bone spicule pattern) in the retinal periphery and/or as areas of focal retinal pigment epithelium clumping. Although some of the patients with retinal abnormalities have been found to have abnormal visual acuity, it is not possible to assess whether POTIGA caused their decreased visual acuity, as baseline assessments are not available for these patients. Two patients with retinal abnormalities have had more extensive diagnostic retinal evaluations. The results of these evaluations were consistent with a retinal dystrophy, including abnormalities in the electroretinogram and electrooculogram of both patients, with abnormal fluorescein angiography and diminished sensitivity on visual field testing in one patient.
The rate of progression of retinal abnormalities and the reversibility after drug discontinuation are unknown. Reversibility of retinal pigmentary abnormalities and partial resolution of vitelliform lesions has been reported after discontinuation of ezogabine in some patients.
Because of the observed ophthalmologic adverse reactions, POTIGA should only be used in patients who have responded inadequately to several alternative treatments and for whom the benefits outweigh the risk of retinal abnormalities and potential vision loss. Patients who fail to show substantial clinical benefit after adequate titration should be discontinued from POTIGA.
Patients should have baseline ophthalmologic testing by an ophthalmic professional and follow-up testing every 6 months. The best method of detection of these abnormalities and the optimal frequency of periodic ophthalmologic monitoring are unknown. Patients who cannot be monitored should usually not be treated with POTIGA. The ophthalmologic monitoring program should include visual acuity testing, dilated fundus photography, and optical coherence tomography. Additional testing may include fluorescein angiograms, perimetry, and electroretinograms. If retinal pigmentary abnormalities or vision changes are detected, POTIGA should be discontinued unless no other suitable treatment options are available and the benefits of treatment outweigh the potential risk of vision loss.
5.2 Urinary Retention
POTIGA caused urinary retention in clinical trials. Urinary retention was generally reported within the first 6 months of treatment, but was also observed later. Urinary retention was reported as an adverse event in 29 of 1,365 (approximately 2%) patients treated with POTIGA in the open-label and placebo-controlled epilepsy database [see Clinical Studies (14)]. Of these 29 patients, 5 (17%) required catheterization, with post-voiding residuals of up to 1,500 mL. POTIGA was discontinued in 3 of the 5 patients who required catheterization, and all were able to void spontaneously; however, 1 of the 3 patients continued intermittent self-catheterization. Two patients continued treatment with POTIGA and were able to void spontaneously after catheter removal. Hydronephrosis occurred in 2 patients, one of whom had associated renal function impairment that resolved upon discontinuation of POTIGA. Hydronephrosis was not reported in placebo patients.
In the placebo-controlled epilepsy trials, “urinary retention,” “urinary hesitation,” and “dysuria” were reported in 0.9%, 2.2%, and 2.3% of patients on POTIGA, respectively, and in 0.5%, 0.9%, and 0.7% of patients on placebo, respectively.
Because of the increased risk of urinary retention on POTIGA, urologic symptoms should be carefully monitored. Closer monitoring is recommended for patients who have other risk factors for urinary retention (e.g., benign prostatic hyperplasia [BPH]), patients who are unable to communicate clinical symptoms (e.g., cognitively impaired patients), or patients who use concomitant medications that may affect voiding (e.g., anticholinergics). In these patients, a comprehensive evaluation of urologic symptoms prior to and during treatment with POTIGA may be appropriate.
5.3 Skin Discoloration
POTIGA can cause skin discoloration. The skin discoloration is generally described as blue, but has also been described as grey-blue or brown. It is predominantly on or around the lips or in the nail beds of the fingers or toes, but more widespread involvement of the face and legs has also been reported. Discoloration of the palate, sclera, and conjunctiva has also been reported.
Approximately 10% of patients in long-term clinical trials developed skin discoloration, generally after 2 or more years of treatment and at higher doses (900 mg or greater) of POTIGA. Among patients in whom the status of both skin, nail, lip, or mucous membrane discoloration and retinal pigmentary abnormalities are reported, approximately a quarter of those with skin, nail, lip, or mucous membrane discoloration had concurrent retinal pigmentary abnormalities [see Warnings and Precautions (5.1)].
Information on the consequences, reversibility, time to onset, and pathophysiology of the skin abnormalities remains incomplete. The possibility of more extensive systemic involvement has not been excluded. If a patient develops skin discoloration, serious consideration should be given to changing to an alternate medication.
5.4 Neuropsychiatric Symptoms
Confusional state, psychotic symptoms, and hallucinations were reported more frequently as adverse reactions in patients treated with POTIGA than in those treated with placebo in placebo-controlled epilepsy trials (see Table 2). Discontinuations resulting from these reactions were more common in the drug-treated group (see Table 2). These effects were dose-related and generally appeared within the first 8 weeks of treatment. Half of the patients in the controlled trials who discontinued POTIGA due to hallucinations or psychosis required hospitalization. Approximately two-thirds of patients with psychosis in controlled trials had no prior psychiatric history. The psychiatric symptoms in the vast majority of patients in both controlled and open-label trials resolved within 7 days of discontinuation of POTIGA. Rapid titration at greater than the recommended doses appeared to increase the risk of psychosis and hallucinations.
Table 2. Major Neuropsychiatric Symptoms in Placebo-Controlled Epilepsy Trials
|
Adverse Reaction |
Number (%) with Adverse Reaction |
Number (%) Discontinuing |
||
|
POTIGA (n = 813) |
Placebo (n = 427) |
POTIGA (n = 813) |
Placebo (n = 427) |
|
|
Confusional state |
75 (9%) |
11 (3%) |
32 (4%) |
4 (<1%) |
|
Psychosis |
9 (1%) |
0 |
6 (<1%) |
0 |
|
Hallucinationsa |
14 (2%) |
2 (<1%) |
6 (<1%) |
0 |
- a Hallucinations includes visual, auditory, and mixed hallucinations.
5.5 Dizziness and Somnolence
POTIGA causes dose-related increases in dizziness and somnolence [see Adverse Reactions (6.1)]. In placebo-controlled trials in patients with epilepsy, dizziness was reported in 23% of patients treated with POTIGA and 9% of patients treated with placebo. Somnolence was reported in 22% of patients treated with POTIGA and 12% of patients treated with placebo. In these trials 6% of patients on POTIGA and 1.2% on placebo discontinued treatment because of dizziness; 3% of patients on POTIGA and <1.0% on placebo discontinued because of somnolence.
Most of these adverse reactions were mild to moderate in intensity and occurred during the titration phase. For those patients continued on POTIGA, dizziness and somnolence appeared to diminish with continued use.
5.6 QT Interval Effect
A study of cardiac conduction showed that POTIGA produced a mean 7.7-msec QT prolongation in healthy volunteers titrated to 400 mg 3 times daily. The QT-prolonging effect occurred within 3 hours. The QT interval should be monitored when POTIGA is prescribed with medicines known to increase QT interval and in patients with known prolonged QT interval, congestive heart failure, ventricular hypertrophy, hypokalemia, or hypomagnesemia [see Clinical Pharmacology (12.2)].
5.7 Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including POTIGA, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive-therapy) of 11 different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted relative risk 1.8, 95% confidence interval [CI]: 1.2, 2.7) of suicidal thinking or behavior compared with patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43% compared with 0.24% among 16,029 placebo-treated patients, representing an increase of approximately 1 case of suicidal thinking or behavior for every 530 patients treated. There were 4 suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as 1 week after starting treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanism of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5 to 100 years) in the clinical trials analyzed.
