Label: FARXIGA- dapagliflozin tablet, film coated


- NDC Code(s): 50090-3481-0, 50090-3482-0
- Packager: A-S Medication Solutions
- This is a repackaged label.
- Source NDC Code(s): 0310-6205, 0310-6210
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated December 12, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FARXIGA safely and effectively. See full prescribing information for FARXIGA.
FARXIGA® (dapagliflozin) tablets, for oral use
Initial U.S. Approval: 2014RECENT MAJOR CHANGES
INDICATIONS AND USAGE
FARXIGA is a sodium-glucose cotransporter 2 (SGLT2) inhibitor indicated:
- •
- To reduce the risk of sustained eGFR decline, end-stage kidney disease, cardiovascular death, and hospitalization for heart failure in adults with chronic kidney disease at risk of progression. (1)
- •
- To reduce the risk of cardiovascular death, hospitalization for heart failure, and urgent heart failure visit in adults with heart failure. (1)
- •
- To reduce the risk of hospitalization for heart failure in adults with type 2 diabetes mellitus and either established cardiovascular disease or multiple cardiovascular risk factors. (1)
- •
- As an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. (1)
Limitations of use:
- •
- Not recommended for use to improve glycemic control in patients with type 1 diabetes mellitus. (1)
- •
- Not recommended for use to improve glycemic control in adults with type 2 diabetes mellitus with an eGFR less than 45 mL/min/1.73 m2. FARXIGA is likely to be ineffective in this setting based upon its mechanism of action. (1)
- •
- Not recommended for the treatment of chronic kidney disease in patients with polycystic kidney disease or patients requiring or with a recent history of immunosuppressive therapy for the treatment of kidney disease. FARXIGA is not expected to be effective in these populations. (1)
DOSAGE AND ADMINISTRATION
- •
- Assess volume status and correct volume depletion before initiating. (2.1)
eGFR
(mL/min/1.73 m2)Recommended Dose
eGFR 45 or greater
To improve glycemic control, the recommended starting dose is 5 mg orally once daily. Dose can be increased to 10 mg orally once daily for additional glycemic control.
For all other indications, the recommended starting dose is 10 mg orally once daily.
eGFR 25 to less than 45
10 mg orally once daily
eGFR less than 25
Initiation is not recommended; however, patients may continue 10 mg orally once daily to reduce the risk of eGFR decline, ESKD, CV death and hHF.
- •
- Withhold FARXIGA for at least 3 days, if possible, prior to major surgery or procedures associated with prolonged fasting. (2.3)
DOSAGE FORMS AND STRENGTHS
- •
- Tablets: 5 mg and 10 mg (3)
CONTRAINDICATIONS
- •
- History of serious hypersensitivity reaction to FARXIGA. (4)
WARNINGS AND PRECAUTIONS
- •
- Diabetic Ketoacidosis in Patients with Type 1 Diabetes Mellitus and Other Ketoacidosis: Consider ketone monitoring in patients with type 1 diabetes mellitus and consider ketone monitoring in others at risk for ketoacidosis, as indicated. Assess for ketoacidosis regardless of presenting blood glucose levels and discontinue FARXIGA if ketoacidosis is suspected. Monitor patients for resolution of ketoacidosis before restarting. (5.1)
- •
- Volume depletion: Before initiating FARXIGA, assess volume status and renal function in the elderly, patients with renal impairment or low systolic blood pressure, and in patients on diuretics. Monitor for signs and symptoms during therapy. (5.2)
- •
- Urosepsis and Pyelonephritis: Evaluate for signs and symptoms of urinary tract infections and treat promptly, if indicated. (5.3)
- •
- Hypoglycemia: Consider a lower dose of insulin or the insulin secretagogue to reduce the risk of hypoglycemia when used in combination with FARXIGA. (5.4)
- •
- Necrotizing Fasciitis of the Perineum (Fournier’s Gangrene): Serious, life-threatening cases have occurred in patients with diabetes, both females and males. Assess patients presenting with pain or tenderness, erythema, or swelling in the genital or perineal area, along with fever or malaise. If suspected, institute prompt treatment. (5.5)
- •
- Genital Mycotic Infections: Monitor and treat if indicated. (5.6)
ADVERSE REACTIONS
- •
- Most common adverse reactions (5% or greater incidence) were female genital mycotic infections, nasopharyngitis, and urinary tract infections. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See full prescribing information for information on drug interactions and interference of FARXIGA with laboratory tests. (7)
USE IN SPECIFIC POPULATIONS
- •
- Pregnancy: Advise females of the potential risk to a fetus especially during the second and third trimesters. (8.1)
- •
- Lactation: Not recommended when breastfeeding. (8.2)
- •
- Geriatrics: Higher incidence of adverse reactions related to hypotension. (8.5)
- •
- Renal Impairment: Higher incidence of adverse reactions related to volume depletion. (8.6)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Prior to Initiation of FARXIGA
2.2 Recommended Dosage
2.3 Temporary Interruption for Surgery
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Diabetic Ketoacidosis in Patients with Type 1 Diabetes Mellitus and Other Ketoacidosis
5.2 Volume Depletion
5.3 Urosepsis and Pyelonephritis
5.4 Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues
5.5 Necrotizing Fasciitis of the Perineum (Fournier’s Gangrene)
5.6 Genital Mycotic Infections
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Glycemic Control in Patients with Type 2 Diabetes Mellitus
14.2 Cardiovascular Outcomes in Patients with Type 2 Diabetes Mellitus
14.3 Chronic Kidney Disease
14.4 Heart Failure
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
FARXIGA (dapagliflozin) is indicated:
- •
- To reduce the risk of sustained eGFR decline, end-stage kidney disease, cardiovascular death, and hospitalization for heart failure in adults with chronic kidney disease at risk of progression.
- •
- To reduce the risk of cardiovascular death, hospitalization for heart failure, and urgent heart failure visit in adults with heart failure.
- •
- To reduce the risk of hospitalization for heart failure in adults with type 2 diabetes mellitus and either established cardiovascular disease or multiple cardiovascular risk factors.
- •
- As an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Limitations of Use
- •
- FARXIGA is not recommended for use to improve glycemic control in patients with type 1 diabetes mellitus [see Warnings and Precautions (5.1)].
- •
- FARXIGA is not recommended for use to improve glycemic control in adults with type 2 diabetes mellitus with an eGFR less than 45 mL/min/1.73 m2. FARXIGA is likely to be ineffective in this setting based upon its mechanism of action.
- •
- FARXIGA is not recommended for the treatment of chronic kidney disease in patients with polycystic kidney disease or patients requiring or with a recent history of immunosuppressive therapy for kidney disease. FARXIGA is not expected to be effective in these populations.
-
2 DOSAGE AND ADMINISTRATION
2.1 Prior to Initiation of FARXIGA
Assess renal function prior to initiation of FARXIGA therapy and then as clinically indicated [see Warnings and Precautions (5.2)].
Assess volume status. In patients with volume depletion, correct this condition before initiating FARXIGA [see Warnings and Precautions (5.2) and Use in Specific Populations (8.5, 8.6)].
2.2 Recommended Dosage
See Table 1 for dosage recommendations based on estimated glomerular filtration rate (eGFR).
Table 1: Recommended Dosage - *
- FARXIGA is not recommended for use to improve glycemic control in adults with type 2 diabetes mellitus with an eGFR less than 45 mL/min/1.73 m2. FARXIGA is likely to be ineffective in this setting based upon its mechanism of action.
eGFR
(mL/min/1.73 m2)Recommended Dose
eGFR 45 or greater
To improve glycemic control, the recommended starting dose is 5 mg orally once daily. Dose can be increased to 10 mg orally once daily for additional glycemic control*.
For all other indications, the recommended starting dose is 10 mg orally once daily.
eGFR 25 to less than 45
10 mg orally once daily*.
eGFR less than 25
Initiation is not recommended; however, patients may continue 10 mg orally once daily to reduce the risk of eGFR decline, ESKD, CV death and hHF.
hHF: hospitalization for heart failure, CV: Cardiovascular, ESKD: End Stage Kidney Disease
2.3 Temporary Interruption for Surgery
Withhold FARXIGA for at least 3 days, if possible, prior to major surgery or procedures associated with prolonged fasting. Resume FARXIGA when the patient is clinically stable and has resumed oral intake [see Warnings and Precautions (5.1) and Clinical Pharmacology (12.2)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
- •
- History of a serious hypersensitivity reaction to FARXIGA, such as anaphylactic reactions or angioedema [see Adverse Reactions (6.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Diabetic Ketoacidosis in Patients with Type 1 Diabetes Mellitus and Other Ketoacidosis
In patients with type 1 diabetes mellitus, FARXIGA significantly increases the risk of diabetic ketoacidosis, a life-threatening event, beyond the background rate. In placebo-controlled trials of patients with type 1 diabetes mellitus, the risk of ketoacidosis was markedly increased in patients who received sodium-glucose cotransporter 2 (SGLT2) inhibitors compared to patients who received placebo. FARXIGA is not indicated for glycemic control in patients with type 1 diabetes mellitus.
Type 2 diabetes mellitus and pancreatic disorders (e.g., history of pancreatitis or pancreatic surgery) are also risk factors for ketoacidosis. There have been postmarketing reports of fatal events of ketoacidosis in patients with type 2 diabetes mellitus using SGLT2 inhibitors, including FARXIGA.
Precipitating conditions for diabetic ketoacidosis or other ketoacidosis include under-insulinization due to insulin dose reduction or missed insulin doses, acute febrile illness, reduced caloric intake, ketogenic diet, surgery, volume depletion, and alcohol abuse.
Signs and symptoms are consistent with dehydration and severe metabolic acidosis and include nausea, vomiting, abdominal pain, generalized malaise, and shortness of breath. Blood glucose levels at presentation may be below those typically expected for diabetic ketoacidosis (e.g., less than 250 mg/dL). Ketoacidosis and glucosuria may persist longer than typically expected. Urinary glucose excretion persists for 3 days after discontinuing FARXIGA [see Clinical Pharmacology (12.2)]; however, there have been postmarketing reports of ketoacidosis and/or glucosuria lasting greater than 6 days and some up to 2 weeks after discontinuation of SGLT2 inhibitors.
Consider ketone monitoring in patients with type 1 diabetes mellitus and consider ketone monitoring in others at risk for ketoacidosis if indicated by the clinical situation. Assess for ketoacidosis regardless of presenting blood glucose levels in patients who present with signs and symptoms consistent with severe metabolic acidosis. If ketoacidosis is suspected, discontinue FARXIGA, promptly evaluate, and treat ketoacidosis, if confirmed.Monitor patients for resolution of ketoacidosis before restarting FARXIGA.
Withhold FARXIGA, if possible, in temporary clinical situations that could predispose patients to ketoacidosis. Resume FARXIGA when the patient is clinically stable and has resumed oral intake [see Dosage and Administration (2.3)].
Educate all patients on the signs and symptoms of ketoacidosis and instruct patients to discontinue FARXIGA and seek medical attention immediately if signs and symptoms occur.
5.2 Volume Depletion
FARXIGA can cause intravascular volume depletion which may sometimes manifest as symptomatic hypotension or acute transient changes in creatinine. There have been post-marketing reports of acute kidney injury, some requiring hospitalization and dialysis, in patients with type 2 diabetes mellitus receiving SGLT2 inhibitors, including FARXIGA. Patients with impaired renal function (eGFR less than 60 mL/min/1.73 m2), elderly patients, or patients on loop diuretics may be at increased risk for volume depletion or hypotension. Before initiating FARXIGA in patients with one or more of these characteristics, assess volume status and renal function. Monitor for signs and symptoms of hypotension, and renal function after initiating therapy.
5.3 Urosepsis and Pyelonephritis
Serious urinary tract infections including urosepsis and pyelonephritis requiring hospitalization have been reported in patients receiving SGLT2 inhibitors, including FARXIGA. Treatment with SGLT2 inhibitors increases the risk for urinary tract infections. Evaluate patients for signs and symptoms of urinary tract infections and treat promptly, if indicated [see Adverse Reactions (6)].
5.4 Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues
Insulin and insulin secretagogues are known to cause hypoglycemia. FARXIGA may increase the risk of hypoglycemia when combined with insulin or an insulin secretagogue [see Adverse Reactions (6.1)]. Therefore, a lower dose of insulin or insulin secretagogue may be required to minimize the risk of hypoglycemia when these agents are used in combination with FARXIGA.
5.5 Necrotizing Fasciitis of the Perineum (Fournier’s Gangrene)
Reports of necrotizing fasciitis of the perineum (Fournier’s Gangrene), a rare but serious and life-threatening necrotizing infection requiring urgent surgical intervention, have been identified in postmarketing surveillance in patients with diabetes mellitus receiving SGLT2 inhibitors, including FARXIGA. Cases have been reported in both females and males. Serious outcomes have included hospitalization, multiple surgeries, and death.
Patients treated with FARXIGA presenting with pain or tenderness, erythema, or swelling in the genital or perineal area, along with fever or malaise, should be assessed for necrotizing fasciitis. If suspected, start treatment immediately with broad-spectrum antibiotics and, if necessary, surgical debridement. Discontinue FARXIGA, closely monitor blood glucose levels, and provide appropriate alternative therapy for glycemic control.
5.6 Genital Mycotic Infections
FARXIGA increases the risk of genital mycotic infections. Patients with a history of genital mycotic infections were more likely to develop genital mycotic infections [see Adverse Reactions (6.1)]. Monitor and treat appropriately.
-
6 ADVERSE REACTIONS
The following important adverse reactions are described below and elsewhere in the labeling:
- •
- Diabetic Ketoacidosis in Patients with Type 1 Diabetes Mellitus and Other Ketoacidosis [see Warnings and Precautions (5.1)]
- •
- Volume Depletion [see Warnings and Precautions (5.2)]
- •
- Urosepsis and Pyelonephritis [see Warnings and Precautions (5.3)]
- •
- Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues [see Warnings and Precautions (5.4)]
- •
- Necrotizing Fasciitis of the Perineum (Fournier’s Gangrene) [see Warnings and Precautions (5.5)]
- •
- Genital Mycotic Infections [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
FARXIGA has been evaluated in clinical trials in patients with type 2 diabetes mellitus, in patients with heart failure, and in patients with chronic kidney disease. The overall safety profile of FARXIGA was consistent across the studied indications. Severe hypoglycemia and diabetic ketoacidosis (DKA) were observed only in patients with diabetes mellitus.
Clinical Trials in Patients with Type 2 Diabetes Mellitus
Pool of 12 Placebo-Controlled Studies for FARXIGA 5 and 10 mg for Glycemic Control
The data in Table 2 is derived from 12 glycemic control placebo-controlled studies in patients with type 2 diabetes mellitus ranging from 12 to 24 weeks. In 4 studies FARXIGA was used as monotherapy, and in 8 studies FARXIGA was used as add-on to background antidiabetic therapy or as combination therapy with metformin [see Clinical Studies (14.1)].
These data reflect exposure of 2338 patients to FARXIGA with a mean exposure duration of 21 weeks. Patients received placebo (N=1393), FARXIGA 5 mg (N=1145), or FARXIGA 10 mg (N=1193) once daily. The mean age of the population was 55 years and 2% were older than 75 years of age. Fifty percent (50%) of the population were male; 81% were White, 14% were Asian, and 3% were Black or African American. At baseline, the population had diabetes for an average of 6 years, had a mean hemoglobin A1c (HbA1c) of 8.3%, and 21% had established microvascular complications of diabetes. Baseline renal function was normal or mildly impaired in 92% of patients and moderately impaired in 8% of patients (mean eGFR 86 mL/min/1.73 m2).
Table 2 shows common adverse reactions associated with the use of FARXIGA. These adverse reactions were not present at baseline, occurred more commonly on FARXIGA than on placebo, and occurred in at least 2% of patients treated with either FARXIGA 5 mg or FARXIGA 10 mg.
Table 2: Adverse Reactions in Placebo-Controlled Studies of Glycemic Control Reported in ≥2% of Patients Treated with FARXIGA Adverse Reaction % of Patients Pool of 12 Placebo-Controlled Studies Placebo
N=1393FARXIGA 5 mg
N=1145FARXIGA 10 mg
N=1193- *
- Genital mycotic infections include the following adverse reactions, listed in order of frequency reported for females: vulvovaginal mycotic infection, vaginal infection, vulvovaginal candidiasis, vulvovaginitis, genital infection, genital candidiasis, fungal genital infection, vulvitis, genitourinary tract infection, vulval abscess, and vaginitis bacterial. (N for females: Placebo=677, FARXIGA 5 mg=581, FARXIGA 10 mg=598).
- †
- Urinary tract infections include the following adverse reactions, listed in order of frequency reported: urinary tract infection, cystitis, Escherichia urinary tract infection, genitourinary tract infection, pyelonephritis, trigonitis, urethritis, kidney infection, and prostatitis.
- ‡
- Increased urination includes the following adverse reactions, listed in order of frequency reported: pollakiuria, polyuria, and urine output increased.
