TURALIO- pexidartinib capsule
Daiichi Sankyo Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use TURALIO safely and effectively. See full prescribing information for TURALIO.
TURALIO® (pexidartinib) capsules, for oral use Initial U.S. Approval: 2019 WARNING: HEPATOTOXICITYSee full prescribing information for complete boxed warning.
INDICATIONS AND USAGETURALIO is a kinase inhibitor indicated for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) associated with severe morbidity or functional limitations and not amenable to improvement with surgery. (1) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSCapsules: 200 mg (3) CONTRAINDICATIONSNone. (4) WARNINGS AND PRECAUTIONSADVERSE REACTIONSMost common adverse reactions (>20%) were increased lactate dehydrogenase, increased aspartate aminotransferase, hair color changes, fatigue, increased alanine aminotransferase, decreased neutrophils, increased cholesterol, increased alkaline phosphatase, decreased lymphocytes, eye edema, decreased hemoglobin, rash, dysgeusia, and decreased phosphate. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact Daiichi Sankyo, Inc. at 1-877-437-7763 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 10/2022 |
FULL PRESCRIBING INFORMATION
WARNING: HEPATOTOXICITY
- TURALIO can cause serious and potentially fatal liver injury [see Warnings and Precautions (5.1)].
- Monitor liver tests prior to initiation of TURALIO and at specified intervals during treatment. Withhold and dose reduce or permanently discontinue TURALIO based on severity of hepatotoxicity [see Dosage and Administration (2.3), Warnings and Precautions (5.1)].
- TURALIO is available only through a restricted program called the TURALIO Risk Evaluation and Mitigation Strategy (REMS) Program [see Warnings and Precautions (5.2)].
1 INDICATIONS AND USAGE
TURALIO is indicated for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) associated with severe morbidity or functional limitations and not amenable to improvement with surgery.
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
Administer TURALIO on an empty stomach, at least one hour before or two hours after a meal or snack [see Warnings and Precautions (5.1), Clinical Pharmacology (12.2, 12.3)].
2.2 Recommended Dosage
The recommended dosage of TURALIO is 400 mg taken twice daily on an empty stomach until disease progression or unacceptable toxicity [see Dosage and Administration (2.1), Clinical Pharmacology (12.3)].
Swallow TURALIO capsules whole. Do not open, break, or chew the capsules.
If a patient vomits or misses a dose of TURALIO, instruct the patient to take the next dose at its scheduled time.
2.3 Dosage Modifications for Adverse Reactions
The recommended dose reductions for adverse reactions are provided in Table 1.
| Dose Reduction | Total Daily Dose | Administration of Total Daily Dose |
|---|---|---|
| First | 600 mg | 200 mg in the morning and 400 mg in the evening |
| Second | 400 mg | 200 mg twice daily |
Permanently discontinue TURALIO in patients who are unable to tolerate 200 mg orally twice daily.
The recommended dosage modifications for adverse reactions are summarized in Table 2.
| Adverse Reaction | Severity | TURALIO Dosage Modifications |
|---|---|---|
| ALT = alanine aminotransferase; ALP = alkaline phosphatase; AST = aspartate aminotransferase; DB = direct bilirubin; GGT = gamma-glutamyl transferase; TB = total bilirubin; ULN = upper limit of normal | ||
|
||
| Hepatotoxicity [see Warnings and Precautions (5.1)] | ||
| Increased ALT and/or AST | Greater than 3 to 5 times ULN |
|
| Greater than 5 to 10 times ULN |
|
|
| Greater than 10 times ULN |
|
|
| Increased ALP* and GGT | ALP greater than 2 times ULN with GGT greater than 2 times ULN |
|
| Increased bilirubin | TB greater than ULN to less than 2 times ULN or DB greater than ULN and less than 1.5 times ULN |
|
| TB greater or equal to 2 times ULN or DB greater than 1.5 times ULN |
|
|
| Adverse Reactions or Other Laboratory Abnormalities [see Adverse Reactions (6.1)] | ||
| Any | Severe or intolerable |
|
2.4 Concomitant Use of Moderate or Strong CYP3A Inhibitors or UGT Inhibitors
Avoid concomitant use of TURALIO with moderate or strong CYP3A inhibitors or UGT inhibitors during treatment with TURALIO. If concomitant use with a moderate or strong CYP3A inhibitor or UGT inhibitor cannot be avoided, reduce the TURALIO dose according to the recommendations in Table 3.