Table 3 shows absolute and relative risk by indication for all evaluated AEDs.
Table 3. Risk of Suicidal Thoughts or Behaviors by Indication for Antiepileptic Drugs in the Pooled Analysis
|
Indication |
Placebo Patients with Events per 1,000 Patients |
Drug Patients with Events per 1,000 Patients |
Relative Risk: Incidence of Events in Drug Patients/Incidence in Placebo Patients |
Risk Difference: Additional Drug Patients with Events per 1,000 Patients |
|
Epilepsy |
1.0 |
3.4 |
3.5 |
2.4 |
|
Psychiatric |
5.7 |
8.5 |
1.5 |
2.9 |
|
Other |
1.0 |
1.8 |
1.9 |
0.9 |
|
Total |
2.4 |
4.3 |
1.8 |
1.9 |
The relative risk for suicidal thoughts or behavior was higher in clinical trials in patients with epilepsy than in clinical trials in patients with psychiatric or other conditions, but the absolute risk differences were similar for epilepsy and psychiatric indications.
Anyone considering prescribing POTIGA or any other AED must balance this risk with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, the prescriber needs to consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
Patients, their caregivers, and families should be informed that AEDs increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of the signs and symptoms of depression; any unusual changes in mood or behavior; or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers.
5.8 Withdrawal Seizures
As with all AEDs, when POTIGA is discontinued, it should be withdrawn gradually when possible to minimize the potential of increased seizure frequency [see Dosage and Administration (2.1)]. The dosage of POTIGA should be reduced over a period of at least 3 weeks, unless safety concerns require abrupt withdrawal.
6 ADVERSE REACTIONS
The following adverse reactions are described in more detail in the Warnings and Precautions section of the label:
- •
- Retinal abnormalities and potential vision loss [see Warnings and Precautions (5.1)]
- •
- Urinary retention [see Warnings and Precautions (5.2)]
- •
- Skin discoloration [see Warnings and Precautions (5.3)]
- •
- Neuropsychiatric symptoms [see Warnings and Precautions (5.4)]
- •
- Dizziness and somnolence [see Warnings and Precautions (5.5)]
- •
- QT interval effect [see Warnings and Precautions (5.6)]
- •
- Suicidal behavior and ideation [see Warnings and Precautions (5.7)]
- •
- Withdrawal seizures [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
POTIGA was administered as adjunctive therapy to 1,365 patients with epilepsy in all controlled and uncontrolled clinical studies during the premarketing development. A total of 801 patients were treated for at least 6 months, 585 patients were treated for 1 year or longer, and 311 patients were treated for at least 2 years.
Adverse Reactions Leading to Discontinuation in All Controlled Clinical Studies
In the 3 randomized, double-blind, placebo-controlled studies, 199 of 813 patients (25%) receiving POTIGA and 45 of 427 patients (11%) receiving placebo discontinued treatment because of adverse reactions. The most common adverse reactions leading to withdrawal in patients receiving POTIGA were dizziness (6%), confusional state (4%), fatigue (3%), and somnolence (3%).
Common Adverse Reactions in All Controlled Clinical Studies
Overall, the most frequently reported adverse reactions in patients receiving POTIGA (≥4% and occurring approximately twice the placebo rate) were dizziness (23%), somnolence (22%), fatigue (15%), confusional state (9%), vertigo (8%), tremor (8%), abnormal coordination (7%), diplopia (7%), disturbance in attention (6%), memory impairment (6%), asthenia (5%), blurred vision (5%), gait disturbance (4%), aphasia (4%), dysarthria (4%), and balance disorder (4%) (see Table 4). In most cases the reactions were of mild or moderate intensity.
Table 4. Adverse Reactions Incidence in Placebo-Controlled Adjunctive Trials in Adult Patients with Partial-Onset Seizures (Adverse reactions in at least 2% of patients treated with POTIGA in any treatment group and numerically more frequent than in the placebo group.)
|
Body System/
|
Placebo |
POTIGA |
|||
|
600 mg/day |
900 mg/day |
1,200 mg/day |
All |
||
|
(N = 427) % |
(n = 281) % |
(n = 273) % |
(n = 259) % |
(N = 813) % |
|
| |||||
|
2 |
8 |
6 |
7 |
7 |
|
2 |
2 |
4 |
10 |
5 |
| |||||
|
5 |
6 |
6 |
9 |
7 |
|
1 |
1 |
4 |
5 |
3 |
|
2 |
3 |
2 |
3 |
2 |
| |||||
|
6 |
16 |
15 |
13 |
15 |
|
2 |
4 |
6 |
4 |
5 |
| |||||
|
2 |
4 |
1 |
5 |
3 |
| |||||
|
1 |
2 |
3 |
3 |
3 |
| |||||
|
9 |
15 |
23 |
32 |
23 |
|
12 |
15 |
25 |
27 |
22 |
|
3 |
3 |
6 |
9 |
6 |
|
3 |
3 |
10 |
12 |
8 |
|
2 |
8 |
8 |
9 |
8 |
|
3 |
5 |
5 |
12 |
7 |
|
<1 |
6 |
6 |
7 |
6 |
|
1 |
2 |
5 |
6 |
4 |
|
<1 |
1 |
3 |
7 |
4 |
|
<1 |
4 |
2 |
8 |
4 |
|
<1 |
3 |
3 |
5 |
4 |
|
2 |
3 |
2 |
5 |
3 |
|
<1 |
<1 |
3 |
3 |
2 |
|
<1 |
1 |
1 |
3 |
2 |
| |||||
|
3 |
4 |
8 |
16 |
9 |
|
2 |
3 |
2 |
5 |
3 |
|
<1 |
<1 |
<1 |
5 |
2 |
|
0 |
0 |
<1 |
2 |
<1 |
| |||||
|
<1 |
1 |
2 |
4 |
2 |
|
<1 |
2 |
1 |
4 |
2 |
|
<1 |
2 |
1 |
2 |
2 |
|
<1 |
<1 |
2 |
3 |
2 |
Other adverse reactions reported in these 3 studies in <2% of patients treated with POTIGA and numerically greater than placebo were increased appetite, hallucinations, myoclonus, peripheral edema, hypokinesia, dry mouth, dysphagia, hyperhydrosis, urinary retention, malaise, and increased liver enzymes.
Most of the adverse reactions appear to be dose related (especially those classified as psychiatric and nervous system symptoms), including dizziness, somnolence, confusional state, tremor, abnormal coordination, memory impairment, blurred vision, gait disturbance, aphasia, balance disorder, constipation, dysuria, and chromaturia.
POTIGA was associated with dose-related weight gain, with mean weight increasing by 0.2 kg, 1.2 kg, 1.6 kg, and 2.7 kg in the placebo, 600 mg per day, 900 mg per day, and 1,200 mg per day groups, respectively.
Additional Adverse Reactions Observed during All Phase 2 and 3 Clinical Trials
Following is a list of adverse reactions reported by patients treated with POTIGA during all clinical trials: rash, nystagmus, dyspnea, leukopenia, muscle spasms, alopecia, nephrolithiasis, syncope, neutropenia, thrombocytopenia, euphoric mood, renal colic, coma, encephalopathy.