- §
- Genital mycotic infections include the following adverse reactions, listed in order of frequency reported for males: balanitis, fungal genital infection, balanitis candida, genital candidiasis, genital infection male, penile infection, balanoposthitis, balanoposthitis infective, genital infection, and posthitis. (N for males: Placebo=716, FARXIGA 5 mg=564, FARXIGA 10 mg=595).
Female genital mycotic infections*
1.5
8.4
6.9
Nasopharyngitis
6.2
6.6
6.3
Urinary tract infections†
3.7
5.7
4.3
Back pain
3.2
3.1
4.2
Increased urination‡
1.7
2.9
3.8
Male genital mycotic infections§
0.3
2.8
2.7
Nausea
2.4
2.8
2.5
Influenza
2.3
2.7
2.3
Dyslipidemia
1.5
2.1
2.5
Constipation
1.5
2.2
1.9
Discomfort with urination
0.7
1.6
2.1
Pain in extremity
1.4
2.0
1.7
Pool of 13 Placebo-Controlled Studies for FARXIGA 10 mg for Glycemic Control
FARXIGA 10 mg was also evaluated in a larger glycemic control placebo-controlled study pool in patients with type 2 diabetes mellitus. This pool combined 13 placebo-controlled studies, including 3 monotherapy studies, 9 add-on to background antidiabetic therapy studies, and an initial combination with metformin study. Across these 13 studies, 2360 patients were treated once daily with FARXIGA 10 mg for a mean duration of exposure of 22 weeks. The mean age of the population was 59 years and 4% were older than 75 years. Fifty-eight percent (58%) of the population were male; 84% were White, 9% were Asian, and 3% were Black or African American. At baseline, the population had diabetes for an average of 9 years, had a mean HbA1c of 8.2%, and 30% had established microvascular disease. Baseline renal function was normal or mildly impaired in 88% of patients and moderately impaired in 11% of patients (mean eGFR 82 mL/min/1.73 m2).
Volume Depletion
FARXIGA causes an osmotic diuresis, which may lead to a reduction in intravascular volume. Adverse reactions related to volume depletion (including reports of dehydration, hypovolemia, orthostatic hypotension, or hypotension) in patients with type 2 diabetes mellitus for the 12-study and 13-study, short-term, placebo-controlled pools and for the DECLARE study are shown in Table 3 [see Warnings and Precautions (5.2)].
Table 3: Adverse Reactions Related to Volume Depletion* in Clinical Studies in Patients with Type 2 Diabetes Mellitus with FARXIGA - *
- Volume depletion includes reports of dehydration, hypovolemia, orthostatic hypotension, or hypotension.
Pool of 12 Placebo-Controlled
StudiesPool of 13 Placebo-Controlled
StudiesDECLARE Study
Placebo
FARXIGA
5 mgFARXIGA
10 mgPlacebo
FARXIGA
10 mgPlacebo
FARXIGA
10 mgOverall population N (%)
N=1393
5
(0.4%)N=1145
7
(0.6%)N=1193
9
(0.8%)N=2295
17
(0.7%)N=2360
27
(1.1%)N=8569
207
(2.4%)N=8574
213
(2.5%)Patient Subgroup n (%)
Patients on loop diuretics
n=55
1
(1.8%)n=40
0
n=31
3
(9.7%)n=267
4
(1.5%)n=236
6
(2.5%)n=934
57
(6.1%)n=866
57
(6.6%)Patients with moderate renal impairment with eGFR ≥30 and <60 mL/min/1.73 m2
n=107
2
(1.9%)n=107
1
(0.9%)n=89
1
(1.1%)n=268
4
(1.5%)n=265
5
(1.9%)n=658
30
(4.6%)n=604
35
(5.8%)Patients ≥65 years of age
n=276
1
(0.4%)n=216
1
(0.5%)n=204
3
(1.5%)n=711
6
(0.8%)n=665
11
(1.7%)n=3950
121
(3.1%)n=3948
117
(3.0%)Hypoglycemia
The frequency of hypoglycemia by study in patients with type 2 diabetes mellitus [see Clinical Studies (14.1)] is shown in Table 4. Hypoglycemia was more frequent when FARXIGA was added to sulfonylurea or insulin [see Warnings and Precautions (5.4)].
Table 4: Incidence of Severe Hypoglycemia* and Hypoglycemia with Glucose < 54 mg/dL† in Controlled Glycemic Control Clinical Studies in Patients with Type 2 Diabetes Mellitus Placebo/Active Control FARXIGA
5 mgFARXIGA
10 mg- *
- Severe episodes of hypoglycemia were defined as episodes of severe impairment in consciousness or behavior, requiring external (third party) assistance, and with prompt recovery after intervention regardless of glucose level.
- †
- Episodes of hypoglycemia with glucose <54 mg/dL (3 mmol/L) were defined as reported episodes of hypoglycemia meeting the glucose criteria that did not also qualify as a severe episode.
- ‡
- OAD = oral antidiabetic therapy.
Monotherapy (24 weeks)
N=75
N=64
N=70
Severe [n (%)]
0
0
0
Glucose <54 mg/dL [n (%)]
0
0
0
Add-on to Metformin (24 weeks)
N=137
N=137
N=135
Severe [n (%)]
0
0
0
Glucose <54 mg/dL [n (%)]
0
0
0
Add-on to Glimepiride (24 weeks)
N=146
N=145
N=151
Severe [n (%)]
0
0
0
Glucose <54 mg/dL [n (%)]
1 (0.7)
3 (2.1)
5 (3.3)
Add-on to Metformin and a Sulfonylurea (24 Weeks)
N=109
-
N=109
Severe [n (%)]
0
-
0
Glucose <54 mg/dL [n (%)]
3 (2.8)
-
7 (6.4)
Add-on to Pioglitazone (24 weeks)
N=139
N=141
N=140
Severe [n (%)]
0
0
0
Glucose <54 mg/dL [n (%)]
0
1 (0.7)
0
Add-on to DPP4 inhibitor (24 weeks)
N=226
–
N=225
Severe [n (%)]
0
–
1 (0.4)
Glucose <54 mg/dL [n (%)]
1 (0.4)
–
1 (0.4)
Add-on to Insulin with or without other OADs‡ (24 weeks)
N=197
N=212
N=196
Severe [n (%)]
1 (0.5)
2 (0.9)
2 (1.0)
Glucose <54 mg/dL [n (%)]
43 (21.8)
55 (25.9)
45 (23.0)
In the DECLARE study [see Clinical Studies (14.2)], severe events of hypoglycemia were reported in 58 (0.7%) out of 8574 patients treated with FARXIGA and 83 (1.0%) out of 8569 patients treated with placebo.
Genital Mycotic Infections
In the glycemic control trials, genital mycotic infections were more frequent with FARXIGA treatment. Genital mycotic infections were reported in 0.9% of patients on placebo, 5.7% on FARXIGA 5 mg, and 4.8% on FARXIGA 10 mg, in the 12-study placebo-controlled pool. Discontinuation from study due to genital infection occurred in 0% of placebo-treated patients and 0.2% of patients treated with FARXIGA 10 mg. Infections were more frequently reported in females than in males (see Table 2). The most frequently reported genital mycotic infections were vulvovaginal mycotic infections in females and balanitis in males. Patients with a history of genital mycotic infections were more likely to have a genital mycotic infection during the study than those with no prior history (10.0%, 23.1%, and 25.0% versus 0.8%, 5.9%, and 5.0% on placebo, FARXIGA 5 mg, and FARXIGA 10 mg, respectively). In the DECLARE study [see Clinical Studies (14.2)], serious genital mycotic infections were reported in <0.1% of patients treated with FARXIGA and <0.1% of patients treated with placebo. Genital mycotic infections that caused study drug discontinuation were reported in 0.9% of patients treated with FARXIGA and <0.1% of patients treated with placebo.
Hypersensitivity Reactions
Hypersensitivity reactions (e.g., angioedema, urticaria, hypersensitivity) were reported with FARXIGA treatment. In glycemic control studies, serious anaphylactic reactions and severe cutaneous adverse reactions and angioedema were reported in 0.2% of comparator-treated patients and 0.3% of FARXIGA-treated patients. If hypersensitivity reactions occur, discontinue use of FARXIGA; treat per standard of care and monitor until signs and symptoms resolve.
Ketoacidosis in Patients with Diabetes Mellitus
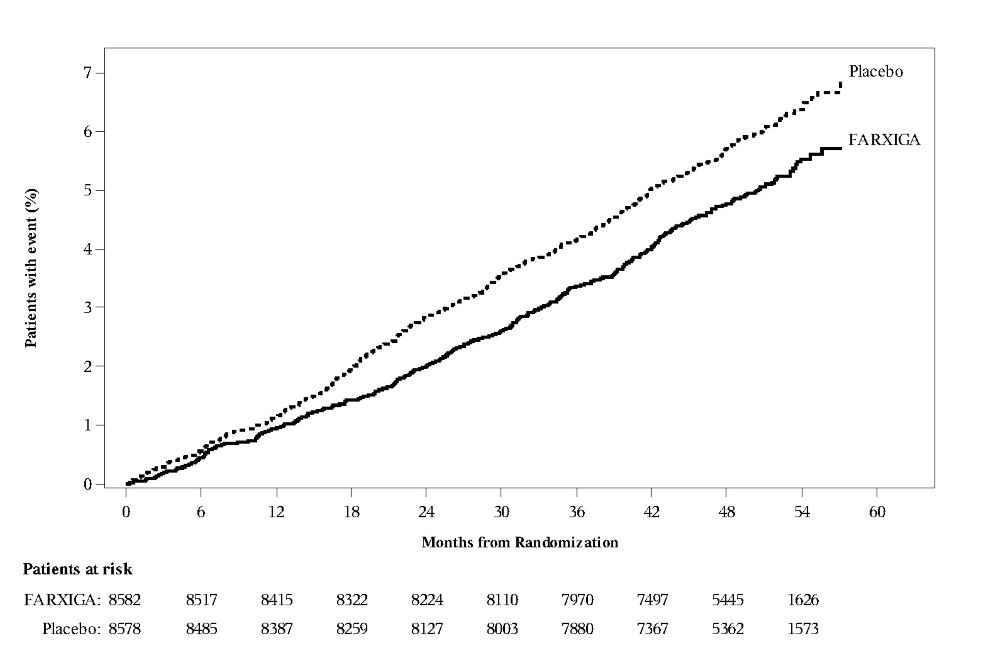
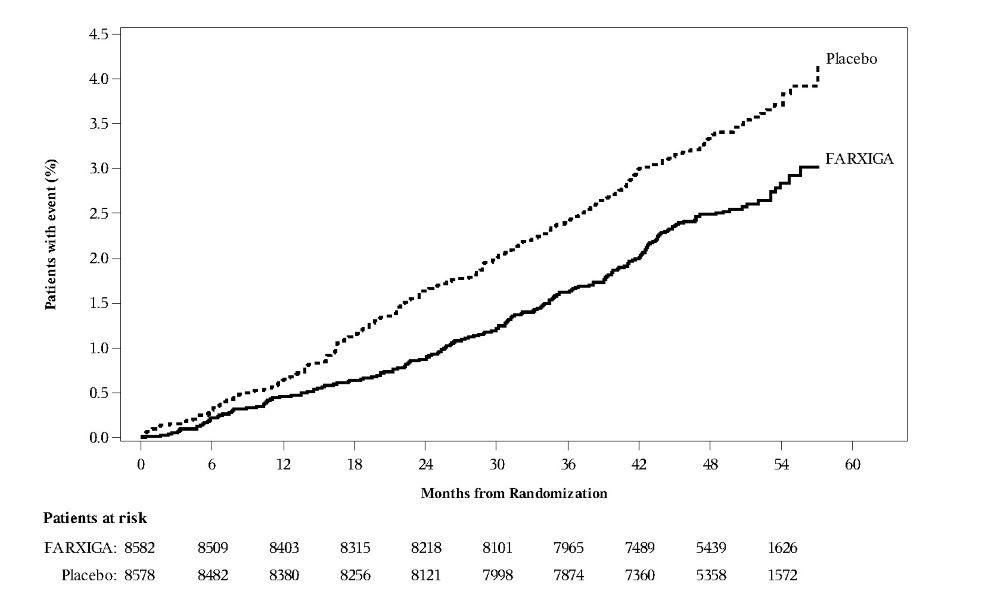
In the DECLARE study [see Clinical Studies (14.2)], events of diabetic ketoacidosis (DKA) were reported in 27 out of 8574 patients in the FARXIGA-treated group and 12 out of 8569 patients in the placebo group. The events were evenly distributed over the study period.
Laboratory Tests
Increases in Serum Creatinine and Decreases in eGFR
Initiation of SGLT2 inhibitors, including FARXIGA causes a small increase in serum creatinine and decrease in eGFR. These changes in serum creatinine and eGFR generally occur within two weeks of starting therapy and then stabilize regardless of baseline kidney function. Changes that do not fit this pattern should prompt further evaluation to exclude the possibility of acute kidney injury [see Warnings and Precautions (5.2)]. In two studies that included patients with type 2 diabetes mellitus with moderate renal impairment, the acute effect on eGFR reversed after treatment discontinuation, suggesting acute hemodynamic changes may play a role in the renal function changes observed with FARXIGA.
Increase in Hematocrit
In the pool of 13 placebo-controlled studies of glycemic control, increases from baseline in mean hematocrit values were observed in FARXIGA-treated patients starting at Week 1 and continuing up to Week 16, when the maximum mean difference from baseline was observed. At Week 24, the mean changes from baseline in hematocrit were −0.33% in the placebo group and 2.30% in the FARXIGA 10 mg group. By Week 24, hematocrit values >55% were reported in 0.4% of placebo-treated patients and 1.3% of FARXIGA 10 mg-treated patients.
Increase in Low-Density Lipoprotein Cholesterol
In the pool of 13 placebo-controlled studies of glycemic control, changes from baseline in mean lipid values were reported in FARXIGA-treated patients compared to placebo-treated patients. Mean percent changes from baseline at Week 24 were 0.0% versus 2.5% for total cholesterol, and -1.0% versus 2.9% for LDL cholesterol in the placebo and FARXIGA 10 mg groups, respectively. In the DECLARE study [see Clinical Studies (14.2)], mean changes from baseline after 4 years were 0.4 mg/dL versus -4.1 mg/dL for total cholesterol, and -2.5 mg/dL versus -4.4 mg/dL for LDL cholesterol, in FARXIGA-treated and the placebo groups, respectively.
Decrease in Serum Bicarbonate
In a study of concomitant therapy of FARXIGA 10 mg with exenatide extended-release (on a background of metformin), four patients (1.7%) on concomitant therapy had a serum bicarbonate value of less than or equal to 13 mEq/L compared to one each (0.4%) in the FARXIGA and exenatide-extended release treatment groups [see Warnings and Precautions (5.1)].