If concomitant use of a moderate or strong CYP3A inhibitor or UGT inhibitor is discontinued, increase the TURALIO dose (after 3 plasma half-lives of the moderate or strong CYP3A inhibitor or UGT inhibitor) to the dose that was used before starting the inhibitor [see Clinical Pharmacology (12.3)].
| Planned Total Daily Dose | Modified Total Daily Dose | Administration of Modified Total Daily Dose |
|---|---|---|
|
||
| 800 mg | 400 mg | 200 mg twice daily |
| 600 mg* | 400 mg | 200 mg twice daily |
| 400 mg* | 200 mg | 200 mg once daily |
2.5 Concomitant Use of Acid-Reducing Agents
Avoid the concomitant use of proton pump inhibitors (PPI) while taking TURALIO. As an alternative to a PPI, administer TURALIO 2 hours before or 2 hours after taking a locally-acting antacid, or if using a histamine 2 (H2)-receptor antagonist, administer TURALIO at least 2 hours before or 10 hours after taking an H2-receptor antagonist [see Clinical Pharmacology (12.3)].
2.6 Dosage Modification for Renal Impairment
The recommended dosage of TURALIO for patients with mild to severe renal impairment (creatinine clearance [CLcr] 15 to 89 mL/min estimated by Cockcroft-Gault using actual body weight) is 200 mg in the morning and 400 mg in the evening [see Clinical Pharmacology (12.3)].
2.7 Dosage Modification for Hepatic Impairment
The recommended dosage of TURALIO for patients with moderate hepatic impairment (total bilirubin greater than 1.5 and up to 3 times upper limit of normal (ULN), not due to Gilbert's syndrome, with any AST) is 200 mg twice daily [see Clinical Pharmacology (12.3)]. TURALIO has not been studied in patients with severe hepatic impairment (total bilirubin greater than 3 to 10 times ULN and any AST).
3 DOSAGE FORMS AND STRENGTHS
Capsules: 200 mg, size 0 with white opaque body and dark green opaque cap with white print "T10"
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
TURALIO can cause serious and potentially fatal liver injury and is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) [see Warnings and Precautions (5.2)].
Hepatotoxicity with ductopenia and cholestasis occurred in patients treated with TURALIO. Across 768 patients who received TURALIO in clinical trials, there were two irreversible cases of cholestatic liver injury. One patient died with advanced cancer and ongoing liver toxicity and one patient required a liver transplant. The mechanism of cholestatic hepatotoxicity is unknown and its occurrence cannot be predicted. It is unknown whether liver injury occurs in the absence of increased transaminases.
In ENLIVEN, 3 of 61 (5%) patients who received TURALIO developed signs of serious liver injury, defined as ALT or AST ≥3 × ULN with total bilirubin ≥2 × ULN. In these patients, peak ALT ranged from 6 to 9 × ULN, peak total bilirubin ranged from 2.5 to 15 × ULN, and alkaline phosphatase (ALP) was ≥2 × ULN. ALT, AST and total bilirubin improved to <2 × ULN in these patients 1 to 7 months after discontinuing TURALIO.