Comparison of Gender, Age, and Race
The overall adverse reaction profile of POTIGA was similar for females and males.
There are insufficient data to support meaningful analyses of adverse reactions by age or race. Approximately 86% of the population studied was Caucasian, and 0.8% of the population was aged 65 years or older.
6.2 Postmarketing Experience
In addition to adverse reactions reported from clinical trials, the following adverse reactions have been identified during postapproval use of POTIGA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Acquired vitelliform lesions.
7 DRUG INTERACTIONS
7.1 Antiepileptic Drugs
The potentially significant interactions between POTIGA and concomitant AEDs are summarized in Table 5.
Table 5. Significant Interactions between POTIGA and Concomitant Antiepileptic Drugs (AEDs)
|
AED |
Dosage of AED (mg/day) |
Dosage of POTIGA (mg/day) |
Influence of POTIGA on AED |
Influence of AED on POTIGA |
Dosage Adjustment |
|
Carbamazepinea,b |
600-2,400 |
300-1,200 |
None |
31% decrease in AUC, 23% decrease in Cmax |
consider an increase in dosage of POTIGA when adding carbamazepinec |
|
Phenytoina,b |
120-600 |
300-1,200 |
None |
34% decrease in AUC, 18% decrease in Cmax |
consider an increase in dosage of POTIGA when adding phenytoinc |
- a Based on results of a Phase 2 study.
- b Inducer for uridine 5'-diphosphate (UDP)-glucuronyltransferases (UGTs).
- c A decrease in dosage of POTIGA should be considered when carbamazepine or phenytoin is discontinued.
[See Clinical Pharmacology (12.3).]
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C. There are no adequate and well-controlled studies in pregnant women. POTIGA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
In animal studies, doses associated with maternal plasma exposures (AUC) to ezogabine and its major circulating metabolite, N-acetyl metabolite of ezogabine (NAMR), similar to or below those expected in humans at the maximum recommended human dose (MRHD) of 1,200 mg per day produced developmental toxicity when administered to pregnant rats and rabbits. The maximum doses evaluated were limited by maternal toxicity (acute neurotoxicity).
Treatment of pregnant rats with ezogabine (oral doses of up to 46 mg/kg/day) throughout organogenesis increased the incidences of fetal skeletal variations. The no-effect dose for embryo-fetal toxicity in rats (21 mg/kg/day) was associated with maternal plasma exposures (AUC) to ezogabine and NAMR less than those in humans at the MRHD. Treatment of pregnant rabbits with ezogabine (oral doses of up to 60 mg/kg/day) throughout organogenesis resulted in decreased fetal body weights and increased incidences of fetal skeletal variations. The no-effect dose for embryo-fetal toxicity in rabbits (12 mg/kg/day) was associated with maternal plasma exposures to ezogabine and NAMR less than those in humans at the MRHD.
Administration of ezogabine (oral doses of up to 61.9 mg/kg/day) to rats throughout pregnancy and lactation resulted in increased pre- and postnatal mortality, decreased body weight gain, and delayed reflex development in the offspring. The no-effect dose for pre- and postnatal developmental effects in rats (17.8 mg/kg/day) was associated with maternal plasma exposures to ezogabine and NAMR less than those in humans at the MRHD.
Pregnancy Registry
To provide information regarding the effects of in utero exposure to POTIGA, physicians are advised to recommend that pregnant patients taking POTIGA enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. This can be done by calling the toll-free number 1-888-233-2334, and must be done by patients themselves. Information on the registry can also be found at the website www.aedpregnancyregistry.org.
8.3 Nursing Mothers
It is not known whether ezogabine is excreted in human milk. However, ezogabine and/or its metabolites are present in the milk of lactating rats. Because of the potential for serious adverse reactions in nursing infants from POTIGA, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
The safety and effectiveness of POTIGA in patients under 18 years of age have not been established.
In juvenile animal studies, increased sensitivity to acute neurotoxicity and urinary bladder toxicity was observed in young rats compared with adults. In studies in which rats were dosed starting on postnatal day 7, ezogabine-related mortality, clinical signs of neurotoxicity, and renal and urinary tract toxicities were observed at doses ≥2 mg/kg/day. The no-effect level was associated with plasma ezogabine exposures (AUC) less than those expected in human adults at the MRHD of 1,200 mg per day. In studies in which dosing began on postnatal day 28, acute central nervous system effects, but no apparent renal or urinary tract effects, were observed at doses of up to 30 mg/kg/day. These doses were associated with plasma ezogabine exposures less than those achieved clinically at the MRHD.
8.5 Geriatric Use
There were insufficient numbers of elderly patients enrolled in partial-onset seizure controlled trials (n = 8 patients on ezogabine) to determine the safety and efficacy of POTIGA in this population.
Dosage adjustment is recommended in patients aged 65 years and older [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)].
POTIGA may cause urinary retention. Elderly men with symptomatic BPH may be at increased risk for urinary retention.
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
A human abuse potential study was conducted in recreational sedative-hypnotic abusers (n = 36) in which single oral doses of ezogabine (300 mg [n = 33], 600 mg [n = 34], 900 mg [n = 6]), the sedative-hypnotic alprazolam (1.5 mg and 3.0 mg), and placebo were administered. Euphoria-type subjective responses to the 300-mg and 600-mg doses of ezogabine were statistically different from placebo but statistically indistinguishable from those produced by either dose of alprazolam. Adverse events reported following administration of single oral doses of 300 mg, 600 mg, and 900 mg ezogabine given without titration included euphoric mood (18%, 21%, and 33%, respectively; 8% from placebo), hallucination (0%, 0%, and 17%, respectively; 0% from placebo) and somnolence (18%, 15%, and 67%, respectively; 15% from placebo).
In Phase 1 clinical studies, healthy individuals who received oral ezogabine (200 mg to 1,650 mg) reported euphoria (8.5%), feeling drunk (5.5%), hallucination (5.1%), disorientation (1.7%), and feeling abnormal (1.5%).
In the 3 randomized, double-blind, placebo-controlled Phase 2 and 3 clinical studies, patients with partial seizures who received oral ezogabine (300 mg to 1,200 mg) reported euphoric mood (0.5%) and feeling drunk (0.9%), while those who received placebo did not report either adverse event (0%).
9.3 Dependence
In a 28-day physical dependence study in which rats received daily ezogabine administration, abrupt drug discontinuation produced behavioral changes that included piloerection, increases in high step gait, and tremors, compared with vehicle-treated animals. These data show that ezogabine produces a withdrawal syndrome indicative of physical dependence.
10 OVERDOSAGE
10.1 Signs, Symptoms, and Laboratory Findings
There is limited experience of overdose with POTIGA. Total daily doses of POTIGA over 2,500 mg were reported during clinical trials. In addition to adverse reactions seen at therapeutic doses, symptoms reported with overdose of POTIGA included agitation, aggressive behavior, and irritability. There were no reported sequelae.
In an abuse potential study, cardiac arrhythmia (asystole or ventricular tachycardia) occurred in 2 volunteers within 3 hours of receiving a single 900-mg dose of POTIGA. The arrhythmias spontaneously resolved and both volunteers recovered without sequelae.