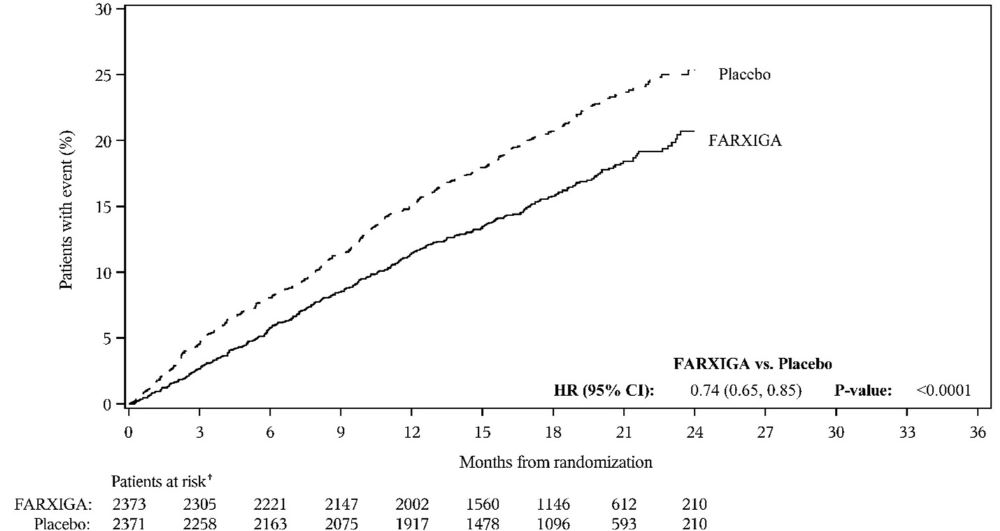
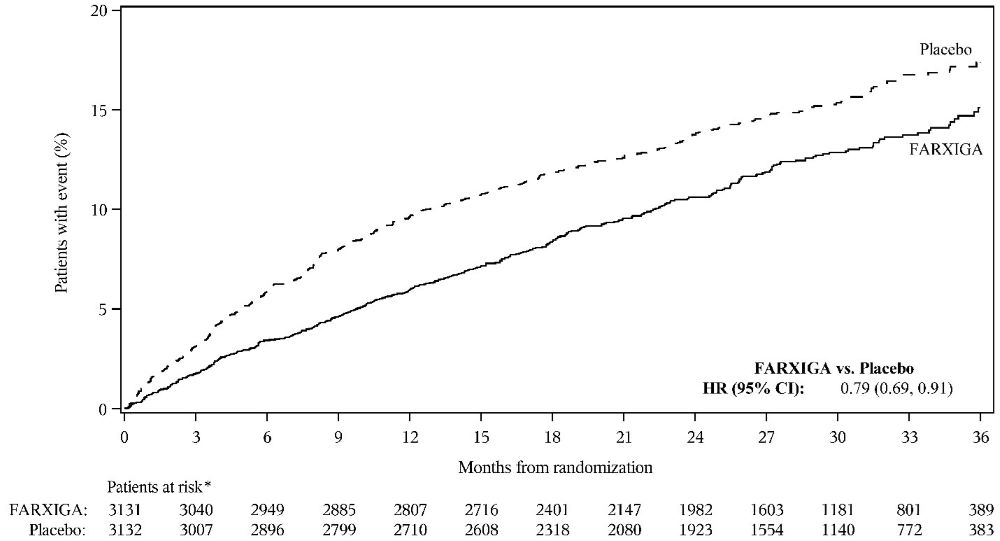
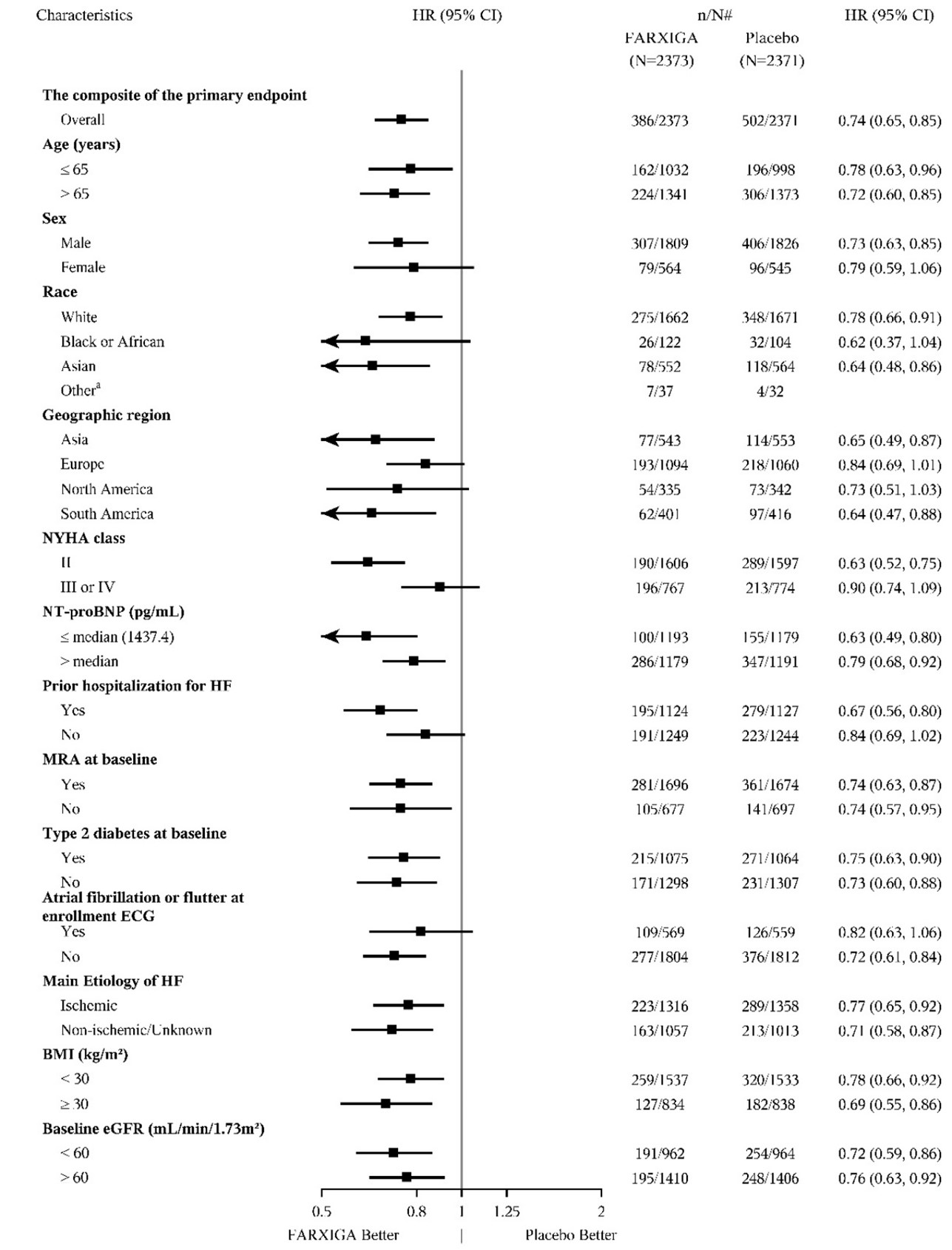
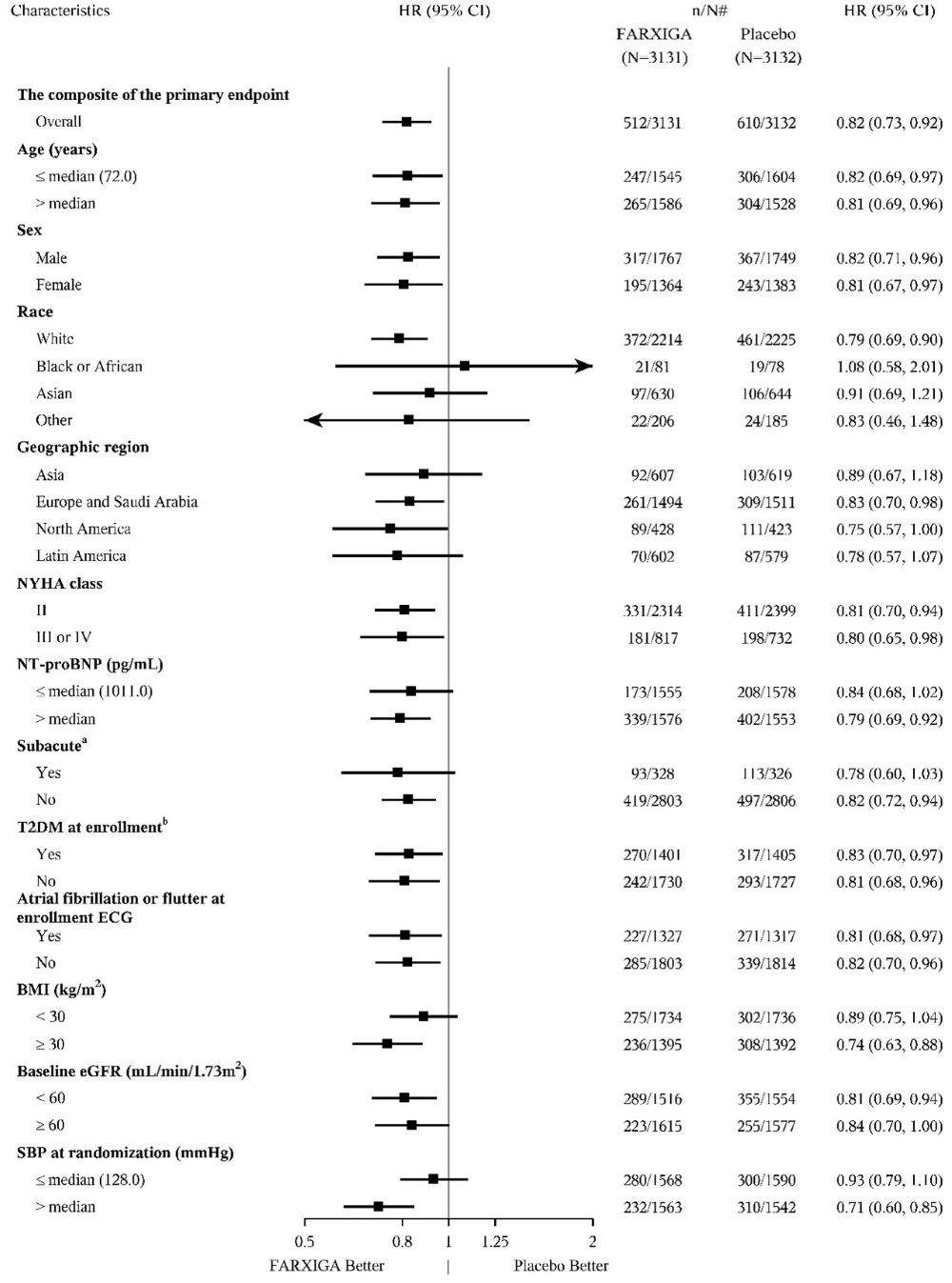
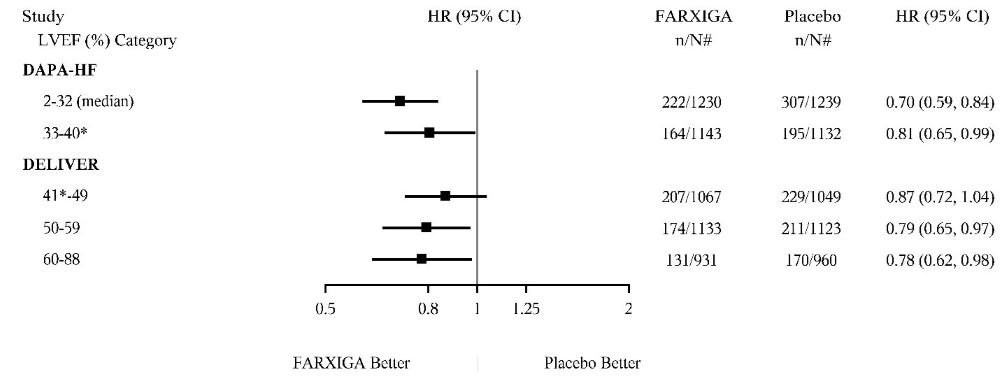
DAPA-HF and DELIVER Heart Failure Studies
No new adverse reactions were identified in the DAPA-HF and DELIVER heart failure studies.
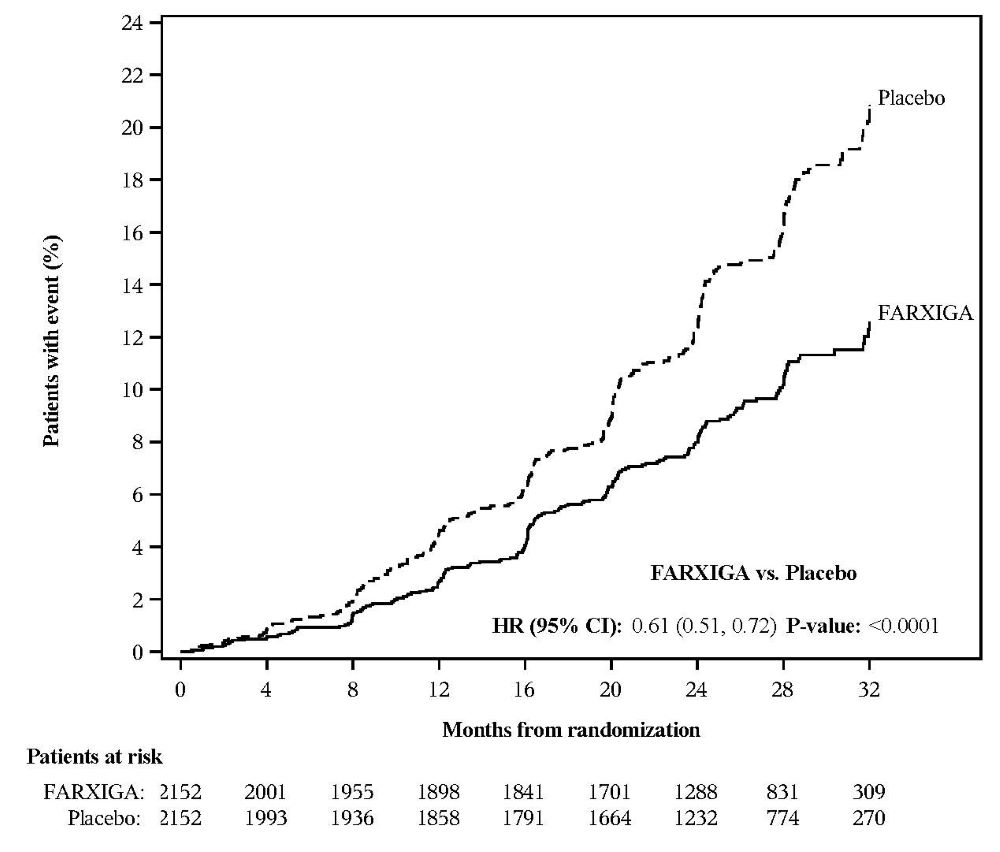
DAPA-CKD Chronic Kidney Disease Study
No new adverse reactions were identified in the DAPA-CKD study in patients with chronic kidney disease.
6.2 Postmarketing Experience
Additional adverse reactions have been identified during post-approval use of FARXIGA in patients with diabetes mellitus. Because these reactions are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Infections: Necrotizing fasciitis of the perineum (Fournier’s Gangrene), urosepsis and pyelonephritis
Metabolism and Nutrition Disorders: Ketoacidosis
Renal and Urinary Disorders: Acute kidney injury
Skin and Subcutaneous Tissue Disorders: Rash
-
7 DRUG INTERACTIONS
Table 5: Clinically Relevant Interactions with FARXIGA Insulin or Insulin Secretagogues
Clinical Impact
The risk of hypoglycemia may be increased when FARXIGA is used concomitantly with insulin or insulin secretagogues (e.g., sulfonylurea) [see Warnings and Precautions (5.4)].
Intervention
Concomitant use may require lower doses of insulin or the insulin secretagogue to reduce the risk of hypoglycemia.
Lithium
Clinical Impact
Concomitant use of an SGLT2 inhibitor with lithium may decrease serum lithium concentrations.
Intervention
Monitor serum lithium concentration more frequently during FARXIGA initiation and dosage changes.
Positive Urine Glucose Test
Clinical Impact
SGLT2 inhibitors increase urinary glucose excretion and will lead to positive urine glucose tests.
Intervention
Monitoring glycemic control with urine glucose tests is not recommended in patients taking SGLT2 inhibitors. Use alternative methods to monitor glycemic control.
Interference with 1,5-anhydroglucitol (1,5-AG) Assay
Clinical Impact
Measurements of 1,5-AG are unreliable in assessing glycemic control in patients taking SGLT2 inhibitors.
Intervention
Monitoring glycemic control with 1,5-AG assay is not recommended. Use alternative methods to monitor glycemic control.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on animal data showing adverse renal effects, FARXIGA is not recommended during the second and third trimesters of pregnancy.
Limited data with FARXIGA in pregnant women are not sufficient to determine drug-associated risk for major birth defects or miscarriage. There are risks to the mother and fetus associated with poorly controlled diabetes and untreated heart failure in pregnancy (see Clinical Considerations).
In animal studies, adverse renal pelvic and tubule dilatations, that were not fully reversible, were observed in rats when dapagliflozin was administered during a period of renal development corresponding to the late second and third trimesters of human pregnancy, at all doses tested; the lowest of which provided an exposure 15-times the 10 mg clinical dose (see Data).
The estimated background risk of major birth defects is 6 to 10% in women with pre-gestational diabetes with a HbA1c greater than 7% and has been reported to be as high as 20 to 25% in women with HbA1c greater than 10%. The estimated background risk of miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryofetal risk
Poorly controlled diabetes in pregnancy increases the maternal risk for diabetic ketoacidosis, preeclampsia, spontaneous abortions, preterm delivery and delivery complications. Poorly controlled diabetes increases the fetal risk for major birth defects, stillbirth, and macrosomia related morbidity.
Data
Animal Data
Dapagliflozin dosed directly to juvenile rats from postnatal day (PND) 21 until PND 90 at doses of 1, 15, or 75 mg/kg/day, increased kidney weights and increased the incidence of renal pelvic and tubular dilatations at all dose levels. Exposure at the lowest dose tested was 15-times the 10 mg clinical dose (based on AUC). The renal pelvic and tubular dilatations observed in juvenile animals did not fully reverse within a 1-month recovery period.
In a prenatal and postnatal development study, dapagliflozin was administered to maternal rats from gestation day 6 through lactation day 21 at doses of 1, 15, or 75 mg/kg/day, and pups were indirectly exposed in utero and throughout lactation. Increased incidence or severity of renal pelvic dilatation was observed in 21-day-old pups offspring of treated dams at 75 mg/kg/day (maternal and pup dapagliflozin exposures were 1415-times and 137-times, respectively, the human values at the 10 mg clinical dose, based on AUC). Dose-related reductions in pup body weights were observed at greater or equal to 29-times the 10 mg clinical dose (based on AUC). No adverse effects on developmental endpoints were noted at 1 mg/kg/day (19-times the 10 mg clinical dose, based on AUC). These outcomes occurred with drug exposure during periods of renal development in rats that corresponds to the late second and third trimester of human development.