Avoid TURALIO in patients with pre-existing increased serum transaminases; total bilirubin or direct bilirubin (>ULN); or active liver or biliary tract disease, including increased ALP. Taking TURALIO with food increases drug exposure by 100% and may increase the risk of hepatotoxicity. Administer TURALIO on an empty stomach, either 1 hour before or 2 hours after a meal or snack [see Dosage and Administration (2.1), Clinical Pharmacology (12.2, 12.3)]. Monitor liver tests, including AST, ALT, total bilirubin, direct bilirubin, ALP and gamma-glutamyl transferase (GGT), prior to initiation of TURALIO, weekly for the first 8 weeks, every 2 weeks for the next month and every 3 months thereafter. Withhold and dose reduce, or permanently discontinue TURALIO based on the severity of the hepatotoxicity [see Dosage and Administration (2.2)]. Rechallenge with a reduced dose of TURALIO may result in a recurrence of increased serum transaminases, bilirubin, or ALP. Monitor liver tests weekly for the first month after rechallenge.
5.2 TURALIO REMS Program
TURALIO is only available through a restricted program under a REMS, because of the risk of hepatotoxicity [see Warnings and Precautions (5.1)].
Notable requirements of the TURALIO REMS Program include the following:
- Prescribers must be certified with the program by enrolling and completing training.
- Patients must complete and sign an enrollment form for inclusion in a patient registry.
- Pharmacies must be certified with the program and must only dispense to patients who are authorized to receive TURALIO.
Further information is available at www.TURALIOREMS.com or 1-833-887-2546.
5.3 Embryo-Fetal Toxicity
Based on animal studies and its mechanism of action, TURALIO may cause fetal harm when administered to a pregnant woman. Oral administration of pexidartinib to pregnant rats and rabbits during the period of organogenesis resulted in malformations, increased post-implantation loss, and abortion at exposures approximately equal to the human exposure at the recommended dose of 800 mg based on area under the curve (AUC).
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective non-hormonal contraception, since TURALIO can render hormonal contraceptives ineffective, during treatment with TURALIO and for 1 month after the final dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with TURALIO and for 1 week after the final dose [see Drug Interactions (7.3), Use in Specific Populations (8.1, 8.3)].
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hepatotoxicity [see Warnings and Precautions (5.1)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of TURALIO was evaluated in ENLIVEN [see Clinical Studies (14.1)]. ENLIVEN excluded patients with ALT, AST, or total bilirubin >1.5 × ULN; and known active or chronic infection with hepatitis B or C virus, or human immunodeficiency virus. Patients received TURALIO without food at a dose of 400 mg in the morning and 600 mg in the evening orally for 2 weeks followed by 400 mg orally twice daily until disease progression or unacceptable toxicity. Seventy-nine percent of patients received TURALIO for 6 months or longer and 66% for greater than one year.
The median age of TURALIO-treated patients was 44 years (range: 22-75), 57% were females, and 85% were White.
Serious adverse reactions were reported in 13% of patients who received TURALIO. Most frequent (occurring in >1 patient) serious adverse reactions included abnormal liver tests (3.3%) and hepatotoxicity (3.3%).
Permanent discontinuation due to an adverse reaction occurred in 13% of patients who received TURALIO. Most frequent adverse reactions (occurring in >1 patient) requiring permanent discontinuation included increased ALT (4.9%), increased AST (4.9%) and hepatotoxicity (3.3%).
Dose reductions or interruptions occurred in 38% of patients who received TURALIO. Most frequent adverse reactions (occurring in >1 patient) requiring a dosage reduction or interruption were increased ALT (13%), increased AST (13%), nausea (8%), increased ALP (7%), vomiting (4.9%), increased bilirubin (3.3%), increased GGT (3.3%), dizziness (3.3%), and abdominal pain (3.3%).
The most common (>20%) adverse reactions, including laboratory abnormalities, in patients who received TURALIO were: increased lactate dehydrogenase (LDH), increased AST, hair color changes, fatigue, increased ALT, decreased neutrophils, increased cholesterol, increased ALP, decreased lymphocytes, eye edema, decreased hemoglobin, rash, dysgeusia and decreased phosphate.