10.2 Management of Overdose
There is no specific antidote for overdose with POTIGA. In the event of overdose, standard medical practice for the management of any overdose should be used. An adequate airway, oxygenation, and ventilation should be ensured; monitoring of cardiac rhythm and vital sign measurement is recommended. A certified poison control center should be contacted for updated information on the management of overdose with POTIGA.
Hemodialysis has been shown to reduce the plasma concentrations of ezogabine and NAMR in subjects with ESRD [see Clinical Pharmacology (12.3)].
11 DESCRIPTION
The chemical name of ezogabine is N-[2-amino-4-(4-fluorobenzylamino)-phenyl] carbamic acid ethyl ester, and it has the following structure:
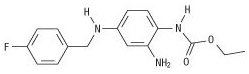
The empirical formula is C16H18FN3O2, representing a molecular weight of 303.3. Ezogabine is a white to slightly colored, odorless, tasteless, crystalline powder. At room temperature, ezogabine is practically insoluble in aqueous media at pH values above 4, while the solubility is higher in polar organic solvents. At gastric pH, ezogabine is sparingly soluble in water (about 16 g/L). The pKa is approximately 3.7 (basic).
POTIGA is supplied for oral administration as 50-mg, 200-mg, 300-mg, and 400-mg film-coated immediate-release tablets. Each tablet contains the labeled amount of ezogabine and the following inactive ingredients: carmine (50-mg and 400-mg tablets), croscarmellose sodium, FD&C Blue No. 2 (50-mg, 300-mg, and 400-mg tablets), hypromellose, iron oxide yellow (200‑mg and 300-mg tablets), lecithin, magnesium stearate, microcrystalline cellulose, polyvinyl alcohol, talc, titanium dioxide, and xanthan gum.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism by which ezogabine exerts its therapeutic effects has not been fully elucidated. In vitro studies indicate that ezogabine enhances transmembrane potassium currents mediated by the KCNQ (Kv7.2 to 7.5) family of ion channels. By activating KCNQ channels, ezogabine is thought to stabilize the resting membrane potential and reduce brain excitability. In vitro studies suggest that ezogabine may also exert therapeutic effects through augmentation of GABA-mediated currents.
12.2 Pharmacodynamics
The QTc prolongation risk of POTIGA was evaluated in healthy subjects. In a randomized, double-blind, active- and placebo-controlled parallel-group study, 120 healthy subjects (40 in each group) were administered POTIGA titrated up to the final dose of 400 mg 3 times daily, placebo, and placebo and moxifloxacin (on day 22). After 22 days of dosing, the maximum mean (upper 1-sided, 95% CI) increase of baseline- and placebo-adjusted QTc interval based on Fridericia correction method (QTcF) was 7.7 msec (11.9 msec) and was observed at 3 hours after dosing in subjects who achieved 1,200 mg per day. No effects on heart rate, PR, or QRS intervals were noted.
Patients who are prescribed POTIGA with medicines known to increase QT interval or who have known prolonged QT interval, congestive heart failure, ventricular hypertrophy, hypokalemia, or hypomagnesemia should be observed closely [see Warnings and Precautions (5.6)].
12.3 Pharmacokinetics
The pharmacokinetic profile is approximately linear in daily doses between 600 mg and 1,200 mg in patients with epilepsy, with no unexpected accumulation following repeated administration. The pharmacokinetics of ezogabine are similar in healthy volunteers and patients with epilepsy.
Absorption
After both single and multiple oral doses, ezogabine is rapidly absorbed with median time to maximum plasma concentration (Tmax) values generally between 0.5 and 2 hours. Absolute oral bioavailability of ezogabine relative to an intravenous dose of ezogabine is approximately 60%. High-fat food does not affect the extent to which ezogabine is absorbed based on plasma AUC values, but it increases peak concentration (Cmax) by approximately 38% and delays Tmax by 0.75 hour.
POTIGA can be taken with or without food.
Distribution
Data from in vitro studies indicate that ezogabine and NAMR are approximately 80% and 45% bound to plasma protein, respectively. Clinically significant interactions with other drugs through displacement from proteins are not anticipated. The steady-state volume of distribution of ezogabine is 2 to 3 L/kg following intravenous dosing, suggesting that ezogabine is well distributed in the body.
Metabolism
Ezogabine is extensively metabolized primarily via glucuronidation and acetylation in humans. A substantial fraction of the ezogabine dose is converted to inactive N-glucuronides, the predominant circulating metabolites in humans. Ezogabine is also metabolized to NAMR that is also subsequently glucuronidated. NAMR has antiepileptic activity, but it is less potent than ezogabine in animal seizure models. Additional minor metabolites of ezogabine are an N-glucoside of ezogabine and a cyclized metabolite believed to be formed from NAMR. In vitro studies using human biomaterials showed that the N-acetylation of ezogabine was primarily carried out by NAT2, while glucuronidation was primarily carried out by UGT1A4, with contributions by UGT1A1, UGT1A3, and UGT1A9.
In vitro studies showed no evidence of oxidative metabolism of ezogabine or NAMR by cytochrome P450 enzymes. Coadministration of ezogabine with medications that are inhibitors or inducers of cytochrome P450 enzymes is therefore unlikely to affect the pharmacokinetics of ezogabine or NAMR.
Elimination
Results of a mass balance study suggest that renal excretion is the major route of elimination for ezogabine and NAMR. About 85% of the dose was recovered in the urine, with the unchanged parent drug and NAMR accounting for 36% and 18% of the administered dose, respectively, and the total N-glucuronides of ezogabine and NAMR accounting for 24% of the administered dose. Approximately 14% of the radioactivity was recovered in the feces, with unchanged ezogabine accounting for 3% of the total dose. Average total recovery in both urine and feces within 240 hours after dosing is approximately 98%.
Ezogabine and its N-acetyl metabolite have similar elimination half-lives (t½) of 7 to 11 hours. The clearance of ezogabine following intravenous dosing was approximately 0.4 to 0.6 L/h/kg. Ezogabine is actively secreted into the urine.
Specific Populations
Race: No study has been conducted to investigate the impact of race on pharmacokinetics of ezogabine. A population pharmacokinetic analysis comparing Caucasians and non-Caucasians (predominately African American and Hispanic patients) showed no significant pharmacokinetic difference. No adjustment of the ezogabine dose for race is recommended.
Gender: The impact of gender on the pharmacokinetics of ezogabine was examined following a single dose of POTIGA to healthy young (aged 21 to 40 years) and elderly (aged 66 to 82 years) subjects. The AUC values were approximately 20% higher in young females compared with young males and approximately 30% higher in elderly females compared with elderly males. The Cmax values were approximately 50% higher in young females compared with young males and approximately 100% higher in elderly females compared with elderly males. There was no gender difference in weight-normalized clearance. Overall, no adjustment of the dosage of POTIGA is recommended based on gender.
Pediatric Patients: The pharmacokinetics of ezogabine in pediatric patients have not been investigated.
Geriatric: The impact of age on the pharmacokinetics of ezogabine was examined following a single dose of ezogabine to healthy young (aged 21 to 40 years) and elderly (aged 66 to 82 years) subjects. Systemic exposure (AUC) of ezogabine was approximately 40% to 50% higher and terminal half-life was prolonged by approximately 30% in the elderly compared with the younger subjects. The peak concentration (Cmax) was similar to that observed in younger subjects. A dosage reduction in the elderly is recommended [see Dosage and Administration (2.3), Use in Specific Populations (8.5)].