In embryofetal development studies in rats and rabbits, dapagliflozin was administered throughout organogenesis, corresponding to the first trimester of human pregnancy. In rats, dapagliflozin was neither embryolethal nor teratogenic at doses up to 75 mg/kg/day (1441-times the 10 mg clinical dose, based on AUC). Dose-related effects on the rat fetus (structural abnormalities and reduced body weight) occurred only at higher dosages, equal to or greater than 150 mg/kg (more than 2344-times the 10 mg clinical dose, based on AUC), which were associated with maternal toxicity. No developmental toxicities were observed in rabbits at doses up to 180 mg/kg/day (1191-times the 10 mg clinical dose, based on AUC).
8.2 Lactation
Risk Summary
There is no information regarding the presence of dapagliflozin in human milk, the effects on the breastfed infant, or the effects on milk production. Dapagliflozin is present in the milk of lactating rats (see Data). However, due to species-specific differences in lactation physiology, the clinical relevance of these data is not clear. Since human kidney maturation occurs in utero and during the first 2 years of life when lactational exposure may occur, there may be risk to the developing human kidney.
Because of the potential for serious adverse reactions in breastfed infants, advise women that use of FARXIGA is not recommended while breastfeeding.
Data
Dapagliflozin was present in rat milk at a milk/plasma ratio of 0.49, indicating that dapagliflozin and its metabolites are transferred into milk at a concentration that is approximately 50% of that in maternal plasma. Juvenile rats directly exposed to dapagliflozin showed risk to the developing kidney (renal pelvic and tubular dilatations) during maturation.
8.4 Pediatric Use
Safety and effectiveness of FARXIGA in pediatric patients under 18 years of age have not been established.
8.5 Geriatric Use
No FARXIGA dosage change is recommended based on age.
A total of 1424 (24%) of the 5936 FARXIGA-treated patients were 65 years and older and 207 (3.5%) patients were 75 years and older in a pool of 21 double-blind, controlled, clinical studies assessing the efficacy of FARXIGA in improving glycemic control in type 2 diabetes mellitus. After controlling for level of renal function (eGFR), efficacy was similar for patients under age 65 years and those 65 years and older. In patients ≥65 years of age, a higher proportion of patients treated with FARXIGA for glycemic control had adverse reactions of hypotension [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
In the DAPA-CKD, DAPA-HF and DELIVER studies, safety and efficacy were similar for patients age 65 years and younger and those older than 65. In the DAPA-HF study, 2714 (57%) out of 4744 patients with HFrEF were older than 65 years. In the DELIVER study, 4759 (76%) out of 6263 patients with heart failure (LVEF >40%) were older than 65 years. In the DAPA-CKD study, 1818 (42%) out of 4304 patients with CKD were older than 65 years.
8.6 Renal Impairment
FARXIGA was evaluated in 4304 patients with chronic kidney disease (eGFR 25 to 75 mL/min/1.73 m2) in the DAPA-CKD study. FARXIGA was also evaluated in 1926 patients with an eGFR of 30 to 60 mL/min/1.73 m2 in the DAPA-HF study. The safety profile of FARXIGA across eGFR subgroups in these studies was consistent with the known safety profile [see Adverse Reactions (6.1) and Clinical Studies (14.3 and 14.4)].
FARXIGA was evaluated in two glycemic control studies that included patients with type 2 diabetes mellitus with moderate renal impairment (an eGFR of 45 to less than 60 mL/min/1.73 m2 [see Clinical Studies (14.1)], and an eGFR of 30 to less than 60 mL/min/1.73 m2, respectively). Patients with diabetes and renal impairment using FARXIGA may be more likely to experience hypotension and may be at higher risk for acute kidney injury secondary to volume depletion. In the study of patients with an eGFR 30 to less than 60 mL/min/1.73 m2, 13 patients receiving FARXIGA experienced bone fractures compared to none receiving placebo. Use of FARXIGA for glycemic control in patients without established CV disease or CV risk factors is not recommended when eGFR is less than 45 mL/min/1.73 m2 [see Dosage and Administration (2.2)].
Efficacy and safety studies with FARXIGA did not enroll patients with an eGFR less than 25 mL/min/1.73 m2 or on dialysis.
8.7 Hepatic Impairment
No dose adjustment is recommended for patients with mild, moderate, or severe hepatic impairment. However, the benefit-risk for the use of dapagliflozin in patients with severe hepatic impairment should be individually assessed since the safety and efficacy of dapagliflozin have not been specifically studied in this population [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There were no reports of overdose during the clinical development program for FARXIGA.
In the event of an overdose, contact the Poison Control Center. It is also reasonable to employ supportive measures as dictated by the patient’s clinical status. The removal of dapagliflozin by hemodialysis has not been studied.
-
11 DESCRIPTION
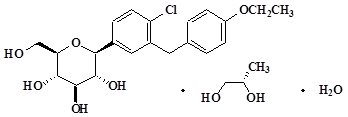
Dapagliflozin, an inhibitor of SGLT2, is described chemically as D-glucitol, 1,5-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-, (1S)-, compounded with (2S)-1,2-propanediol, hydrate (1:1:1). The empirical formula is C21H25ClO6•C3H8O2•H2O and the molecular weight is 502.98. The structural formula is:
FARXIGA is available as a film-coated tablet for oral administration containing the equivalent of 5 mg dapagliflozin as dapagliflozin propanediol or the equivalent of 10 mg dapagliflozin as dapagliflozin propanediol, and the following inactive ingredients: microcrystalline cellulose, anhydrous lactose, crospovidone, silicon dioxide, and magnesium stearate. In addition, the film coating contains the following inactive ingredients: polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and yellow iron oxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Sodium-glucose cotransporter 2 (SGLT2), expressed in the proximal renal tubules, is responsible for the majority of the reabsorption of filtered glucose from the tubular lumen. Dapagliflozin is an inhibitor of SGLT2. By inhibiting SGLT2, dapagliflozin reduces reabsorption of filtered glucose and thereby promotes urinary glucose excretion.
Dapagliflozin also reduces sodium reabsorption and increases the delivery of sodium to the distal tubule. This may influence several physiological functions including, but not restricted to, lowering both pre- and afterload of the heart and downregulation of sympathetic activity, and decreased intraglomerular pressure which is believed to be mediated by increased tubuloglomerular feedback.
12.2 Pharmacodynamics
General
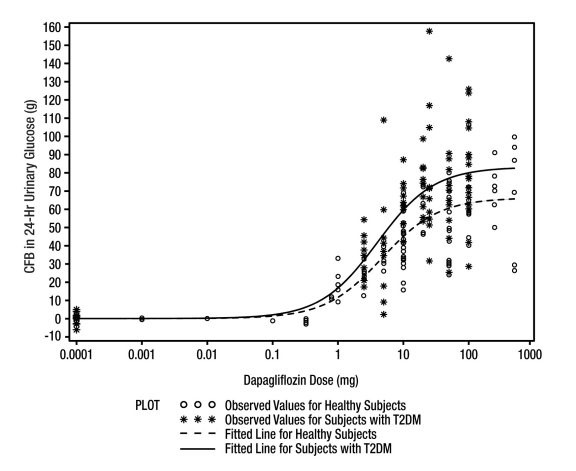
Increases in the amount of glucose excreted in the urine were observed in healthy subjects and in patients with type 2 diabetes mellitus following the administration of dapagliflozin (see Figure 1). Dapagliflozin doses of 5 or 10 mg per day in patients with type 2 diabetes mellitus for 12 weeks resulted in excretion of approximately 70 grams of glucose in the urine per day at Week 12. A near maximum glucose excretion was observed at the dapagliflozin daily dose of 20 mg. This urinary glucose excretion with dapagliflozin also results in increases in urinary volume [see Adverse Reactions (6.1)]. After discontinuation of dapagliflozin, on average, the elevation in urinary glucose excretion approaches baseline by about 3 days for the 10 mg dose.
Figure 1: Scatter Plot and Fitted Line of Change from Baseline in 24-Hour Urinary Glucose Amount versus Dapagliflozin Dose in Healthy Subjects and Subjects with Type 2 Diabetes Mellitus (T2DM) (Semi-Log Plot)
Cardiac Electrophysiology
Dapagliflozin was not associated with clinically meaningful prolongation of QTc interval at daily doses up to 150 mg (15-times the recommended maximum dose) in a study of healthy subjects. In addition, no clinically meaningful effect on QTc interval was observed following single doses of up to 500 mg (50-times the recommended maximum dose) of dapagliflozin in healthy subjects.
12.3 Pharmacokinetics
Absorption
Following oral administration of dapagliflozin, the maximum plasma concentration (Cmax) is usually attained within 2 hours under fasting state. The Cmax and AUC values increase dose proportionally with increase in dapagliflozin dose in the therapeutic dose range. The absolute oral bioavailability of dapagliflozin following the administration of a 10 mg dose is 78%. Administration of dapagliflozin with a high-fat meal decreases its Cmax by up to 50% and prolongs Tmax by approximately 1 hour but does not alter AUC as compared with the fasted state. These changes are not considered to be clinically meaningful and dapagliflozin can be administered with or without food.
Distribution
Dapagliflozin is approximately 91% protein bound. Protein binding is not altered in patients with renal or hepatic impairment.
Metabolism
The metabolism of dapagliflozin is primarily mediated by UGT1A9; CYP-mediated metabolism is a minor clearance pathway in humans. Dapagliflozin is extensively metabolized, primarily to yield dapagliflozin 3-O-glucuronide, which is an inactive metabolite. Dapagliflozin 3-O-glucuronide accounted for 61% of a 50 mg [14C]-dapagliflozin dose and is the predominant drug-related component in human plasma.
Elimination
Dapagliflozin and related metabolites are primarily eliminated via the renal pathway. Following a single 50 mg dose of [14C]-dapagliflozin, 75% and 21% total radioactivity is excreted in urine and feces, respectively. In urine, less than 2% of the dose is excreted as parent drug. In feces, approximately 15% of the dose is excreted as parent drug. The mean plasma terminal half-life (t½) for dapagliflozin is approximately 12.9 hours following a single oral dose of FARXIGA 10 mg.
Specific Populations
Renal Impairment
At steady-state (20 mg once daily dapagliflozin for 7 days), patients with type 2 diabetes with mild, moderate, or severe renal impairment (as determined by eGFR) had geometric mean systemic exposures of dapagliflozin that were 45%, 100%, and 200% higher, respectively, as compared to patients with type 2 diabetes mellitus with normal renal function. There was no meaningful difference in exposure between patients with chronic kidney disease with and without type 2 diabetes. Higher systemic exposure of dapagliflozin in patients with type 2 diabetes mellitus with renal impairment did not result in a correspondingly higher 24-hour urinary glucose excretion. The steady-state 24-hour urinary glucose excretion in patients with type 2 diabetes mellitus and mild, moderate, and severe renal impairment was 42%, 80%, and 90% lower, respectively, than in patients with type 2 diabetes mellitus with normal renal function.
The impact of hemodialysis on dapagliflozin exposure is not known [see Dosage and Administration (2.2), Warnings and Precautions (5.2), Use in Specific Populations (8.6), and Clinical Studies (14)].
Hepatic Impairment
In subjects with mild and moderate hepatic impairment (Child-Pugh classes A and B), mean Cmax and AUC of dapagliflozin were up to 12% and 36% higher, respectively, as compared to healthy matched control subjects following single-dose administration of 10 mg dapagliflozin. These differences were not considered to be clinically meaningful. In patients with severe hepatic impairment (Child-Pugh class C), mean Cmax and AUC of dapagliflozin were up to 40% and 67% higher, respectively, as compared to healthy matched controls [see Use in Specific Populations (8.7)].
Drug Interactions
In Vitro Assessment of Drug Interactions
In in vitro studies, dapagliflozin and dapagliflozin 3-O-glucuronide neither inhibited CYP 1A2, 2C9, 2C19, 2D6, or 3A4, nor induced CYP 1A2, 2B6, or 3A4. Dapagliflozin is a weak substrate of the P-glycoprotein (P-gp) active transporter, and dapagliflozin 3-O-glucuronide is a substrate for the OAT3 active transporter. Dapagliflozin or dapagliflozin 3-O-glucuronide did not meaningfully inhibit P-gp, OCT2, OAT1, or OAT3 active transporters. Overall, dapagliflozin is unlikely to affect the pharmacokinetics of concurrently administered medications that are P-gp, OCT2, OAT1, or OAT3 substrates.
Effects of Other Drugs on Dapagliflozin
Table 6 shows the effect of coadministered drugs on the pharmacokinetics of dapagliflozin. No dose adjustments are recommended for dapagliflozin.
Table 6: Effects of Coadministered Drugs on Dapagliflozin Systemic Exposure Coadministered Drug
(Dose Regimen)*Dapagliflozin
(Dose Regimen)*Effect on Dapagliflozin Exposure
(% Change [90% CI])Cmax
AUC†
No dosing adjustments required for the following:
Oral Antidiabetic Agents
Metformin (1000 mg)
20 mg
↔
↔
Pioglitazone (45 mg)
50 mg
↔
↔
Sitagliptin (100 mg)
20 mg
↔
↔
Glimepiride (4 mg)
20 mg
↔
↔
Voglibose (0.2 mg three times daily)
10 mg
↔
↔
Other Medications
Hydrochlorothiazide (25 mg)
50 mg
↔
↔
Bumetanide (1 mg)
10 mg once daily
for 7 days↔
↔
Valsartan (320 mg)
20 mg
↓12%
[↓3%, ↓20%]↔
Simvastatin (40 mg)
20 mg
↔
↔
Anti-infective Agent
Rifampin (600 mg once daily for 6 days)
10 mg
↓7%
[↓22%, ↑11%]↓22%
[↓27%, ↓17%]Nonsteroidal Anti-inflammatory Agent
Mefenamic Acid (loading dose of 500 mg followed by 14 doses of 250 mg every 6 hours)
10 mg
↑13%
[↑3%, ↑24%]↑51%
[↑44%, ↑58%]↔ = no change (geometric mean ratio of test: reference within 0.80 to 1.25); ↓ or ↑ = parameter was lower or higher, respectively, with coadministration compared to dapagliflozin administered alone (geometric mean ratio of test: reference was lower than 0.80 or higher than 1.25)
Effects of Dapagliflozin on Other Drugs
Table 7 shows the effect of dapagliflozin on other coadministered drugs. Dapagliflozin did not meaningfully affect the pharmacokinetics of the coadministered drugs.
Table 7: Effects of Dapagliflozin on the Systemic Exposures of Coadministered Drugs Coadministered Drug
(Dose Regimen)*Dapagliflozin
(Dose Regimen)*Effect on Coadministered Drug Exposure
(% Change [90% CI])Cmax
AUC†
No dosing adjustments required for the following:
Oral Antidiabetic Agents
Metformin (1000 mg)
20 mg
↔
↔
Pioglitazone (45 mg)
50 mg
↓7%
[↓25%, ↑15%]↔
Sitagliptin (100 mg)
20 mg
↔
↔
Glimepiride (4 mg)
20 mg
↔
↑13%
[0%, ↑29%]Other Medications
Hydrochlorothiazide (25 mg)
50 mg
↔
↔
Bumetanide (1 mg)
10 mg once daily
for 7 days↑13%
[↓2%, ↑31%]↑13%
[↓1%, ↑30%]Valsartan (320 mg)
20 mg
↓6%
[↓24%, ↑16%]↑5%
[↓15%, ↑29%]Simvastatin (40 mg)
20 mg
↔
↑19%
Digoxin (0.25 mg)
20 mg loading dose
then 10 mg once daily
for 7 days↔
↔
Warfarin (25 mg)
20 mg loading dose
then 10 mg once daily
for 7 days↔
↔
↔ = no change (geometric mean ratio of test: reference within 0.80 to 1.25); ↓ or ↑ = parameter was lower or higher, respectively, with coadministration compared to the other medicine administered alone (geometric mean ratio of test: reference was lower than 0.80 or higher than 1.25).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Dapagliflozin did not induce tumors in either mice or rats at any of the doses evaluated in 2-year carcinogenicity studies. Oral doses in mice consisted of 5, 15, and 40 mg/kg/day in males and 2, 10, and 20 mg/kg/day in females, and oral doses in rats were 0.5, 2, and 10 mg/kg/day for both males and females. The highest doses evaluated in mice were approximately 72-times (males) and 105-times (females) the clinical dose of 10 mg per day, based on AUC exposure. In rats, the highest dose was approximately 131-times (males) and 186-times (females) the clinical dose of 10 mg per day, based on AUC exposure.
Dapagliflozin was negative in the Ames mutagenicity assay and was positive in a series of in vitro clastogenicity assays in the presence of S9 activation and at concentrations greater than or equal to 100 μg/mL. Dapagliflozin was negative for clastogenicity in a series of in vivo studies evaluating micronuclei or DNA repair in rats at exposure multiples greater than 2100-times the clinical dose.
There was no carcinogenicity or mutagenicity signal in animal studies, suggesting that dapagliflozin does not represent a genotoxic risk to humans.