Tables 4, 5 and 6 summarize the adverse reactions and laboratory abnormalities in ENLIVEN during the randomized phase (Week 25).
| TURALIO N=61 | Placebo N=59 |
|||
|---|---|---|---|---|
| Adverse Reaction | All Grades (%) | Grade ≥ 3 (%) | All Grades (%) | Grade ≥ 3 (%) |
|
||||
| Skin and subcutaneous tissue | ||||
| Hair color changes | 67 | 0 | 3.4 | 0 |
| Rash* | 28 | 1.6 | 7 | 0 |
| Pruritus† | 18 | 0 | 3.4 | 0 |
| General | ||||
| Fatigue‡ | 64 | 0 | 41 | 0 |
| Peripheral edema§ | 20 | 0 | 7 | 0 |
| Eye | ||||
| Eye edema¶ | 30 | 1.6 | 5 | 0 |
| Nervous system | ||||
| Dysgeusia# | 26 | 0 | 1.7 | 0 |
| NeuropathyÞ | 10 | 0 | 5 | 0 |
| Gastrointestinal | ||||
| Vomiting | 20 | 1.6 | 5 | 0 |
| Constipation | 12 | 0 | 5 | 0 |
| Metabolism and nutrition | ||||
| Decreased appetite | 16 | 0 | 10 | 0 |
| Vascular | ||||
| Hypertension | 15 | 4.9 | 10 | 0 |
| TURALIO* | Placebo* | |||||
|---|---|---|---|---|---|---|
| Laboratory Abnormality† | Grade 1 (%) | Grade 2 (%) | Grade ≥ 3 (%) | Grade 1 (%) | Grade 2 (%) | Grade ≥ 3 (%) |
| ALT = alanine aminotransferase; AST = aspartate aminotransferase; ALP = alkaline phosphatase | ||||||
| Liver Tests | ||||||
| Increased AST | 61 | 15 | 12 | 15 | 0 | 0 |
| Increased ALT | 31 | 13 | 20 | 22 | 0 | 0 |
| Increased ALP | 31 | 3.3 | 4.9 | 1.7 | 0 | 0 |
| Increased bilirubin | 3.3 | 3.3 | 3.3 | 0 | 0 | 0 |
| TURALIO* | Placebo* | |||
|---|---|---|---|---|
| Laboratory Abnormality† | All Grades (%) | Grade ≥3 (%) | All Grades (%) | Grade ≥3 (%) |
| LDH=Lactate Dehydrogenase | ||||
|
||||
| Chemistry | ||||
| Increased LDH‡ | 92 | 0 | 5 | 0 |
| Increased cholesterol | 44 | 4.9 | 25 | 0 |
| Decreased phosphate | 25 | 3.3 | 5 | 0 |
| Hematology | ||||
| Decreased neutrophils | 44 | 3.3 | 9 | 0 |
| Decreased lymphocytes | 38 | 1.6 | 3.4 | 0 |
| Decreased hemoglobin | 30 | 0 | 14 | 1.7 |
| Decreased platelets | 15 | 0 | 5 | 0 |
Clinically relevant adverse reactions occurring in <10% of patients were:
Eye: blurred vision, photophobia, diplopia, reduced visual acuity
Gastrointestinal: dry mouth, stomatitis, mouth ulceration
General: pyrexia
Hepatobiliary: cholangitis, hepatotoxicity, liver disorder
Neurological: cognitive disorders (memory impairment, amnesia, confusional state, disturbance in attention, attention deficit/hyperactivity disorder)
Skin and subcutaneous tissue: alopecia, skin pigment changes (hypopigmentation, depigmentation, discoloration, hyperpigmentation)
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of TURALIO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Investigations: Blood creatine phosphokinase increased
7 DRUG INTERACTIONS
7.1 Use with Hepatotoxic Products
TURALIO can cause hepatotoxicity. In patients with increased serum transaminases, total bilirubin, or direct bilirubin (>ULN) or active liver or biliary tract disease, avoid coadministration of TURALIO with other products known to cause hepatotoxicity [see Warnings and Precautions (5.1)].