Renal Impairment: The pharmacokinetics of ezogabine were studied following a single 100-mg dose of POTIGA in subjects with normal (CrCL >80 mL/min), mild (CrCL ≥50 to ≤80 mL/min), moderate (CrCL ≥30 to <50 mL/min), or severe renal impairment (CrCL <30 mL/min) (n = 6 in each cohort) and in subjects with ESRD requiring hemodialysis (n = 6). The ezogabine AUC was increased by approximately 30% in patients with mild renal impairment and doubled in patients with moderate impairment to ESRD (CrCL <50 mL/min) relative to healthy subjects. Similar increases in NAMR exposure were observed in the various degrees of renal impairment.
In a second single-dose study in patients with ESRD (N = 8) receiving chronic hemodialysis, initiation of dialysis at approximately 4 hours after a single dose of ezogabine 100 mg resulted in a median reduction in plasma concentrations of ezogabine by 52% (range: 17% to 59%) and NAMR by 51% (range: 27% to 72%) from the start to the end of dialysis. This compares with a median reduction of 19% in ezogabine plasma concentrations and 13% for NAMR over the same time period off dialysis.
Dosage reduction is recommended in patients with creatinine clearance <50 mL/min and in patients with ESRD receiving hemodialysis. Immediately following hemodialysis a supplemental dose is recommended [see Dosage and Administration (2.3), Use in Specific Populations (8.6)].
Hepatic Impairment: The pharmacokinetics of ezogabine were studied following a single 100-mg dose of POTIGA in subjects with normal, mild (Child-Pugh score 5 to 6), moderate (Child-Pugh score 7 to 9), or severe hepatic (Child-Pugh score >9) impairment (n = 6 in each cohort). Relative to healthy subjects, ezogabine AUC was not affected by mild hepatic impairment, but was increased by approximately 50% in subjects with moderate hepatic impairment and doubled in subjects with severe hepatic impairment. There was an increase of approximately 30% in exposure to NAMR in patients with moderate to severe impairment. Dosage reduction is recommended in patients with moderate or severe hepatic impairment [see Dosage and Administration (2.3), Use in Specific Populations (8.7)].
Drug Interactions
In vitro studies using human liver microsomes indicated that ezogabine does not inhibit enzyme activity for CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4/5. In addition, in vitro studies in human primary hepatocytes showed that ezogabine and NAMR did not induce CYP1A2 or CYP3A4/5 activity. Therefore, ezogabine is unlikely to affect the pharmacokinetics of substrates of the major cytochrome P450 isoenzymes through inhibition or induction mechanisms.
Ezogabine is not an inhibitor of the transporters P-glycoprotein (P-gp), organic anion transporter 1 (OAT1), or organic cation transporter 2 (OCT2) or a substrate for P-gp efflux. Ezogabine is a weak inhibitor of the transporter organic anion transporter 3 (OAT3). Ezogabine’s metabolite, NAMR, is also a weak inhibitor of P-gp efflux. In vitro studies showed that ezogabine is not a substrate for OAT1, OCT2, or OAT3.
Interactions with Antiepileptic Drugs: The interactions between POTIGA and concomitant AEDs are summarized in Table 6.
Table 6. Interactions between POTIGA and Concomitant Antiepileptic Drugs (AEDs)
|
AED |
Dose of AED (mg/day) |
Dose of POTIGA (mg/day) |
Influence of POTIGA on AED |
Influence of AED on POTIGA |
Dosage Adjustment |
|
Carbamazepinea,b |
600-2,400 |
300-1,200 |
None |
31% decrease in AUC, 23% decrease in Cmax, 28% increase in clearance |
consider an increase in dosage of POTIGA when adding carbamazepinec |
|
Phenytoina,b |
120-600 |
300-1,200 |
None |
34% decrease in AUC, 18% decrease in Cmax, 33% increase in clearance |
consider an increase in dosage of POTIGA when adding phenytoinc |
|
Topiramatea |
250-1,200 |
300-1,200 |
None |
None |
None |
|
Valproatea |
750-2,250 |
300-1,200 |
None |
None |
None |
|
Phenobarbital |
90 |
600 |
None |
None |
None |
|
Lamotrigine |
200 |
600 |
18% decrease in AUC, 22% increase in clearance |
None |
None |
|
Othersd |
None |
None |
None |
- a Based on results of a Phase 2 study.
- b Inducer for uridine 5'-diphosphate (UDP)-glucuronyltransferases (UGTs).
- c A decrease in dose of POTIGA should be considered when carbamazepine or phenytoin is discontinued.
- d Zonisamide, valproic acid, clonazepam, gabapentin, levetiracetam, oxcarbazepine, phenobarbital, pregabalin, topiramate, clobazam, and lamotrigine, based on a population pharmacokinetic analysis using pooled data from Phase 3 clinical trials.
Digoxin: A repeat-dose trial of ezogabine 600 mg to 1,200 mg administered to healthy subjects (n = 29) resulted in a small increase of the systemic exposure of digoxin (AUC range of 8% to 18%) that did not appear to be dose related. No dose adjustment is necessary for digoxin when coadministered with POTIGA.
Oral Contraceptives: In one study examining the potential interaction between ezogabine (150 mg 3 times daily for 3 days) and the combination oral contraceptive norgestrel/ethinyl estradiol (0.3 mg/0.03 mg) tablets in 20 healthy females, no significant alteration in the pharmacokinetics of either drug was observed.
In a second study examining the potential interaction of repeated ezogabine dosing (250 mg 3 times daily for 14 days) and the combination oral contraceptive norethindrone/ethinyl estradiol (1 mg/0.035 mg) tablets in 25 healthy females, no significant alteration in the pharmacokinetics of either drug was observed.
Alcohol: In a healthy volunteer study, the coadministration of ethanol 1g/kg (5 standard alcohol drinks) over 20 minutes and ezogabine (200 mg) resulted in an increase in the ezogabine Cmax and AUC by 23% and 37%, respectively [see Drug Interactions (7.2)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 1-year neonatal mouse study of ezogabine (2 single-dose oral administrations of up to 96 mg/kg on postnatal days 8 and 15), a dose-related increase in the frequency of lung neoplasms (bronchioalveolar carcinoma and/or adenoma) was observed in treated males. No evidence of carcinogenicity was observed in rats following oral administration of ezogabine (oral gavage doses of up to 50 mg/kg/day) for 2 years. Plasma exposure (AUC) to ezogabine at the highest doses tested was less than that in humans at the maximum recommended human dose (MRHD) of 1,200 mg per day.
Mutagenesis
Highly purified ezogabine was negative in the in vitro Ames assay, the in vitro Chinese hamster ovary (CHO) Hprt gene mutation assay, and the in vivo mouse micronucleus assay. Ezogabine was positive in the in vitro chromosomal aberration assay in human lymphocytes. The major circulating metabolite of ezogabine, NAMR, was negative in the in vitro Ames assay, but positive in the in vitro chromosomal aberration assay in CHO cells.
Impairment of Fertility
Ezogabine had no effect on fertility, general reproductive performance, or early embryonic development when administered to male and female rats at doses of up to 46.4 mg/kg/day (associated with a plasma ezogabine exposure [AUC] less than that in humans at the MRHD) prior to and during mating, and continuing in females through gestation day 7.