Dapagliflozin had no effects on mating, fertility, or early embryonic development in treated male or female rats at exposure multiples less than or equal to 1708-times and 998-times the maximum recommended human dose in males and females, respectively.
-
14 CLINICAL STUDIES
14.1 Glycemic Control in Patients with Type 2 Diabetes Mellitus
Overview of Clinical Studies of FARXIGA for Type 2 Diabetes Mellitus
FARXIGA has been studied as monotherapy, in combination with metformin, pioglitazone, sulfonylurea (glimepiride), sitagliptin (with or without metformin), metformin plus a sulfonylurea, or insulin (with or without other oral antidiabetic therapy), compared to a sulfonylurea (glipizide), and in combination with a GLP-1 receptor agonist (exenatide extended-release) added-on to metformin. FARXIGA has also been studied in patients with type 2 diabetes mellitus and moderate renal impairment.
Treatment with FARXIGA as monotherapy and in combination with metformin, glimepiride, pioglitazone, sitagliptin, or insulin produced statistically significant improvements in mean change from baseline at Week 24 in HbA1c compared to control. Reductions in HbA1c were seen across subgroups including gender, age, race, duration of disease, and baseline body mass index (BMI).
Monotherapy
A total of 840 treatment-naive patients with inadequately controlled type 2 diabetes mellitus participated in 2 placebo-controlled studies to evaluate the safety and efficacy of monotherapy with FARXIGA.
In 1 monotherapy study, a total of 558 treatment-naive patients with inadequately controlled diabetes participated in a 24-week study (NCT00528372). Following a 2-week diet and exercise placebo lead-in period, 485 patients with HbA1c ≥7% and ≤10% were randomized to FARXIGA 5 mg or FARXIGA 10 mg once daily in either the morning (QAM, main cohort) or evening (QPM), or placebo.
At Week 24, treatment with FARXIGA 10 mg QAM provided significant improvements in HbA1c and the fasting plasma glucose (FPG) compared with placebo (see Table 8).
Table 8: Results at Week 24 (LOCF*) in a Placebo-Controlled Study of FARXIGA Monotherapy in Patients with Type 2 Diabetes Mellitus (Main Cohort AM Doses) Efficacy Parameter FARXIGA
10 mg
N=70†FARXIGA
5 mg
N=64†Placebo
N=75†- *
- LOCF: last observation (prior to rescue for rescued patients) carried forward.
- †
- All randomized patients who took at least one dose of double-blind study medication during the short-term double-blind period.
- ‡
- Least squares mean adjusted for baseline value.
- §
- p-value <0.0001 versus placebo. Sensitivity analyses yielded smaller estimates of treatment difference with placebo.
- ¶
- Not evaluated for statistical significance as a result of the sequential testing procedure for the secondary endpoints.
HbA1c (%)
Baseline (mean)
8.0
7.8
7.8
Change from baseline (adjusted mean‡)
−0.9
−0.8
−0.2
Difference from placebo (adjusted mean‡)
(95% CI)−0.7§
(−1.0, −0.4)−0.5
(−0.8, −0.2)Percent of patients achieving HbA1c <7%
adjusted for baseline50.8%¶
44.2%¶
31.6%
FPG (mg/dL)
Baseline (mean)
166.6
157.2
159.9
Change from baseline (adjusted mean‡)
−28.8
−24.1
−4.1
Difference from placebo (adjusted mean‡)
(95% CI)−24.7§
(−35.7, −13.6)−19.9
(−31.3, −8.5)Initial Combination Therapy with Metformin XR
A total of 1236 treatment-naive patients with inadequately controlled type 2 diabetes mellitus (HbA1c ≥7.5% and ≤12%) participated in 2 active-controlled studies of 24-week duration to evaluate initial therapy with FARXIGA 5 mg (NCT00643851) or 10 mg (NCT00859898) in combination with metformin extended-release (XR) formulation.
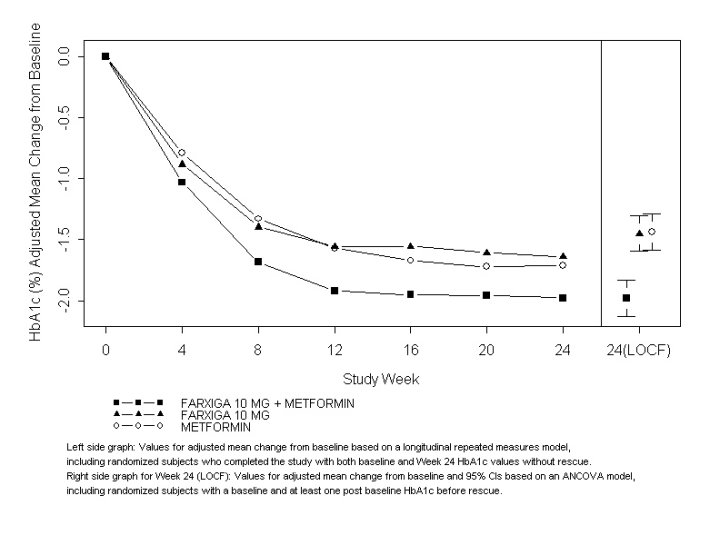
In 1 study, 638 patients randomized to 1 of 3 treatment arms following a 1-week lead-in period received: FARXIGA 10 mg plus metformin XR (up to 2000 mg per day), FARXIGA 10 mg plus placebo, or metformin XR (up to 2000 mg per day) plus placebo. Metformin XR dose was up-titrated weekly in 500 mg increments, as tolerated, with a median dose achieved of 2000 mg.
The combination treatment of FARXIGA 10 mg plus metformin XR provided statistically significant improvements in HbA1c and FPG compared with either of the monotherapy treatments and statistically significant reduction in body weight compared with metformin XR alone (see Table 9 and Figure 2). FARXIGA 10 mg as monotherapy also provided statistically significant improvements in FPG and statistically significant reduction in body weight compared with metformin alone and was non-inferior to metformin XR monotherapy in lowering HbA1c.
Table 9: Results at Week 24 (LOCF*) in an Active-Controlled Study of FARXIGA Initial Combination Therapy with Metformin XR Efficacy Parameter FARXIGA
10 mg
+ Metformin XRFARXIGA
10 mgMetformin
XRN=211† N=219† N=208† - *
- LOCF: last observation (prior to rescue for rescued patients) carried forward.
- †
- All randomized patients who took at least one dose of double-blind study medication during the short-term double-blind period.
- ‡
- Least squares mean adjusted for baseline value.
- §
- p-value <0.0001.
- ¶
- Non-inferior versus metformin XR.
- #
- p-value <0.05.
HbA1c (%)
Baseline (mean)
9.1
9.0
9.0
Change from baseline (adjusted mean‡)
−2.0
−1.5
−1.4
Difference from FARXIGA (adjusted mean‡)
(95% CI)−0.5§
(−0.7, −0.3)Difference from metformin XR (adjusted mean‡)
(95% CI)−0.5§
(−0.8, −0.3)0.0¶
(−0.2, 0.2)Percent of patients achieving HbA1c <7%
adjusted for baseline46.6%#
31.7%
35.2%
FPG (mg/dL)
Baseline (mean)
189.6
197.5
189.9
Change from baseline (adjusted mean‡)
−60.4
−46.4
−34.8
Difference from FARXIGA (adjusted mean‡)
(95% CI)−13.9§
(−20.9, −7.0)Difference from metformin XR (adjusted mean‡)
(95% CI)−25.5§
(−32.6, −18.5)−11.6#
(−18.6, −4.6)Body Weight (kg)
Baseline (mean)
88.6
88.5
87.2
Change from baseline (adjusted mean‡)
−3.3
−2.7
−1.4
Difference from metformin XR (adjusted mean‡)
(95% CI)−2.0§
(−2.6, −1.3)−1.4§
(−2.0, −0.7)
Figure 2: Adjusted Mean Change from Baseline Over Time in HbA1c (%) in a 24-Week Active-Controlled Study of FARXIGA Initial Combination Therapy with Metformin XR
In a second study, 603 patients were randomized to 1 of 3 treatment arms following a 1-week lead-in period: FARXIGA 5 mg plus metformin XR (up to 2000 mg per day), FARXIGA 5 mg plus placebo, or metformin XR (up to 2000 mg per day) plus placebo. Metformin XR dose was up-titrated weekly in 500 mg increments, as tolerated, with a median dose achieved of 2000 mg.
The combination treatment of FARXIGA 5 mg plus metformin XR provided statistically significant improvements in HbA1c and FPG compared with either of the monotherapy treatments and statistically significant reduction in body weight compared with metformin XR alone (see Table 10).
Table 10: Results at Week 24 (LOCF*) in an Active-Controlled Study of FARXIGA Initial Combination Therapy with Metformin XR Efficacy Parameter
FARXIGA
5 mg
+ Metformin XRFARXIGA
5 mgMetformin
XR
N=194†
N=203†
N=201†
HbA1c (%)
Baseline (mean)
9.2
9.1
9.1
Change from baseline (adjusted mean‡)
−2.1
−1.2
−1.4
Difference from FARXIGA (adjusted mean‡)
(95% CI)−0.9§
(−1.1, −0.6)Difference from metformin XR (adjusted mean‡)
(95% CI)−0.7§
(-0.9, -0.5)Percent of patients achieving HbA1c <7%
adjusted for baseline52.4%¶
22.5%
34.6%
FPG (mg/dL)
Baseline (mean)
193.4
190.8
196.7
Change from baseline (adjusted mean‡)
-61.0
-42.0
-33.6
Difference from FARXIGA (adjusted mean‡)
(95% CI)-19.1§
(-26.7, -11.4)Difference from metformin XR (adjusted mean‡)
(95% CI)-27.5§
(-35.1, -19.8)Body Weight (kg)
Baseline (mean)
84.2
86.2
85.8
Change from baseline (adjusted mean‡)
-2.7
-2.6
-1.3
Difference from metformin XR (adjusted mean‡)
(95% CI)-1.4§
(-2.0, -0.7)Add-On to Metformin
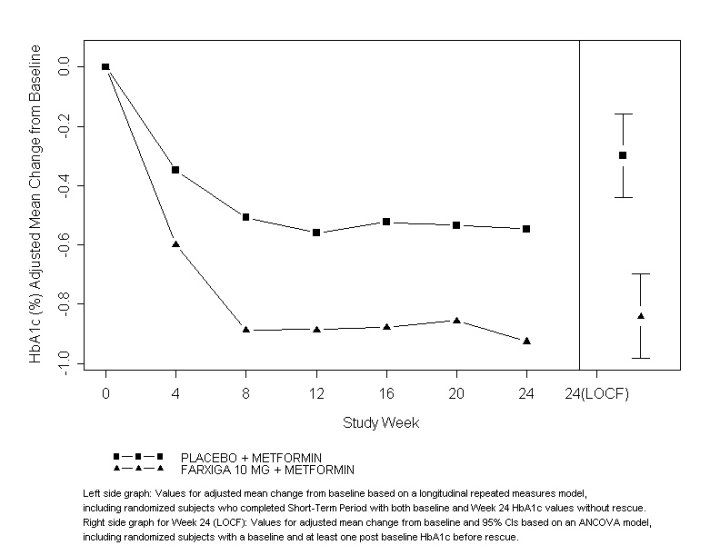
A total of 546 patients with type 2 diabetes mellitus with inadequate glycemic control (HbA1c ≥7% and ≤10%) participated in a 24-week, placebo-controlled study to evaluate FARXIGA in combination with metformin (NCT00528879). Patients on metformin at a dose of at least 1500 mg per day were randomized after completing a 2-week, single-blind, placebo lead-in period. Following the lead-in period, eligible patients were randomized to FARXIGA 5 mg, FARXIGA 10 mg, or placebo in addition to their current dose of metformin.
As add-on treatment to metformin, FARXIGA 10 mg provided statistically significant improvements in HbA1c and FPG, and statistically significant reduction in body weight compared with placebo at Week 24 (see Table 11 and Figure 3). Statistically significant (p <0.05 for both doses) mean changes from baseline in systolic blood pressure relative to placebo plus metformin were -4.5 mmHg and -5.3 mmHg with FARXIGA 5 mg and 10 mg plus metformin, respectively.
Table 11: Results of a 24-Week (LOCF*) Placebo-Controlled Study of FARXIGA in Add-On Combination with Metformin - *
- LOCF: last observation (prior to rescue for rescued patients) carried forward.
- †
- All randomized patients who took at least one dose of double-blind study medication during the short-term double-blind period.
- ‡
- Least squares mean adjusted for baseline value.
- §
- p-value <0.0001 versus placebo + metformin.
- ¶
- p-value <0.05 versus placebo + metformin.
Efficacy Parameter
FARXIGA 10 mg
+ Metformin
N=135†FARXIGA 5 mg
+ Metformin
N=137†Placebo
+ Metformin
N=137†HbA1c (%)
Baseline (mean)
7.9
8.2
8.1
Change from baseline (adjusted mean‡)
-0.8
-0.7
-0.3
Difference from placebo (adjusted mean‡)
(95% CI)-0.5§
(-0.7, -0.3)
-0.4§
(-0.6, -0.2)
Percent of patients achieving HbA1c <7%
adjusted for baseline40.6%¶
37.5%¶
25.9%
FPG (mg/dL)
Baseline (mean)
156.0
169.2
165.6
Change from baseline at Week 24 (adjusted mean‡)
-23.5
-21.5
-6.0
Difference from placebo (adjusted mean‡)
(95% CI)-17.5§
(-25.0, -10.0)-15.5§
(-22.9, -8.1)Change from baseline at Week 1 (adjusted mean‡)
-16.5§
(N=115)-12.0§
(N=121)1.2
(N=126)Body Weight (kg)
Baseline (mean)
86.3
84.7
87.7
Change from baseline (adjusted mean‡)
-2.9
-3.0
-0.9
Difference from placebo (adjusted mean‡)
(95% CI)
-2.0§
(-2.6, -1.3)-2.2§
(-2.8, -1.5)
Figure 3: Adjusted Mean Change from Baseline Over Time in HbA1c (%) in a 24-Week Placebo-Controlled Study of FARXIGA in Combination with Metformin
Active Glipizide-Controlled Study Add-On to Metformin
A total of 816 patients with type 2 diabetes mellitus with inadequate glycemic control (HbA1c >6.5% and ≤10%) were randomized in a 52-week, glipizide-controlled, non-inferiority study to evaluate FARXIGA as add-on therapy to metformin (NCT00660907). Patients on metformin at a dose of at least 1500 mg per day were randomized following a 2-week placebo lead-in period to glipizide or dapagliflozin (5 mg or 2.5 mg, respectively) and were up-titrated over 18 weeks to optimal glycemic effect (FPG <110 mg/dL, <6.1 mmol/L) or to the highest dose level (up to glipizide 20 mg and FARXIGA 10 mg) as tolerated by patients. Thereafter, doses were kept constant, except for down-titration to prevent hypoglycemia.
At the end of the titration period, 87% of patients treated with FARXIGA had been titrated to the maximum study dose (10 mg) versus 73% treated with glipizide (20 mg). FARXIGA led to a similar mean reduction in HbA1c from baseline at Week 52 (LOCF), compared with glipizide, thus demonstrating non-inferiority (see Table 12). FARXIGA treatment led to a statistically significant mean reduction in body weight from baseline at Week 52 (LOCF) compared with a mean increase in body weight in the glipizide group. Statistically significant (p<0.0001) mean change from baseline in systolic blood pressure relative to glipizide plus metformin was -5.0 mmHg with FARXIGA plus metformin.
Table 12: Results at Week 52 (LOCF*) in an Active-Controlled Study Comparing FARXIGA to Glipizide as Add-On to Metformin Efficacy Parameter FARXIGA
+ Metformin
N=400†Glipizide
+ Metformin
N=401†HbA1c (%)
Baseline (mean)
7.7
7.7
Change from baseline (adjusted mean‡)
-0.5
-0.5
Difference from glipizide + metformin (adjusted mean‡)
(95% CI)0.0§
(-0.1, 0.1)Body Weight (kg)
Baseline (mean)
88.4
87.6
Change from baseline (adjusted mean‡)
-3.2
1.4
Difference from glipizide + metformin (adjusted mean‡)
(95% CI)-4.7¶
(-5.1, -4.2)Add-On Combination Therapy with Other Antidiabetic Agents
Add-On Combination Therapy with a Sulfonylurea
A total of 597 patients with type 2 diabetes mellitus and inadequate glycemic control (HbA1c ≥7% and ≤10%) were randomized in this 24-week, placebo-controlled study to evaluate FARXIGA in combination with glimepiride (a sulfonylurea) (NCT00680745).
Patients on at least half the maximum recommended dose of glimepiride as monotherapy (4 mg) for at least 8 weeks lead-in were randomized to FARXIGA 5 mg, FARXIGA 10 mg, or placebo in addition to glimepiride 4 mg per day. Down-titration of glimepiride to 2 mg or 0 mg was allowed for hypoglycemia during the treatment period; no up-titration of glimepiride was allowed.