7.2 Effect of Other Drugs on TURALIO
| Moderate or Strong CYP3A Inhibitors | |
| Clinical Impact |
|
| Management |
|
| Strong CYP3A Inducers | |
| Clinical Impact |
|
| Management |
|
| UGT Inhibitors | |
| Clinical Impact |
|
| Management |
|
| Acid-Reducing Agents | |
| Clinical Impact |
|
| Management |
|
7.3 Effect of TURALIO on Other Drugs
| CYP3A Substrates | |
| Clinical Impact |
|
| Management |
|
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], TURALIO may cause embryo-fetal harm when administered to a pregnant woman. The available human data do not establish the presence or absence of major birth defects or miscarriage related to the use of TURALIO. Oral administration of pexidartinib to pregnant animals during the period of organogenesis resulted in malformations, post-implantation loss, and abortion at maternal exposures that were approximately equal to the human exposure at the recommended dose of 800 mg (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Embryo-fetal development studies investigating the administration of pexidartinib during the period of organogenesis were conducted in rats and rabbits. In rats, pexidartinib resulted in increased post-implantation loss and fetal malformations including localized fetal edema, absence of kidney and ureter, abnormalities of the reproductive tract, and developmental variations including misshapen kidney, decreased skeletal ossification and higher mean litter proportions of slightly or moderately malaligned sternebrae at doses of 40 mg/kg (approximately equal to the human exposure at the recommended dose of 800 mg). In rabbits, administration of pexidartinib resulted in increased post-implantation loss, abortion, and fetal malformations including absence of kidney or ureter, rudimentary, misshapen or malpositioned kidney, rib abnormalities, and skeletal variations of accessory skull bones at doses of 60 mg/kg (approximately equal to the human exposure at the recommended dose of 800 mg).
8.2 Lactation
Risk Summary
There are no data on the presence of pexidartinib or its metabolites in either human or animal milk or its effects on a breastfed child or on milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with TURALIO and for at least 1 week after the final dose.
8.3 Females and Males of Reproductive Potential
TURALIO may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to the initiation of TURALIO [see Use in Specific Populations (8.1)].
Contraception
Females
Advise females of reproductive potential to use effective non-hormonal contraception during treatment with TURALIO and for 1 month after the final dose. Counsel patients to use non-hormonal method(s) of contraception, since TURALIO can render hormonal contraceptives ineffective [see Drug Interactions (7.3), Nonclinical Toxicology (13.1)].
Males
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TURALIO and for 1 week after the final dose [see Nonclinical Toxicology (13.1)].
Infertility
Based on findings from animal studies, TURALIO may impair both male and female fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of TURALIO in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of TURALIO did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
8.6 Renal Impairment
Reduce the dose when administering TURALIO to patients with mild to severe renal impairment (CLcr 15 to 89 mL/min, estimated by Cockcroft-Gault [C-G]) [see Dosage and Administration (2.6), Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dosage adjustment is recommended for patients with mild hepatic impairment (total bilirubin less than or equal to upper limit of normal [ULN] with AST greater than ULN or total bilirubin greater than 1 and up to 1.5 times ULN with any AST) [see Clinical Pharmacology (12.3)].
Reduce the dosage of TURALIO for patients with moderate hepatic impairment (total bilirubin greater than 1.5 and up to 3 times ULN, not due to Gilbert's syndrome, with any AST) [see Dosage and Administration (2.7), Clinical Pharmacology (12.3)].
TURALIO has not been studied in patients with severe hepatic impairment (total bilirubin greater than 3 to 10 times ULN and any AST).
10 OVERDOSAGE
Due to the high plasma protein binding, TURALIO is not expected to be dialyzable [see Clinical Pharmacology (12.3)].
11 DESCRIPTION
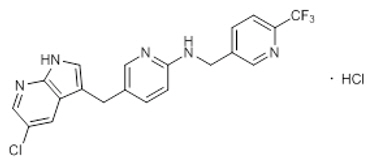
Pexidartinib is a kinase inhibitor. The chemical name of pexidartinib hydrochloride is 5-[(5-Chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)methyl]-N-{[6-(trifluoromethyl)pyridin-3-yl]methyl}pyridin-2-amine monohydrochloride. Pexidartinib hydrochloride is an off-white to white solid. The molecular formula for pexidartinib hydrochloride is C20H15ClF3N5∙HCl. The molecular weight is 454.28 for the hydrochloride salt and 417.81 for the free base. The chemical structure is:
The solubility of pexidartinib hydrochloride in aqueous solutions decreases with increasing pH. The pKa1 and pKa2 were determined to be 2.6 and 5.4 respectively for the conjugate acids. Pexidartinib hydrochloride is soluble in methanol, slightly soluble in water and ethanol, and practically insoluble in heptane.