14 CLINICAL STUDIES
The efficacy of POTIGA as adjunctive therapy in partial-onset seizures was established in 3 multicenter, randomized, double-blind, placebo-controlled studies in 1,239 adult patients. The primary endpoint consisted of the percent change in seizure frequency from baseline to the double-blind treatment phase (titration through maintenance).
Patients enrolled in the studies had partial-onset seizures with or without secondary generalization and were not adequately controlled with 1 to 3 concomitant AEDs, with or without concomitant vagus nerve stimulation. More than 75% of patients were taking 2 or more concomitant AEDs. During an 8-week baseline period, patients experienced at least 4 partial-onset seizures per 28 days on average with no seizure-free period exceeding 3 to 4 weeks. Patients had a mean duration of epilepsy of 22 years. Across the 3 studies, the median baseline seizure frequency ranged from 8 to 12 seizures per month. The criteria for statistical significance was P<0.05.
Patients were randomized to the total daily maintenance dosages of 600 mg per day, 900 mg per day, or 1,200 mg per day, each administered in 3 equally divided doses. During the titration phase of all 3 studies, treatment was initiated at 300 mg per day (100 mg 3 times per day) and increased in weekly increments of 150 mg per day to the target maintenance dosage.
Figure 1 shows the median percent reduction in 28-day seizure frequency (baseline to double-blind phase) as compared with placebo across all 3 studies. A statistically significant effect was observed with POTIGA at doses of 600 mg per day (Study 1), at 900 mg per day (Studies 1 and 3), and at 1,200 mg per day (Studies 2 and 3).
Figure 1. Median Percent Reduction from Baseline in Seizure Frequency per 28 Days by Dose
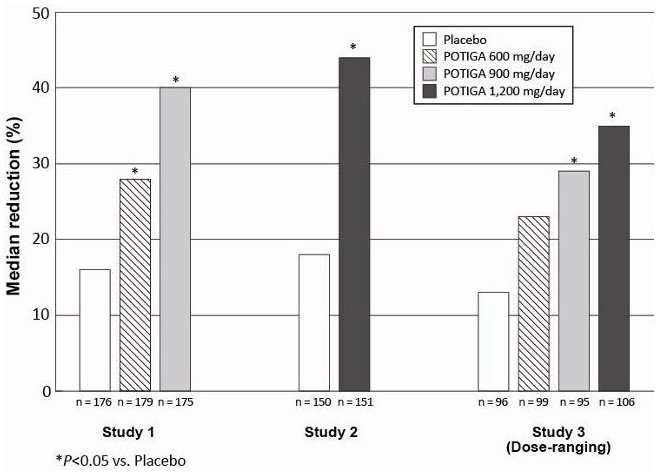
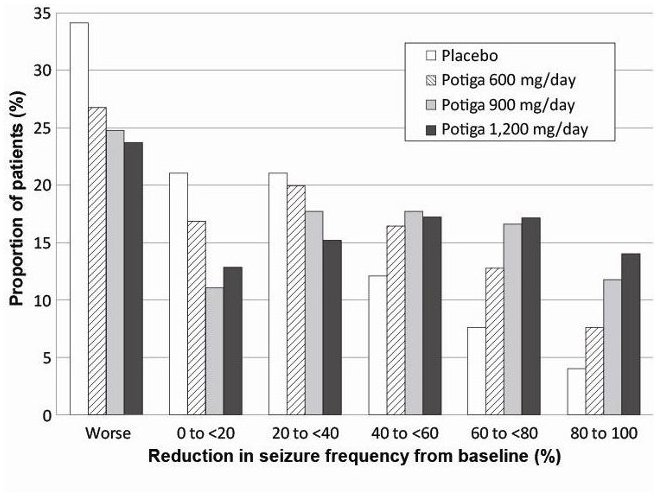
Figure 2 shows changes from baseline in the 28-day total partial seizure frequency by category for patients treated with POTIGA and placebo in an integrated analysis across the 3 clinical trials. Patients in whom the seizure frequency increased are shown at left as “worse.” Patients in whom the seizure frequency decreased are shown in five categories.
Figure 2. Proportion of Patients by Category of Seizure Response for POTIGA and Placebo across All Three Double-blind Trials
16 HOW SUPPLIED/STORAGE AND HANDLING
POTIGA is supplied as film-coated immediate-release tablets for oral administration containing 50 mg, 200 mg, 300 mg, or 400 mg of ezogabine in the following packs:
- •
- 50-mg Tablets: purple, round, film-coated tablets debossed with “RTG 50” on one side in bottles of 90 tablets with desiccant (NDC 0173-0810-59).
- •
- 200-mg Tablets: yellow, oblong, film-coated tablets debossed with “RTG-200” on one side in bottles of 90 tablets with desiccant (NDC 0173-0812-59).
- •
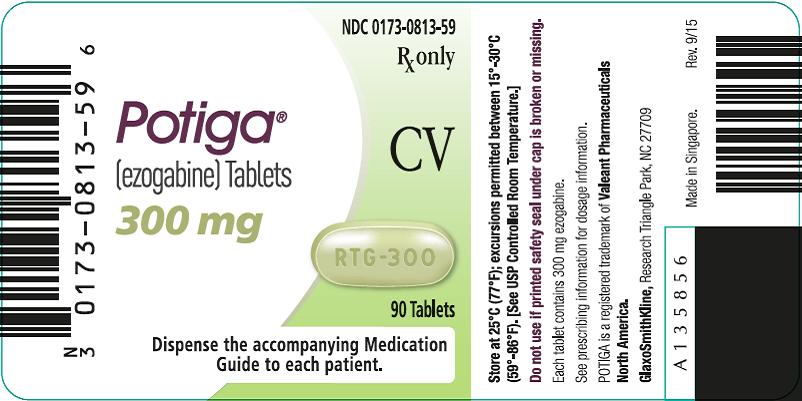
- 300-mg Tablets: green, oblong, film-coated tablets debossed with “RTG-300” on one side in bottles of 90 tablets with desiccant (NDC 0173-0813-59).
- •
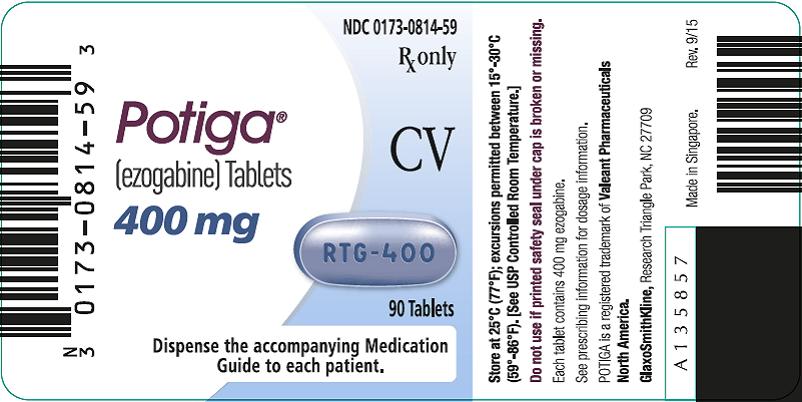
- 400-mg Tablets: purple, oblong, film-coated tablets debossed with “RTG-400” on one side in bottles of 90 tablets with desiccant (NDC 0173-0814-59).