In combination with glimepiride, FARXIGA 10 mg provided statistically significant improvement in HbA1c, FPG, and 2-hour PPG, and statistically significant reduction in body weight compared with placebo plus glimepiride at Week 24 (see Table 13). Statistically significant (p<0.05 for both doses) mean changes from baseline in systolic blood pressure relative to placebo plus glimepiride were -2.8 mmHg and -3.8 mmHg with FARXIGA 5 mg and 10 mg plus glimepiride, respectively.
Add-on Combination Therapy with Metformin and a Sulfonylurea
A total of 218 patients with type 2 diabetes mellitus and inadequate glycemic control (HbA1c ≥7% and ≤10.5%) participated in a 24-week, placebo-controlled study to evaluate FARXIGA in combination with metformin and a sulfonylurea (NCT01392677). Patients on a stable dose of metformin (immediate- or extended-release formulations) ≥1500 mg/day plus maximum tolerated dose, which must be at least half the maximum dose, of a sulfonylurea for at least 8 weeks prior to enrollment were randomized after an 8-week placebo lead-in period to FARXIGA 10 mg or placebo. Dose-titration of FARXIGA or metformin was not permitted during the 24-week treatment period. Down-titration of the sulfonylurea was permitted to prevent hypoglycemia, but no up-titration was permitted. As add-on treatment to combined metformin and a sulfonylurea, treatment with FARXIGA 10 mg provided statistically significant improvements in HbA1c and FPG and statistically significant reduction in body weight compared with placebo at Week 24 (Table 13). A statistically significant (p <0.05) mean change from baseline in systolic blood pressure relative to placebo in combination with metformin and a sulfonylurea was -3.8 mmHg with FARXIGA 10 mg in combination with metformin and a sulfonylurea at Week 8.
Add-On Combination Therapy with a Thiazolidinedione
A total of 420 patients with type 2 diabetes mellitus with inadequate glycemic control (HbA1c ≥7% and ≤10.5%) participated in a 24-week, placebo-controlled study to evaluate FARXIGA in combination with pioglitazone (a thiazolidinedione [TZD]) alone (NCT00683878). Patients on a stable dose of pioglitazone of 45 mg per day (or 30 mg per day, if 45 mg per day was not tolerated) for 12 weeks were randomized after a 2-week lead-in period to 5 or 10 mg of FARXIGA or placebo in addition to their current dose of pioglitazone. Dose titration of FARXIGA or pioglitazone was not permitted during the study.
In combination with pioglitazone, treatment with FARXIGA 10 mg provided statistically significant improvements in HbA1c, 2-hour PPG, FPG, the proportion of patients achieving HbA1c <7%, and a statistically significant reduction in body weight compared with the placebo plus pioglitazone treatment groups (see Table 13) at Week 24. A statistically significant (p <0.05) mean change from baseline in systolic blood pressure relative to placebo in combination with pioglitazone was -4.5 mmHg with FARXIGA 10 mg in combination with pioglitazone.
Add-On Combination Therapy with a DPP4 Inhibitor
A total of 452 patients with type 2 diabetes mellitus who were drug naive, or who were treated at entry with metformin or a DPP4 inhibitor alone or in combination, and had inadequate glycemic control (HbA1c ≥7.0% and ≤10.0% at randomization), participated in a 24-week, placebo-controlled study to evaluate FARXIGA in combination with sitagliptin (a DPP4 inhibitor) with or without metformin (NCT00984867).
Eligible patients were stratified based on the presence or absence of background metformin (≥1500 mg per day), and within each stratum were randomized to either FARXIGA 10 mg plus sitagliptin 100 mg once daily, or placebo plus sitagliptin 100 mg once daily. Endpoints were tested for FARXIGA 10 mg versus placebo for the total study group (sitagliptin with and without metformin) and for each stratum (sitagliptin alone or sitagliptin with metformin). Thirty-seven percent (37%) of patients were drug naive, 32% were on metformin alone, 13% were on a DPP4 inhibitor alone, and 18% were on a DPP4 inhibitor plus metformin. Dose titration of FARXIGA, sitagliptin, or metformin was not permitted during the study.
In combination with sitagliptin (with or without metformin), FARXIGA 10 mg provided statistically significant improvements in HbA1c, FPG, and a statistically significant reduction in body weight compared with the placebo plus sitagliptin (with or without metformin) group at Week 24 (see Table 13). These improvements were also seen in the stratum of patients who received FARXIGA 10 mg plus sitagliptin alone (placebo-corrected mean change for HbA1c -0.56%; n=110) compared with placebo plus sitagliptin alone (n=111), and the stratum of patients who received FARXIGA 10 mg plus sitagliptin and metformin (placebo-corrected mean change for HbA1c -0.40; n=113) compared with placebo plus sitagliptin with metformin (n=113).
Add-On Combination Therapy with Insulin
A total of 808 patients with type 2 diabetes mellitus who had inadequate glycemic control (HbA1c ≥7.5% and ≤10.5%) were randomized in a 24-week, placebo-controlled study to evaluate FARXIGA as add-on therapy to insulin (NCT00673231). Patients on a stable insulin regimen, with a mean dose of at least 30 IU of injectable insulin per day, for a period of at least 8 weeks prior to enrollment and on a maximum of 2 oral antidiabetic medications (OADs), including metformin, were randomized after completing a 2-week enrollment period to receive either FARXIGA 5 mg, FARXIGA 10 mg, or placebo in addition to their current dose of insulin and other OADs, if applicable. Patients were stratified according to the presence or absence of background OADs. Up- or down-titration of insulin was only permitted during the treatment phase in patients who failed to meet specific glycemic goals. Dose modifications of blinded study medication or OAD(s) were not allowed during the treatment phase, with the exception of decreasing OAD(s) where there were concerns over hypoglycemia after cessation of insulin therapy.
In this study, 50% of patients were on insulin monotherapy at baseline, while 50% were on 1 or 2 OADs in addition to insulin. At Week 24, FARXIGA 10 mg dose provided statistically significant improvement in HbA1c and reduction in mean insulin dose, and a statistically significant reduction in body weight compared with placebo in combination with insulin, with or without up to 2 OADs (see Table 13); the effect of FARXIGA on HbA1c was similar in patients treated with insulin alone and patients treated with insulin plus OAD. Statistically significant (p<0.05) mean change from baseline in systolic blood pressure relative to placebo in combination with insulin was -3.0 mmHg with FARXIGA 10 mg in combination with insulin.
At Week 24, FARXIGA 5 mg (-5.7 IU, difference from placebo) and 10 mg (-6.2 IU, difference from placebo) once daily resulted in a statistically significant reduction in mean daily insulin dose (p<0.0001 for both doses) compared to placebo in combination with insulin, and a statistically significantly higher proportion of patients on FARXIGA 10 mg (19.6%) reduced their insulin dose by at least 10% compared to placebo (11.0%).
Table 13: Results of 24-Week (LOCF*) Placebo-Controlled Studies of FARXIGA in Combination with Antidiabetic Agents - *
- LOCF: last observation (prior to rescue for rescued patients) carried forward.
- †
- Randomized and treated patients with baseline and at least 1 post-baseline efficacy measurement.
- ‡
- Least squares mean adjusted for baseline value based on an ANCOVA model.
- §
- p-value <0.0001 versus placebo.
- ¶
- 2-hour PPG level as a response to a 75-gram oral glucose tolerance test (OGTT).
- #
- Least squares mean adjusted for baseline value based on a longitudinal repeated measures model.
- Þ
- All randomized patients who took at least one dose of double-blind study medication during the short-term, double-blind period.
- ß
- p-value <0.05 versus placebo.
- à
- NT: Not formally tested because of failing to achieve a statistically significant difference in an endpoint that was earlier in the testing sequence.
Efficacy Parameter
FARXIGA 10 mg
FARXIGA 5 mg
Placebo
In Combination with Sulfonylurea (Glimepiride)
Intent-to-Treat Population
N=151†
N=142†
N=145†
HbA1c (%)
Baseline (mean)
8.1
8.1
8.2
Change from baseline (adjusted mean‡)
-0.8
-0.6
-0.1
Difference from placebo (adjusted mean‡)
(95% CI)-0.7§
(-0.9, -0.5)
-0.5§
(-0.7, -0.3)
Percent of patients achieving HbA1c <7% adjusted for baseline
31.7%§
30.3%§
13.0%
FPG (mg/dL)
Baseline (mean)
172.4
174.5
172.7
Change from baseline (adjusted mean‡)
-28.5
-21.2
-2.0
Difference from placebo (adjusted mean‡)
(95% CI)
-26.5§
(-33.5, -19.5)
-19.3§
(-26.3, -12.2)
2-hour PPG¶ (mg/dL)
Baseline (mean)
329.6
322.8
324.1
Change from baseline (adjusted mean‡)
-60.6
–54.5
-11.5
Difference from placebo (adjusted mean‡)
(95% CI)
-49.1§
(-64.1, -34.1)
-43.0§
(–58.4, -27.5)
Body Weight (kg)
Baseline (mean)
80.6
81.0
80.9
Change from baseline (adjusted mean‡)
-2.3
-1.6
-0.7
Difference from placebo (adjusted mean‡)
(95% CI)
-1.5§
(-2.2, -0.9)
-0.8§
(-1.5, -0.2)
In Combination with Metformin and a Sulfonylurea
Intent-to-Treat Population
N=108†
-
N=108†
HbA1c (%)
Baseline (mean)
8.08
-
8.24
-0.86
-
-0.17
Difference from placebo (adjusted mean‡#)
(95% CI)
-0.69§
(-0.89, -0.49)
-
Percent of patients achieving HbA1c <7%
adjusted for baseline31.8%§
-
11.1%
FPG (mg/dL)
Baseline (mean)
167.4
-
180.3
Change from baseline (adjusted mean‡)
-34.2
-
-0.8
Difference from placebo (adjusted mean‡)
(95% CI)
-33.5§
(-43.1, -23.8)
-
Body Weight (kg)
Baseline (mean)
88.57
-
90.07
Change from baseline (adjusted mean‡)
-2.65
-
-0.58
Difference from placebo (adjusted mean‡)
(95% CI)
-2.07§
(-2.79, -1.35)
-
In Combination with Thiazolidinedione (Pioglitazone)
Intent-to-Treat Population
N=140Þ
N=141Þ
N=139Þ
HbA1c (%)
Baseline (mean)
8.4
8.4
8.3
Change from baseline (adjusted mean‡)
-1.0
-0.8
-0.4
Difference from placebo (adjusted mean‡)
(95% CI)
-0.6§
(-0.8, -0.3)
-0.4§
(-0.6, -0.2)
Percent of patients achieving HbA1c <7%
adjusted for baseline38.8%ß
32.5%ß
22.4%
FPG (mg/dL)
Baseline (mean)
164.9
168.3
160.7
Change from baseline (adjusted mean‡)
-29.6
-24.9
-5.5
Difference from placebo (adjusted mean‡)
(95% CI)
-24.1§
(-32.2, -16.1)
-19.5§
(-27.5, -11.4)
2-hour PPG¶ (mg/dL)
Baseline (mean)
308.0
284.8
293.6
Change from baseline (adjusted mean‡)
-67.5
-65.1
-14.1
Difference from placebo (adjusted mean‡)
(95% CI)
-53.3§
(-71.1, -35.6)
-51.0§
(-68.7, -33.2)
Body Weight (kg)
Baseline (mean)
84.8
87.8
86.4
Change from baseline (adjusted mean‡)
-0.1
0.1
1.6
Difference from placebo (adjusted mean‡)
(95% CI)
-1.8§
(-2.6, -1.0)
-1.6§
(-2.3, -0.8)
In Combination with DPP4 Inhibitor (Sitagliptin) with or without Metformin
Intent-to-Treat Population
N=223†
–
N=224†
HbA1c (%)
Baseline (mean)
7.90
–
7.97
Change from baseline (adjusted mean‡)
-0.45
–
0.04
Difference from placebo (adjusted mean‡)
(95% CI)
-0.48§
(-0.62, -0.34)
–
Patients with HbA1c decrease ≥0.7% (adjusted percent)
35.4%
–
16.6%
FPG (mg/dL)
Baseline (mean)
161.7
–
163.1
Change from baseline at Week 24 (adjusted mean‡)
-24.1
–
3.8
Difference from placebo (adjusted mean‡)
(95% CI)
-27.9§
(-34.5, -21.4)
–
Body Weight (kg)
Baseline (mean)
91.02
–
89.23
Change from baseline (adjusted mean‡)
-2.14
–
-0.26
Difference from placebo (adjusted mean‡)
(95% CI)
-1.89§
(-2.37, -1.40)
–
In Combination with Insulin with or without up to 2 Oral Antidiabetic Therapies
Intent-to-Treat Population
N=194†
N=211†
N=193†
HbA1c (%)
Baseline (mean)
8.6
8.6
8.5
Change from baseline (adjusted mean‡)
-0.9
-0.8
-0.3
Difference from placebo (adjusted mean‡)
(95% CI)
-0.6§
(-0.7, -0.5)
-0.5§
(-0.7, -0.4)
FPG (mg/dL)
Baseline (mean)
173.7
NTà
170.0
Change from baseline (adjusted mean‡)
-21.7
NTà
3.3
Difference from placebo (adjusted mean‡)
(95% CI)
-25.0§
(-34.3, -15.8)
NTà
Body Weight (kg)
Baseline (mean)
94.6
93.2
94.2
Change from baseline (adjusted mean‡)
-1.7
-1.0
0.0
Difference from placebo (adjusted mean‡)
(95% CI)
-1.7§
(-2.2, -1.2)
-1.0§
(-1.5, -0.5)
Combination Therapy with Exenatide-Extended Release as Add-On to Metformin
A total of 694 adult patients with type 2 diabetes mellitus and inadequate glycemic control (HbA1c ≥8.0 and ≤12.0%) on metformin, were evaluated in a 28-week double-blind, active-controlled study to compare FARXIGA in combination with exenatide extended-release (a GLP-1 receptor agonist) to FARXIGA alone and exenatide extended-release alone, as add-on to metformin (NCT02229396). Patients on metformin at a dose of at least 1,500 mg per day were randomized following a 1-week placebo lead-in period to receive either FARXIGA 10 mg once daily (QD) in combination with exenatide extended-release 2 mg once weekly (QW), FARXIGA 10 mg QD, or exenatide extended–release 2 mg QW.
At Week 28, FARXIGA in combination with exenatide extended-release provided statistically significantly greater reductions in HbA1c (-1.77%) compared to FARXIGA alone (-1.32%, p=0.001) and exenatide extended-release alone (-1.42%, p=0.012). FARXIGA in combination with exenatide extended-release provided statistically significantly greater reductions in FPG (-57.35 mg/dL) compared to FARXIGA alone (-44.72 mg/dL, p=0.006) and exenatide extended-release alone (-40.53, p <0.001).
Use in Patients with Type 2 Diabetes Mellitus and Moderate Renal Impairment
FARXIGA was assessed in two placebo-controlled studies of patients with type 2 diabetes mellitus and moderate renal impairment.
Patients with type 2 diabetes mellitus and an eGFR between 45 to less than 60 mL/min/1.73 m2 inadequately controlled on current diabetes therapy participated in a 24-week, double-blind, placebo-controlled clinical study (NCT02413398). Patients were randomized to either FARXIGA 10 mg or placebo, administered orally once daily. At Week 24, FARXIGA provided statistically significant reductions in HbA1c compared with placebo (Table 14).
Table 14: Results at Week 24 of Placebo-Controlled Study for FARXIGA in Patients with Type 2 Diabetes Mellitus and Renal Impairment (eGFR 45 to less than 60 mL/min/1.73 m2) FARXIGA 10 mg Placebo Number of patients: N=160 N=161 - *
- Least squares mean adjusted for baseline value; at Week 24, HbA1c was missing for 5.6% and 6.8% of individuals treated with FARXIGA and placebo, respectively. Retrieved dropouts, i.e. observed HbA1c at Week 24 from subjects who discontinued treatment, were used to impute missing values in HbA1c.
- †
- p-value =0.008 versus placebo.
HbA1c (%)
Baseline (mean)
8.3
8.0
Change from baseline (adjusted mean*)
-0.4
-0.1
Difference from placebo (adjusted mean*)
(95% CI)
-0.3†
(-0.5, - 0.1)
14.2 Cardiovascular Outcomes in Patients with Type 2 Diabetes Mellitus
Dapagliflozin Effect on Cardiovascular Events (DECLARE, NCT01730534) was an international, multicenter, randomized, double-blind, placebo-controlled, clinical study conducted to determine the effect of FARXIGA relative to placebo on cardiovascular (CV) outcomes when added to current background therapy. All patients had type 2 diabetes mellitus and either established CV disease or two or more additional CV risk factors (age ≥55 years in men or ≥60 years in women and one or more of dyslipidemia, hypertension, or current tobacco use). Concomitant antidiabetic and atherosclerotic therapies could be adjusted, at the discretion of investigators, to ensure participants were treated according to the standard care for these diseases.
Of 17160 randomized patients, 6974 (40.6%) had established CV disease and 10186 (59.4%) did not have established CV disease. A total of 8582 patients were randomized to FARXIGA 10 mg, 8578 to placebo, and patients were followed for a median of 4.2 years.