TURALIO (pexidartinib) capsules are for oral use. Each capsule contains 200 mg pexidartinib which is equivalent to 217.5 mg pexidartinib hydrochloride. The capsule contains the following inactive ingredients: poloxamer 407, mannitol, crospovidone, and magnesium stearate. The hypromellose capsule shell contains hypromellose, titanium dioxide, black iron oxide and yellow iron oxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pexidartinib is a small molecule tyrosine kinase inhibitor that targets colony stimulating factor 1 receptor (CSF1R), KIT proto-oncogene receptor tyrosine kinase (KIT), and FMS-like tyrosine kinase 3 (FLT3) harboring an internal tandem duplication (ITD) mutation. Overexpression of the CSF1R ligand promotes cell proliferation and accumulation in the synovium. In vitro, pexidartinib inhibited proliferation of cell lines dependent on CSF1R and ligand-induced autophosphorylation of CSF1R. Pexidartinib also inhibited the proliferation of a CSF1R dependent cell line in vivo.
12.2 Pharmacodynamics
Exposure-Response Relationships
There is an exposure response relationship between pexidartinib steady state exposure and serum transaminase levels (ALT and AST) with a higher risk of increased serum transaminases at higher exposure. Additionally, increased transaminases occurred more frequently with higher pexidartinib doses (200 to 1200 mg per day).
12.3 Pharmacokinetics
The pharmacokinetics of TURALIO was evaluated following single doses in healthy subjects and following multiple doses in patients as summarized in Table 9.
|
||
| General Information | ||
| Steady state exposure [Mean (SD)]* | Cmax | 8625 (2746) ng/mL |
| AUC0-12h | 77465 (24975) ng∙h/mL | |
| Dose proportionality | Pexidartinib exposure (Cmax and AUC0-INF) increased linearly over the single oral dose range of 200 to 2400 mg (0.5 to 6 times the recommended dose). | |
| Time to steady state† | Approximately 7 days | |
| Accumulation ratio (AUC) [Median] † | 3.6 | |
| Absorption | ||
| Tmax [Median] | 2.5 hours | |
| Effect of food | ||
| Administration with high-fat meal‡ |
|
|
| Administration with low-fat meal§ |
|
|
| Distribution | ||
| In vitro plasma protein binding |
|
|
| Apparent volume of distribution (Vz/F) [Mean (CV%)]¶ |
|
|
| Elimination | ||
| Apparent clearance [Mean (CV%)]¶ |
|
|
| t1/2 [Mean (SD)] |
|
|
| Metabolism | ||
| Primary pathway |
|
|
| N-glucuronide metabolite |
|
|
| Excretion# | ||
|
||
Specific Populations
No clinically meaningful differences in the pharmacokinetics of pexidartinib were observed based on age (18 to 84 years), sex, race (White and Black), or mild hepatic impairment (total bilirubin ≤ ULN with AST > ULN or total bilirubin > 1 to 1.5 × ULN with any AST).
Patients with Renal Impairment
Mild (CLcr 60 to 89 mL/min), moderate (CLcr 30 to 59 mL/min) and severe (CLcr 15 to 29 mL/min) renal impairment increased pexidartinib exposure (AUC) by approximately 30%, relative to that in patients with normal renal function (CLcr ≥90 mL/min).
Patients with Hepatic Impairment
Moderate hepatic impairment (total bilirubin > 1.5 and up to 3 × ULN, not due to Gilbert's syndrome, with any AST) increased pexidartinib exposure (AUC) by 43% relative to exposure in patients with normal hepatic function (total bilirubin and AST ≤ ULN).