Store at 25°C (77°F); excursions permitted to 15°-30°C (59°-86°F) [See USP Controlled Room Temperature.]
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Retinal Abnormalities and Potential Vision Loss
Inform patients of the risk of retinal abnormalities and possible risk of vision loss, which may be permanent [see Warnings and Precautions 5.1]. All patients taking POTIGA should participate in baseline and periodic ophthalmologic monitoring of vision by an ophthalmic professional. Inform patients that if they suspect any vision changes, they should notify their physician immediately.
Urinary Retention
Inform patients that POTIGA can cause urinary retention (including urinary hesitation and dysuria). Instruct patients to seek immediate medical assistance if they experience any symptoms of urinary retention, inability to urinate, and/or pain with urination [see Warnings and Precautions (5.2)]. Urologic consultation may be helpful for patients who cannot reliably report symptoms of urinary retention (for example, patients with cognitive impairment).
Skin Discoloration
Inform patients that POTIGA can cause discoloration of nails, lips, skin, palate, and parts of the eye and that it is not known if the discoloration is reversible upon drug discontinuation [see Warnings and Precautions 5.3]. Most skin discoloration has been reported after at least 2 years of treatment with POTIGA, but may happen earlier. Inform patients that the possibility of more extensive systemic involvement has not been excluded. Instruct patients to notify their physician if they develop skin discoloration.
Psychiatric Symptoms
Inform patients that POTIGA can cause psychiatric symptoms such as confusional state, disorientation, hallucinations, and other symptoms of psychosis. Instruct patients and their caregivers to notify their physicians if they experience psychotic symptoms [see Warnings and Precautions (5.4)].
Central Nervous System Effects
Inform patients that POTIGA may cause dizziness, somnolence, memory impairment, abnormal coordination/balance, disturbance in attention, and ophthalmological effects such as diplopia or blurred vision. Advise patients taking POTIGA not to drive, operate complex machinery, or engage in other hazardous activities until they have become accustomed to any such effects associated with POTIGA [see Warnings and Precautions (5.5)].
Suicidal Thinking and Behavior
Inform patients, their caregivers, and families that AEDs, including POTIGA, may increase the risk of suicidal thoughts and behavior and advise them of the need to be alert for the emergence or worsening of symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Instruct them to immediately report behaviors of concern to healthcare providers [see Warnings and Precautions (5.7)].
Pregnancy
Advise patients to notify their physicians if they become pregnant or intend to become pregnant during therapy. Advise patients to notify their physicians if they intend to breastfeed or are breastfeeding an infant.
Encourage patients to enroll in the NAAED Pregnancy Registry if they become pregnant. This registry collects information about the safety of AEDs during pregnancy. To enroll, patients can call the toll-free number 1-888-233-2334 [see Use in Specific Populations (8.1)].
POTIGA is a registered trademark of Valeant Pharmaceuticals North America LLC.
GlaxoSmithKline
Research Triangle Park, NC 27709
©2016 the GSK group of companies. All rights reserved.
PTG:9PI
MEDICATION GUIDE
POTIGA® (po-TEE-ga) tablets, CV
(ezogabine)
Read this Medication Guide before you start taking POTIGA and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your healthcare provider about your medical condition or treatment. If you have questions about POTIGA, ask your healthcare provider or pharmacist.
What is the most important information I should know about POTIGA?
Do not stop POTIGA without first talking to a healthcare provider. Stopping POTIGA suddenly can cause serious problems. Stopping POTIGA suddenly can cause you to have more seizures more often.
- 1.
- POTIGA can cause changes to your retina, which is located in the back of your eye and is needed for vision. These types of changes can cause vision loss.
- •
- If a decrease in your vision happens, it is not known if it will get better.
- •
- You and your healthcare provider should decide if the benefit of taking POTIGA is more important than the possible risk of vision loss.
- •
- You should have a complete eye exam if you are currently taking POTIGA or before starting treatment, and then every 6 months while taking POTIGA.
- •
- Tell your healthcare provider right away if you notice any changes in your vision.
- 2.
- POTIGA can make it hard for you to urinate (empty your bladder) and may cause you to be unable to urinate. Call your healthcare provider right away if you:
- •
- are unable to start urinating.
- •
- have trouble emptying your bladder.
- •
- have a weak urine stream.
- •
- have pain with urination.
- 3.
- POTIGA can cause changes in the color of your skin, nails, lips, roof of your mouth, and whites of your eyes or insides of your eyelids.
- •
- The changes in color may be blue, grey-blue, or brown.
- •
- Most changes in color have happened in people who have taken POTIGA for at least 2 years, but may happen earlier.
- •
- It is not known if the changes in color go away after stopping POTIGA.
- •
- Tell your healthcare provider if you notice any changes in color to your body.
- 4.
- POTIGA can cause mental (psychiatric) problems, including:
- •
- confusion
- •
- new or worse aggressive behavior, hostility, anger, or irritability
- •
- new or worse psychosis (hearing or seeing things that are not real)
- •
- being suspicious or distrustful (believing things that are not true)
- •
- other unusual or extreme changes in behavior or mood
- Tell your healthcare provider right away if you have any new or worsening mental problems while using POTIGA.
- 5.
- Like other antiepileptic drugs, POTIGA may cause suicidal thoughts or actions in a very small number of people, about 1 in 500.
- Call a healthcare provider right away if you have any of these symptoms, especially if they are new, worse, or worry you:
- •
- thoughts about suicide or dying
- •
- attempt to commit suicide
- •
- new or worse depression
- •
- new or worse anxiety
- •
- feeling agitated or restless
- •
- panic attacks
- •
- trouble sleeping (insomnia)
- •
- new or worse irritability
- •
- acting aggressive, being angry, or violent
- •
- acting on dangerous impulses
- •
- an extreme increase in activity and talking (mania)
- •
- other unusual changes in behavior or mood
- Suicidal thoughts or actions can be caused by things other than medicines. If you have suicidal thoughts or actions, your healthcare provider may check for other causes.
How can I watch for early symptoms of suicidal thoughts and actions?
- •
- Pay attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings.
- •
- Keep all follow-up visits with your healthcare provider as scheduled.
Call your healthcare provider between visits as needed, especially if you are worried about symptoms.
What is POTIGA?
POTIGA is a prescription medicine that is used with other medicines to treat partial-onset seizures in adults with epilepsy when several other medicines have not worked well. POTIGA is used when the benefit of taking it is more important than the possible risk of vision loss.
POTIGA is a controlled substance (CV) because it can be abused or lead to drug dependence. Keep your POTIGA in a safe place to protect it from theft. Never give your POTIGA to anyone else because it may harm them. Selling or giving away this medicine is against the law.
It is not known if POTIGA is safe and effective in children under 18 years of age.
What should I tell my healthcare provider before taking POTIGA?
Before you take POTIGA, tell your healthcare provider if you:
- •
- have trouble urinating.
- •
- have an enlarged prostate.
- •
- have or have had depression, mood problems, or suicidal thoughts or behavior.
- •
- have heart problems, including a condition called long QT Syndrome, or have low potassium or magnesium in your blood.
- •
- have liver problems.
- •
- have kidney problems.