Approximately 80% of the trial population was White, 4% Black or African American, and 13% Asian. The mean age was 64 years, and approximately 63% were male.
Mean duration of diabetes was 11.9 years and 22.4% of patients had diabetes for less than 5 years. Mean eGFR was 85.2 mL/min/1.73 m2. At baseline, 23.5% of patients had microalbuminuria (UACR ≥30 to ≤300 mg/g) and 6.8% had macroalbuminuria (UACR >300 mg/g). Mean HbA1c was 8.3% and mean BMI was 32.1 kg/m2. At baseline, 10% of patients had a history of heart failure.
Most patients (98.1%) used one or more antihyperglycemic medications at baseline. 82.0% of the patients were being treated with metformin, 40.9% with insulin, 42.7% with a sulfonylurea, 16.8% with a DPP4 inhibitor, and 4.4% with a GLP-1 receptor agonist.
Approximately 81.3% of patients were treated with angiotensin converting enzyme inhibitors or angiotensin receptor blockers, 75.0% with statins, 61.1% with antiplatelet therapy, 55.5% with acetylsalicylic acid, 52.6% with beta-blockers, 34.9% with calcium channel blockers, 22.0% with thiazide diuretics, and 10.5% with loop diuretics.
A Cox proportional hazards model was used to test for non-inferiority against the pre-specified risk margin of 1.3 for the hazard ratio (HR) of the composite of CV death, myocardial infarction (MI), or ischemic stroke (MACE) and if non-inferiority was demonstrated, to test for superiority on the two primary endpoints: 1) the composite of hospitalization for heart failure or CV death, and 2) MACE.
The incidence rate of MACE was similar in both treatment arms: 2.30 MACE events per 100 patient-years on dapagliflozin vs 2.46 MACE events per 100 patient-years on placebo. The estimated hazard ratio of MACE associated with dapagliflozin relative to placebo was 0.93 with a 95% CI of (0.84, 1.03). The upper bound of this confidence interval, 1.03, excluded the pre-specified non-inferiority margin of 1.3.
FARXIGA was superior to placebo in reducing the incidence of the primary composite endpoint of hospitalization for heart failure or CV death (HR 0.83 [95% CI 0.73, 0.95]).
The treatment effect was due to a significant reduction in the risk of hospitalization for heart failure in subjects randomized to FARXIGA (HR 0.73 [95% CI 0.61, 0.88]), with no change in the risk of CV death (Table 15 and Figures 4 and 5).
Table 15: Treatment Effects for the Primary Endpoints* and their Components* in the DECLARE Study Patients with events n (%)
Efficacy Variable
(time to first occurrence)
FARXIGA 10 mg
N=8582
Placebo
N=8578
Hazard ratio
(95% CI)
Primary Endpoints
Composite of Hospitalization for Heart Failure, CV Death†
417 (4.9)
496 (5.8)
0.83 (0.73, 0.95)
Composite Endpoint of CV Death, MI, Ischemic Stroke
756 (8.8)
803 (9.4)
0.93 (0.84, 1.03)
Components of the composite endpoints‡
Hospitalization for Heart Failure
212 (2.5)
286 (3.3)
0.73 (0.61, 0.88)
CV Death
245 (2.9)
249 (2.9)
0.98 (0.82, 1.17)
Myocardial Infarction
393 (4.6)
441 (5.1)
0.89 (0.77, 1.01)
Ischemic Stroke
235 (2.7)
231 (2.7)
1.01 (0.84, 1.21)
N=Number of patients, CI=Confidence interval, CV=Cardiovascular, MI=Myocardial infarction.
Figure 4: Time to First Occurrence of Hospitalization for Heart Failure or CV Death in the DECLARE Study
Figure 5: Time to First Occurrence of Hospitalization for Heart Failure in the DECLARE Study
14.3 Chronic Kidney Disease
The Study to Evaluate the Effect of Dapagliflozin on Renal Outcomes and Cardiovascular Mortality in Patients with Chronic Kidney Disease (DAPA-CKD, NCT03036150) was an international, multicenter, randomized, double-blind, placebo-controlled study in patients with chronic kidney disease (CKD) (eGFR between 25 and 75 mL/min/1.73 m2) and albuminuria (urine albumin creatinine ratio [UACR] between 200 and 5000 mg/g) who were receiving standard of care background therapy, including a maximally tolerated, labeled daily dose of an angiotensin-converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB). The trial excluded patients with autosomal dominant or autosomal recessive polycystic kidney disease, lupus nephritis, or ANCA-associated vasculitis and patients requiring cytotoxic, immunosuppressive, or immunomodulatory therapies in the preceding 6 months.
The primary objective was to determine whether FARXIGA reduces the incidence of the composite endpoint of ≥50% sustained decline in eGFR, progression to end-stage kidney disease (ESKD) (defined as sustained eGFR <15 mL/min/1.73 m2, initiation of chronic dialysis treatment or renal transplant), CV or renal death.
A total of 4304 patients were randomized equally to FARXIGA 10 mg or placebo and were followed for a median of 28.5 months.
The mean age of the study population was 62 years and 67% were male. The population was 53% White, 4% Black or African American, and 34% Asian; 25% were of Hispanic or Latino ethnicity.
At baseline, mean eGFR was 43 mL/min/1.73 m2, 44% of patients had an eGFR 30 mL/min/1.73 m2 to less than 45 mL/min/1.73 m2, and 15% of patients had an eGFR less than 30 mL/min/1.73 m2. Median UACR was 950 mg/g. A total of 68% of the patients had type 2 diabetes mellitus at randomization. The most common etiologies of CKD were diabetic nephropathy (58%), ischemic/hypertensive nephropathy (16%), and IgA nephropathy (6%).
At baseline, 97% of patients were treated with ACEi or ARB. Approximately 44% were taking antiplatelet agents, and 65% were on a statin.
FARXIGA reduced the incidence of the primary composite endpoint of ≥50% sustained decline in eGFR, progression to ESKD, CV or renal death (HR 0.61 [95% CI 0.51,0.72]; p<0.0001). The FARXIGA and placebo event curves separate by Month 4 and continue to diverge over the study period. The treatment effect reflected a reduction in ≥50% sustained decline in eGFR, progression to ESKD, and CV death. There were few renal deaths during the trial (Table 16, Figure 6).
FARXIGA also reduced the incidence of the composite endpoint of CV death or hospitalization for heart failure (HR 0.71 [95% CI 0.55, 0.92], p=0.0089) and all-cause mortality (HR 0.69 [95% CI 0.53, 0.88], p=0.0035).
Table 16: Treatment Effect for the Primary Composite Endpoint, its Components, and Secondary Composite Endpoints, in the DAPA-CKD Study - *
- ESKD is defined as sustained eGFR <15 mL/min/1.73 m2, initiation of chronic dialysis treatment, or transplant.
Patients with events (event rate)
Efficacy Variable
(time to first occurrence)
FARXIGA 10 mg
N=2152Placebo
N=2152Hazard ratio
(95% CI)p-value
Composite of ≥50% sustained eGFR decline, ESKD, CV or renal death
197 (4.6)
312 (7.5)
0.61
(0.51, 0.72)<0.0001
≥50% sustained eGFR decline
112 (2.6)
201 (4.8)
0.53
(0.42, 0.67)ESKD*
109 (2.5)
161 (3.8)
0.64
(0.50, 0.82)CV Death
65 (1.4)
80 (1.7)
0.81
(0.58, 1.12)Renal Death
2 (<0.1)
6 (0.1)
≥50% sustained eGFR decline, ESKD or renal death
142 (3.3)
243 (5.8)
0.56
(0.45, 0.68)<0.0001
CV death or Hospitalization for Heart Failure
100 (2.2)
138 (3.0)
0.71
(0.55, 0.92)0.0089
Hospitalization for Heart Failure
37 (0.8)
71 (1.6)
0.51
(0.34, 0.76)All-Cause Mortality
101 (2.2)
146 (3.1)
0.69
(0.53, 0.88)0.0035
N=Number of patients, CI=Confidence interval, CV=Cardiovascular, ESKD=End stage kidney disease.
NOTE: Time to first event was analyzed in a Cox proportional hazards model. Event rates are presented as the number of subjects with event per 100 patient years of follow-up.
There were too few events of renal death to compute a reliable hazard ratio.
Figure 6: Time to First Occurrence of the Primary Composite Endpoint, ≥50% Sustained Decline in eGFR, ESKD, CV or Renal Death (DAPA-CKD Study)
Patients at risk is the number of subjects at risk at the beginning of the period. 1 month corresponds to 30 days. 2-sided p-value is displayed. HR, CI and p-value are from the Cox proportional hazard model.
HR=hazard ratio; CI=confidence interval; eGFR=estimated glomerular filtration rate; ESKD=end stage kidney disease; CV=cardiovascular; vs=versus.
The results of the primary composite endpoint were consistent across the subgroups examined, including CKD patients with and without type 2 diabetes mellitus, causes of CKD, age, biological sex, race, UACR, and eGFR.
DAPA-CKD enrolled a population with relatively advanced CKD at high risk of progression. Exploratory analyses of a randomized, double-blind, placebo-controlled trial conducted to determine the effect of FARXIGA on CV outcomes (the DECLARE trial) support the conclusion that FARXIGA is also likely to be effective in patients with less advanced CKD.
14.4 Heart Failure
The efficacy and safety of FARXIGA 10 mg were assessed independently in two Phase 3 studies in patients with heart failure.
Dapagliflozin And Prevention of Adverse outcomes in Heart Failure (DAPA-HF, NCT03036124) was an international, multicenter, randomized, double-blind, placebo-controlled study in patients with heart failure (New York Heart Association [NYHA] functional class II-IV) with reduced ejection fraction (left ventricular ejection fraction [LVEF] 40% or less) to determine whether FARXIGA reduces the risk of cardiovascular death and hospitalization for heart failure. Of 4744 patients, 2373 were randomized to FARXIGA 10 mg and 2371 to placebo and were followed for a median of 18 months.
Dapagliflozin Evaluation to Improve the LIVEs of Patients with PReserved Ejection Fraction Heart Failure (DELIVER, NCT03619213) was an international, multicenter, randomized, double-blind, placebo-controlled study in patients aged ≥40 years with heart failure (NYHA class II-IV) with LVEF >40% and evidence of structural heart disease to determine whether FARXIGA reduces the risk of cardiovascular death, hospitalization for heart failure or urgent heart failure visits. Of 6263 patients, 3131 were randomized to FARXIGA 10 mg and 3132 to placebo and were followed for a median of 28 months. The study included 654 (10%) heart failure patients who were randomized during hospitalization for heart failure or within 30 days of discharge.
In DAPA-HF, at baseline, 94% of patients were treated with ACEi, ARB or angiotensin receptor-neprilysin inhibitor (ARNI, including sacubitril/valsartan 11%), 96% with beta-blocker, 71% with mineralocorticoid receptor antagonist (MRA), 93% with diuretic, and 26% had an implantable device.
In DELIVER, at baseline, 77% of patients were treated with ACEi, ARB or ARNI, 83% with beta-blocker, 43% with MRA, 98% with diuretic.
In both studies, FARXIGA reduced the incidence of the primary composite endpoint of CV death, hospitalization for heart failure or urgent heart failure visit (see Table 17).
Table 17: Treatment Effect for the Primary Composite Endpoint*, its Components* in the DAPA-HF and DELIVER Studies DAPA-HF Study
DELIVER Study
Patients with events
(event rate)Hazard ratio
(95% CI)p-value†
Patients with events
(event rate)Hazard ratio
(95% CI)p-value†
Efficacy Variable
(Time to first occurrence)
FARXIGA 10 mg
N=2373Placebo
N=2371FARXIGA 10 mg
N=3131Placebo
N=3132Composite of Hospitalization for Heart Failure, CV Death‡ or Urgent Heart Failure Visit
386
(11.6)502
(15.6)0.74
(0.65, 0.85)<0.0001
512
(7.8)610
(9.6)0.82
(0.73, 0.92)0.0008
Components of the composite endpoints
CV Death‡
227
(6.5)273
(7.9)0.82
(0.69, 0.98)231
(3.3)261
(3.8)0.88
(0.74, 1.05)Hospitalization for Heart Failure or Urgent Heart Failure Visit
237
(7.1)326
(10.1)0.70
(0.59, 0.83)368
(5.6)455
(7.2)0.79
(0.69, 0.91)Hospitalization for Heart Failure
231
(6.9)318
(9.8)0.70
(0.59, 0.83)329
(5.0)418
(6.5)0.77
(0.67, 0.89)Urgent Heart Failure Visit
10
(0.3)23
(0.7)0.43
(0.20, 0.90)60
(0.9)78
(1.1)0.76
(0.55, 1.07)N=Number of patients, CI=Confidence interval, CV=Cardiovascular.
NOTE: Time to first event was analyzed in a Cox proportional hazards model. The number of first events for the single components are the actual number of first events for each component and does not add up to the number of events in the composite endpoint. Event rates are presented as the number of subjects with event per 100 patient years of follow-up.
In both studies, all three components of the primary composite endpoint individually contributed to the treatment effect. In both studies, the FARXIGA and placebo event curves separated early and continued to diverge over the study period (see Figures 7 and 9).
Figure 7: Time to the First Occurrence of the Composite of Cardiovascular Death*, Hospitalization for Heart Failure or Urgent Heart Failure Visit
A) DAPA-HF Study
B) DELIVER Study
NOTE: An urgent heart failure visit was defined as an urgent, unplanned, assessment by a physician, e.g., in an Emergency Department, and requiring treatment for worsening heart failure (other than just an increase in oral diuretics).
* In DAPA-HF, the CV death component of the primary endpoint included death of undetermined cause. In DELIVER, the CV death component of the primary endpoint excluded death of undetermined cause.
† Patients at risk is the number of patients at risk at the beginning of the period.
HR=Hazard ratio, CI=Confidence interval, CV=Cardiovascular.
Figure 8: Time to Cardiovascular Death*
A) DAPA-HF Study
B) DELIVER Study
* In DAPA-HF, the CV death component of the primary endpoint included death of undetermined cause. In DELIVER, the CV death component of the primary endpoint excluded death of undetermined cause.
† Patients at risk is the number of patients at risk at the beginning of the period.
HR=Hazard ratio, CI=Confidence interval, CV=Cardiovascular.
Figure 9: Time to the First Occurrence of Hospitalization for Heart Failure or Urgent Heart Failure Visit
A) DAPA-HF Study
B) DELIVER Study
* Patients at risk is the number of patients at risk at the beginning of the period.
HR=Hazard ratio, CI=Confidence interval.
In DAPA-HF, FARXIGA reduced the total number of hospitalizations for heart failure (first and recurrent) events and CV death, with 567 and 742 total events in the FARXIGA-treated vs placebo group (Rate Ratio 0.75 [95% CI 0.65, 0.88]; p=0.0002).
In DELIVER, FARXIGA reduced the total number of heart failure events (first and recurrent hospitalization for heart failure or urgent heart failure visit) and CV death, with 815 and 1057 total events in the FARXIGA treated vs placebo group (Rate Ratio 0.77 [95% CI 0.67, 0.89]; p=0.0003).
In both studies, the results of the primary composite endpoint were consistent across the subgroups examined (see Figure 10).
Figure 10: Treatment Effects for Primary Composite Endpoint (Cardiovascular Death and Heart Failure Events) Subgroup Analysis
A) DAPA-HF Study
a Hazard ratio estimates are not presented for subgroups with less than 15 events in total, both arms combined.
n/N# Number of subjects with event/number of subjects in the subgroup.
NT-proBNP = N-terminal pro b-type natriuretic peptide, HF = Heart failure, MRA = mineralocorticoid receptor antagonist,
ECG = electrocardiogram, eGFR = estimated glomerular filtration rate.
Note: The figure above presents effects in various subgroups, all of which are baseline characteristics. The 95% confidence limits that are shown do not take into account the number of comparisons made and may not reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
B) DELIVER Study
a Subacute patient defined as randomized during hospitalization for heart failure or within 30 days of discharge.
b Defined as history of type 2 diabetes mellitus. This analysis does not include type 2 diabetes mellitus as a stratification factor.
n/N# Number of subjects with event/number of subjects in the subgroup.
NT-proBNP = N-terminal pro b-type natriuretic peptide, HF = Heart failure, ECG = electrocardiogram, eGFR = estimated glomerular filtration rate, BMI = body mass index, SBP = systolic blood pressure, T2DM = type 2 diabetes mellitus.
NOTE: The figure above presents effects in various subgroups, all of which are baseline characteristics. The 95% confidence limits that are shown do not take into account the number of comparisons made and may not reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
The treatment effect of FARXIGA on the composite endpoint of cardiovascular death, hospitalization for heart failure or urgent heart failure was consistent across the LVEF range as evaluated in DAPA-HF and DELIVER studies (Figure 11).
Figure 11: Treatment Effects for Primary Composite Endpoint (Cardiovascular Death and Heart Failure Events) by LVEF (DAPA-HF and DELIVER Studies)
* 1 patient in DAPA-HF study had LVEF >40. 4 patients in DELIVER study had LVEF≤40.
In DAPA-HF study, the 5% and 95% percentiles of LVEF were 20 and 40 respectively. In DELIVER study, the 5% and 95% percentiles of LVEF were 42 and 70, respectively.
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Diabetic Ketoacidosis in Patients with Type 1 Diabetes Mellitus and Other Ketoacidosis
In patients with type 1 diabetes mellitus, inform them that using FARXIGA can increase their risk of life-threatening diabetic ketoacidosis. For all other patients, inform them that FARXIGA can cause potentially fatal ketoacidosis and that type 2 diabetes mellitus and pancreatic disorders (e.g., history of pancreatitis or pancreatic surgery) are risk factors.
Educate all patients on precipitating factors (such as insulin dose reduction or missed insulin doses, infection, reduced caloric intake, ketogenic diet, surgery, dehydration, and alcohol abuse) and symptoms of ketoacidosis (including nausea, vomiting, abdominal pain, tiredness, and labored breathing). Inform patients that blood glucose may be normal even in the presence of ketoacidosis.
Advise patients that they may be asked to monitor ketones. If symptoms of ketoacidosis occur, instruct patients to discontinue FARXIGA and seek medical attention immediately [see Warnings and Precautions (5.1)].
Volume Depletion
Inform patients that symptomatic hypotension may occur with FARXIGA and advise them to contact their healthcare provider if they experience such symptoms [see Warnings and Precautions (5.2)]. Inform patients that dehydration may increase the risk for hypotension, and to have adequate fluid intake.
Serious Urinary Tract Infections
Inform patients of the potential for urinary tract infections, which may be serious. Provide them with information on the symptoms of urinary tract infections. Advise them to seek medical advice promptly if such symptoms occur [see Warnings and Precautions (5.3)].
Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues
Inform patients that the incidence of hypoglycemia may increase when FARXIGA is added to an insulin secretagogue (e.g., sulfonylurea) and/or insulin. Educate patients on the signs and symptoms of hypoglycemia [see Warnings and Precautions (5.4)].
Necrotizing Fasciitis of the Perineum (Fournier’s Gangrene)
Inform patients that necrotizing infections of the perineum (Fournier’s Gangrene) have occurred with FARXIGA in patients with diabetes mellitus. Counsel patients to promptly seek medical attention if they develop pain or tenderness, redness, or swelling of the genitals or the area from the genitals back to the rectum, along with a fever above 100.4°F or malaise [see Warnings and Precautions (5.5)].
Genital Mycotic Infections in Females (e.g., Vulvovaginitis)
Inform female patients that vaginal yeast infections may occur and provide them with information on the signs and symptoms of vaginal yeast infections. Advise them of treatment options and when to seek medical advice [see Warnings and Precautions (5.6)].
Genital Mycotic Infections in Males (e.g., Balanitis)
Inform male patients that yeast infections of the penis (e.g., balanitis or balanoposthitis) may occur, especially in patients with prior history. Provide them with information on the signs and symptoms of balanitis and balanoposthitis (rash or redness of the glans or foreskin of the penis). Advise them of treatment options and when to seek medical advice [see Warnings and Precautions (5.6)].
Hypersensitivity Reactions
Inform patients that serious hypersensitivity reactions (e.g., urticaria, anaphylactic reactions, and angioedema) have been reported with FARXIGA. Advise patients to immediately report any signs or symptoms suggesting allergic reaction or angioedema, and to take no more of the drug until they have consulted prescribing physicians.
Pregnancy
Advise pregnant patients of the potential risk to a fetus with treatment with FARXIGA. Instruct patients to immediately inform their healthcare provider if pregnant or planning to become pregnant [see Use in Specific Populations (8.1)].
Lactation
Advise patients that use of FARXIGA is not recommended while breastfeeding [see Use in Specific Populations (8.2)].
Laboratory Tests
Due to its mechanism of action, patients taking FARXIGA will test positive for glucose in their urine.
Missed Dose
If a dose is missed, advise patients to take it as soon as it is remembered unless it is almost time for the next dose, in which case patients should skip the missed dose and take the medicine at the next regularly scheduled time. Advise patients not to take two doses of FARXIGA at the same time.
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
MEDICATION GUIDE
FARXIGA® [FAR-SEE-GUH]
(dapagliflozin)
tablets, for oral useWhat is the most important information I should know about FARXIGA?
FARXIGA can cause serious side effects, including:
- •
-
Diabetic ketoacidosis (increased ketones in your blood or urine) in people with type 1 diabetes and other ketoacidosis. FARXIGA can cause ketoacidosis that can be life-threatening and may lead to death. Ketoacidosis is a serious condition which needs to be treated in a hospital. People with type 1 diabetes have a high risk of getting ketoacidosis. People with type 2 diabetes or pancreas problems also have an increased risk of getting ketoacidosis. Ketoacidosis can also happen in people who: are sick, cannot eat or drink as usual, skip meals, are on a diet high in fat and low in carbohydrates (ketogenic diet), take less than the usual amount of insulin or miss insulin doses, drink too much alcohol, have a loss of too much fluid from the body (volume depletion), or who have surgery. Ketoacidosis can happen even if your blood sugar is less than 250 mg/dL. Your healthcare provider may ask you to periodically check ketones in your urine or blood.
Stop taking FARXIGA and call your healthcare provider or get medical help right away if you get any of the following. If possible, check for ketones in your urine or blood, even if your blood sugar is less than 250 mg/dL.
- o
- nausea
- o
- vomiting
- o
- stomach area (abdominal) pain
- o
- tiredness
- o
- trouble breathing
- o
- ketones in your urine or blood
- •
-
Dehydration. FARXIGA can cause some people to become dehydrated (the loss of body water and salt). Dehydration may cause you to feel dizzy, faint, lightheaded, or weak, especially when you stand up (orthostatic hypotension). There have been reports of sudden kidney injury in people with Type 2 diabetes who are taking FARXIGA. You may be at a higher risk of dehydration if you:
- ∘
- take medicines to lower your blood pressure, including water pills (diuretics)
- ∘
- are on a low salt diet
- ∘
- have kidney problems
- ∘
- are 65 years of age or older
- Talk to your healthcare provider about what you can do to prevent dehydration including how much fluid you should drink on a daily basis. Call your healthcare provider right away if you reduce the amount of food or liquid you drink, for example if you cannot eat or you start to lose liquids from your body, for example from vomiting, diarrhea, or being in the sun too long.
- •
-
Vaginal yeast infection. Women who take FARXIGA may get vaginal yeast infections. Symptoms of a vaginal yeast infection include:
- ∘
- vaginal odor
- ∘
- white or yellowish vaginal discharge (discharge may be lumpy or look like cottage cheese)
- ∘
- vaginal itching
- •
-
Yeast infection of the penis (balanitis). Swelling of an uncircumcised penis may develop that makes it difficult to pull back the skin around the tip of the penis. Other symptoms of yeast infection of the penis include:
- ∘
- redness, itching, or swelling of the penis
- ∘
- foul smelling discharge from the penis
- ∘
- rash of the penis
- ∘
- pain in the skin around the penis
Talk to your healthcare provider about what to do if you get symptoms of a yeast infection of the vagina or penis. Your healthcare provider may suggest you use an over-the-counter antifungal medicine. Talk to your healthcare provider right away if you use an over-the-counter antifungal medication and your symptoms do not go away.
What is FARXIGA?
- •
- FARXIGA is a prescription medicine used:
- ∘
- to reduce the risk of further worsening of your kidney disease, end-stage kidney disease (ESKD), death due to cardiovascular disease, and hospitalization for heart failure in adults with chronic kidney disease
- ∘
- to reduce the risk of cardiovascular death, hospitalization for heart failure and urgent heart failure visit in adults with heart failure, when the heart cannot pump enough blood to the rest of your body
- ∘
- to reduce the risk of hospitalization for heart failure in adults with type 2 diabetes who also have known cardiovascular disease or multiple cardiovascular risk factors
- ∘
- along with diet and exercise to improve blood sugar (glucose) control in adults with type 2 diabetes.
- •
- FARXIGA is not for use to improve blood sugar (glucose) control in people with type 1 diabetes.
- •
- FARXIGA is not for use to improve blood sugar (glucose) control in adults with type 2 diabetes who have moderate to severe kidney problems, because it may not work.
- •
- FARXIGA is not for people with certain genetic forms of polycystic kidney disease, or who are taking or have recently received immunosuppressive therapy to treat kidney disease. FARXIGA is not expected to work if you have these conditions.
- •
- It is not known if FARXIGA is safe and effective in children younger than 18 years of age.
Who should not take FARXIGA?
Do not take FARXIGA if you:
- •
- are allergic to dapagliflozin or any of the ingredients in FARXIGA. See the end of this Medication Guide for a list of ingredients in FARXIGA. Symptoms of a serious allergic reaction to FARXIGA may include:
- ∘
- rash
- ∘
- raised red patches on your skin (hives)
- ∘
- swelling of the face, lips, tongue, and throat that may cause difficulty in breathing or swallowing.
- If you have any of these symptoms, stop taking FARXIGA and contact your healthcare provider or go to the nearest hospital emergency room right away.
What should I tell my healthcare provider before taking FARXIGA?
Before you take FARXIGA, tell your healthcare provider if you:
- •
- have type 1 diabetes or have had diabetic ketoacidosis.
- •
- have a decrease in your insulin dose.
- •
- have a serious infection.
- •
- have a history of infection of the vagina or penis.
- •
- have liver problems.
- •
- have a history of urinary tract infections or problems with urination.
- •
- are on a low sodium (salt) diet. Your healthcare provider may ask you to change your diet.
- •
- are going to have surgery. Your healthcare provider may stop your FARXIGA before you have surgery. Talk to your healthcare provider if you are having surgery about when to stop taking FARXIGA and when to start it again.
- •
- are eating less or there is a change in your diet.
- •
- are dehydrated.
- •
- have or have had problems with your pancreas, including pancreatitis or surgery on your pancreas.
- •
- drink alcohol very often or drink a lot of alcohol in the short term (“binge” drinking).
- •
- are pregnant or plan to become pregnant. FARXIGA may harm your unborn baby. If you become pregnant while taking FARXIGA, your healthcare provider may switch you to a different medicine to control your blood sugar. Talk to your healthcare provider about the best way to control your blood sugar if you plan to become pregnant or while you are pregnant.
- •
- are breastfeeding or plan to breastfeed. It is not known if FARXIGA passes into your breast milk. You should not breastfeed if you take FARXIGA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
FARXIGA may affect the way other medicines work, and other medicines may affect how FARXIGA works.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take FARXIGA?
- •
- Take FARXIGA exactly as your healthcare provider tells you to take it.
- •
- Take FARXIGA by mouth 1 time each day, with or without food.
- •
- Your healthcare provider will tell you how much FARXIGA to take and when to take it. Your healthcare provider may change your dose if needed.
- •
- If you miss a dose, take it as soon as you remember. If it is almost time for your next dose, skip the missed dose and take the medicine at the next regularly scheduled time. Do not take 2 doses of FARXIGA at the same time. Talk with your healthcare provider if you have questions about a missed dose.
- •
- If you take too much FARXIGA, call your healthcare provider or go to the nearest emergency room right away.
- •
- If you have diabetes:
- ∘
- When your body is under some types of stress, such as fever, trauma (such as a car accident), infection, or surgery, the amount of diabetes medicine you need may change. Tell your healthcare provider right away if you have any of these conditions and follow your healthcare provider’s instructions.
- ∘
- Your healthcare provider may tell you to take FARXIGA along with other diabetes medicines. Low blood sugar can happen more often when FARXIGA is taken with certain other diabetes medicines. See “What are the possible side effects of FARXIGA?”.
- •
- FARXIGA will cause your urine to test positive for glucose.
- •
- Your healthcare provider may do certain blood tests before you start FARXIGA and during treatment as needed. Your healthcare provider may change your dose of FARXIGA based on the results of your blood tests.
What are the possible side effects of FARXIGA?
FARXIGA may cause serious side effects, including:
See “What is the most important information I should know about FARXIGA?”.
- •
- Serious urinary tract infections. Serious urinary tract infections that may lead to hospitalization have happened in people who are taking FARXIGA. Tell your healthcare provider if you have any signs or symptoms of a urinary tract infection such as a burning feeling when passing urine, a need to urinate often, the need to urinate right away, pain in the lower part of your stomach (pelvis), or blood in the urine. Sometimes people also may have a fever, back pain, nausea or vomiting.
- •
- Low blood sugar (hypoglycemia) in patients with diabetes mellitus. If you take FARXIGA with another medicine that can cause low blood sugar, such as a sulfonylurea or insulin, your risk of getting low blood sugar is higher. The dose of your sulfonylurea medicine or insulin may need to be lowered while you take FARXIGA. Signs and symptoms of low blood sugar may include:
- ∘
- headache
- ∘
- confusion
- ∘
- hunger
- ∘
- shaking or feeling jittery
- ∘
- drowsiness
- ∘
- dizziness
- ∘
- fast heartbeat
- ∘
- weakness
- ∘
- sweating
- ∘
- irritibility
- •
- A rare but serious bacterial infection that causes damage to the tissue under the skin (necrotizing fasciitis) in the area between and around the anus and genitals (perineum). Necrotizing fasciitis of the perineum has happened in women and men with diabetes mellitus who take FARXIGA. Necrotizing fasciitis of the perineum may lead to hospitalization, may require multiple surgeries, and may lead to death. Seek medical attention right away if you have fever or you are feeling very weak, tired, or uncomfortable (malaise) and you develop any of the following symptoms in the area between and around the anus and genitals:
-
- ∘
- pain or tenderness
- ∘
- swelling
- ∘
- redness of skin (erythema)
- •
- Serious allergic reaction. If you have any symptoms of a serious allergic reaction, stop taking FARXIGA and call your healthcare provider right away or go to the nearest hospital emergency room. See “Who should not take FARXIGA?”. Your healthcare provider may give you a medicine for your allergic reaction and prescribe a different medicine for your diabetes.
The most common side effects of FARXIGA include:
- •
- vaginal yeast infections and yeast infections of the penis
- •
- stuffy or runny nose and sore throat
- •
- changes in urination, including urgent need to urinate more often, in larger amounts, or at night
These are not all the possible side effects of FARXIGA. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store FARXIGA?
Store FARXIGA at room temperature between 68°F and 77°F (20°C and 25°C).
General information about the safe and effective use of FARXIGA
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use FARXIGA for a condition for which it is not prescribed. Do not give FARXIGA to other people, even if they have the same symptoms you have. It may harm them.
This Medication Guide summarizes the most important information about FARXIGA. If you would like more information, talk to your healthcare provider. You can ask your pharmacist or healthcare provider for information about FARXIGA that is written for healthcare professionals.
For more information about FARXIGA, go to www.farxiga.com or call 1-800-236-9933.
What are the ingredients in FARXIGA?
Active ingredient: dapagliflozin.
Inactive ingredients: microcrystalline cellulose, anhydrous lactose, crospovidone, silicon dioxide, and magnesium stearate. The film coating contains: polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and yellow iron oxide.
Distributed by: AstraZeneca Pharmaceuticals LP Wilmington, DE 19850FARXIGA is a registered trademark of the AstraZeneca group of companies.
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised 09/2023
- DAPAGLIFLOZIN
- DAPAGLIFLOZIN
-
INGREDIENTS AND APPEARANCE
FARXIGA
dapagliflozin tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50090-3481(NDC:0310-6210) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DAPAGLIFLOZIN PROPANEDIOL (UNII: 887K2391VH) (DAPAGLIFLOZIN - UNII:1ULL0QJ8UC) DAPAGLIFLOZIN 10 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) anhydrous lactose (UNII: 3SY5LH9PMK) CROSPOVIDONE (UNII: 2S7830E561) silicon dioxide (UNII: ETJ7Z6XBU4) magnesium stearate (UNII: 70097M6I30) Product Characteristics Color YELLOW Score no score Shape DIAMOND (biconvenx) Size 11mm Flavor Imprint Code 10;1428 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50090-3481-0 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 06/15/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA202293 01/14/2008 FARXIGA
dapagliflozin tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50090-3482(NDC:0310-6205) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DAPAGLIFLOZIN PROPANEDIOL (UNII: 887K2391VH) (DAPAGLIFLOZIN - UNII:1ULL0QJ8UC) DAPAGLIFLOZIN 5 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) anhydrous lactose (UNII: 3SY5LH9PMK) CROSPOVIDONE (UNII: 2S7830E561) silicon dioxide (UNII: ETJ7Z6XBU4) magnesium stearate (UNII: 70097M6I30) Product Characteristics Color YELLOW Score no score Shape ROUND (biconvex) Size 7mm Flavor Imprint Code 5;1427 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50090-3482-0 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 06/15/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA202293 01/14/2008 Labeler - A-S Medication Solutions (830016429) Establishment Name Address ID/FEI Business Operations A-S Medication Solutions 830016429 RELABEL(50090-3481, 50090-3482)