- •
- drink alcohol.
- •
- have any other medical conditions.
- •
- are pregnant or plan to become pregnant. It is not known if POTIGA will harm your unborn baby.
- •
- If you become pregnant while taking POTIGA, talk to your healthcare provider about registering with the North American Antiepileptic Drug Pregnancy Registry. The purpose of this registry is to collect information about the safety of medicines used to treat seizures during pregnancy. You can enroll in this registry by calling 1-888-233-2334.
- •
- are breastfeeding or plan to breastfeed. It is not known if POTIGA passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby if you take POTIGA. You and your healthcare provider should decide if you will take POTIGA or breastfeed. You should not do both.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Taking POTIGA with certain other medicines can affect each other, causing side effects.
Especially tell your healthcare provider if you take:
- •
- phenytoin (DILANTIN®, PHENYTEK®)
- •
- carbamazepine (CARBATROL®, TEGRETOL®, TEGRETOL®-XR, EQUETRO®, EPITOL®)
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take POTIGA?
- •
- Take POTIGA exactly as your healthcare provider tells you to take it. Your healthcare provider will tell you how much POTIGA to take and when to take it.
- •
- Your healthcare provider may change your dose of POTIGA. Do not change your dose without talking to your healthcare provider.
- •
- POTIGA can be taken with or without food.
- •
- Swallow POTIGA tablets whole. Do not break, crush, dissolve, or chew POTIGA tablets before swallowing.
- •
- If you take too much POTIGA, call your local Poison Control Center or go to the nearest hospital emergency room right away.
What should I avoid while taking POTIGA?
Do not drive, operate machinery, or do other dangerous activities until you know how POTIGA affects you. POTIGA can cause dizziness, sleepiness, double-vision, and blurred vision.
What are the possible side effects of POTIGA?
POTIGA may cause serious side effects, including:
- •
- See “What is the most important information I should know about POTIGA?”
- •
- Dizziness and sleepiness. These symptoms can increase when your dose of POTIGA is increased. See “What should I avoid while taking POTIGA?”
- •
- Changes in your heart rhythm and the electrical activity of your heart. Your healthcare provider should monitor your heart during treatment if you have a certain type of heart disease or take certain medications.
- •
- Drinking alcohol during treatment with POTIGA may increase the side effects that you get with POTIGA.
The most common side effects of POTIGA include:
- •
- dizziness
- •
- somnolence
- •
- sleepiness
- •
- tiredness
- •
- confusion
- •
- spinning sensation (vertigo)
- •
- tremor
- •
- problems with balance and muscle coordination, including trouble with walking and moving
- •
- blurred or double vision
- •
- trouble concentrating
- •
- memory problems
- •
- weakness
Tell your healthcare provider about any side effect that bothers you or that does not go away.
These are not all the possible side effects of POTIGA. Ask your healthcare provider or pharmacist for more information.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store POTIGA?
- •
- Store POTIGA at room temperature between 68°F and 77°F (20°C and 25°C).
- •
- Keep POTIGA and all medicines out of the reach of children.
General information about the safe and effective use of POTIGA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use POTIGA for a condition for which it was not prescribed. Do not give POTIGA to other people, even if they have the same symptoms you have. It may harm them.
This Medication Guide summarizes the most important information about POTIGA. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about POTIGA that is written for healthcare professionals.
For more information, go to www.potiga.com or call 1-877-3POTIGA (1-877-376-8442).
What are the ingredients in POTIGA?
Active ingredient: ezogabine
Inactive ingredients in all strengths: croscarmellose sodium, hypromellose, lecithin, magnesium stearate, microcrystalline cellulose, polyvinyl alcohol, talc, titanium dioxide, and xanthan gum
50-mg and 400-mg tablets also contain: carmine
50-mg, 300-mg, and 400-mg tablets also contain: FD&C Blue No 2
200-mg and 300-mg tablets also contain: iron oxide yellow
This Medication Guide has been approved by the U.S. Food and Drug Administration.
POTIGA is a registered trademark of Valeant Pharmaceuticals North America LLC.
The other brands listed are trademarks of their respective owners and are not trademarks of the GSK group of companies. The makers of these brands are not affiliated with and do not endorse the GSK group of companies or its products.
GlaxoSmithKline
Research Triangle Park, NC 27709
©2016 the GSK group of companies. All rights reserved.
May 2016
PTG:7MG
PRINCIPAL DISPLAY PANEL
NDC 0173-0810-59
Potiga®
(ezogabine) Tablets
50 mg
Rx only
CV
90 Tablets
Dispense the accompanying Medication Guide to each patient.
Store at 25°C (77°F); excursions permitted between 15°-30°C (59°-86°F). [See USP Controlled Room Temperature.]
Do not use if printed safety seal under cap is broken or missing.
Each tablet contains 50 mg ezogabine.
See prescribing information for dosage information.
POTIGA is a trademark of Valeant Pharmaceuticals North America.
GlaxoSmithKline, Research Triangle Park, NC 27709
Made in Singapore.
Rev. 9/15
A135854

PRINCIPAL DISPLAY PANEL
NDC 0173-0812-59
Potiga®
(ezogabine) Tablets
200 mg
Rx only
CV
90 Tablets
Dispense the accompanying Medication Guide to each patient.
Store at 25°C (77°F); excursions permitted between 15°-30°C (59°-86°F). [See USP Controlled Room Temperature.]
Do not use if printed safety seal under cap is broken or missing.
Each tablet contains 200 mg ezogabine.
See prescribing information for dosage information.
POTIGA is a trademark of Valeant Pharmaceuticals North America.
GlaxoSmithKline, Research Triangle Park, NC 27709
Made in Singapore.
Rev. 9/15
A135855
PRINCIPAL DISPLAY PANEL
NDC 0173-0813-59
Potiga®
(ezogabine) Tablets
300 mg
Rx only
CV
90 Tablets
Dispense the accompanying Medication Guide to each patient.
Store at 25°C (77°F); excursions permitted between 15°-30°C (59°-86°F). [See USP Controlled Room Temperature.]
Do not use if printed safety seal under cap is broken or missing.
Each tablet contains 300 mg ezogabine.
See prescribing information for dosage information.
POTIGA is a trademark of Valeant Pharmaceuticals North America.
GlaxoSmithKline, Research Triangle Park, NC 27709
Made in Singapore.
Rev. 9/15
A135856
PRINCIPAL DISPLAY PANEL
NDC 0173-0814-59
Potiga®
(ezogabine) Tablets
400 mg
Rx only
CV
90 Tablets
Dispense the accompanying Medication Guide to each patient.
Store at 25°C (77°F); excursions permitted between 15°-30°C (59°-86°F). [See USP Controlled Room Temperature.]
Do not use if printed safety seal under cap is broken or missing.
Each tablet contains 400 mg ezogabine.
See prescribing information for dosage information.
POTIGA is a trademark of Valeant Pharmaceuticals North America.
GlaxoSmithKline, Research Triangle Park, NC 27709
Made in Singapore.
Rev. 9/15
A135857
| POTIGA
ezogabine tablet, film coated |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| POTIGA
ezogabine tablet, film coated |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| POTIGA
ezogabine tablet, film coated |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| POTIGA
ezogabine tablet, film coated |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| Labeler - GlaxoSmithKline LLC (167380711) |