The pharmacokinetics of pexidartinib have not been studied in patients with severe hepatic impairment (total bilirubin > 3 to 10 × ULN with any AST).
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Effects of Other Drugs on Pexidartinib
Strong CYP3A Inducers: Coadministration of rifampicin (strong CYP3A inducer) decreased pexidartinib Cmax by 33% and AUC0-INF by 65%.
Moderate CYP3A Inducers: Coadministration of efavirenz (moderate CYP3A inducer) is predicted to decrease pexidartinib Cmax by 27% and AUC by 38% at steady state.
Strong CYP3A Inhibitors: Coadministration of itraconazole (strong CYP3A inhibitor) increased pexidartinib Cmax by 48% and AUC0-INF by 70%.
Moderate CYP3A Inhibitors: Coadministration of fluconazole (moderate CYP3A inhibitor) is predicted to increase pexidartinib Cmax by 41% and AUC by 67% at steady state.
Effects of Pexidartinib on Other Drugs
CYP2C19 Substrates: Coadministration of a single oral dose of TURALIO 1800 mg (4.5 times the approved recommended dose of 400 mg) decreased omeprazole (CYP2C19 substrate) Cmax by 37% and AUC0-INF by 17%.
CYP3A Substrates: Coadministration of TURALIO 400 mg twice daily decreased midazolam (CYP3A substrate) Cmax by 28% and AUC0-INF by 59%.
CYP2C9 Substrates: Coadministration of TURALIO 400 mg twice daily increased tolbutamide (CYP2C9 substrate) AUC0-INF by 28%. The effect of pexidartinib on CYP2C9 substrates is not considered clinically relevant.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies were performed in mice and rats. Both studies were negative for carcinogenic findings at exposures up to 9 times the human exposure at the recommended daily dose of 800 mg based on AUC.
Pexidartinib was not mutagenic in an in vitro bacterial reverse mutation (AMES) assay or clastogenic in either an in vitro human peripheral blood lymphocyte chromosomal aberrations assay or in an in vivo mouse bone marrow micronucleus assay.
Based on nonclinical findings, TURALIO may impair male and female fertility. In a fertility study in which pexidartinib was administered orally to male and female rats, there were reductions in pregnancy, as well as increases in pre- and post-implantation loss with a corresponding reduction in viable embryos at 40 mg/kg (approximately 1.3 times the human exposure at the recommended dose of 800 mg). Males at this dose level displayed reductions in spermatogenic parameters and adverse effects on sperm concentration, production, motility, and morphology. Lower testicular and epididymal weights occurred in this study at doses of ≥10 mg/kg/day (approximately 0.3 times the human exposure at the recommended dose of 800 mg). This is consistent with findings in chronic toxicology studies of germ cell depletion of the testes and hypospermia and cellular debris in the epididymis in male reproductive tissues of both rats and dogs at respective doses as low as 20 and 30 mg/kg/day (approximately 0.6 and 0.1 times the human exposure at the recommended dose of 800 mg). In rats, these changes persisted following a 16-week recovery period at the 60 mg/kg/day dose level (approximately 1.5 times the human exposure at the recommended dose of 800 mg).
In female rats, necrosis of corpora lutea occurred at doses ≥0.5 mg/kg/day (approximately 0.01 times the human exposure at the recommended dose of 800 mg) with pigment deposition within the interstitium of the ovaries, an increased incidence of luteal cysts and incidence/severity of hemorrhage of corpora lutea, and a decreased incidence of retained antral follicles and decreased corpora lutea at 60 mg/kg (approximately 1.8 times the human exposure at the recommended dose of 800 mg). In female dogs there were decreased follicle numbers and moderate atrophy of the oviduct, uterus, and cervix at doses as low as 1 mg/kg (approximately 0.01 times the human exposure at the recommended dose of 800 mg).
13.2 Animal Toxicology and/or Pharmacology
In repeat dose toxicity studies of up to 26 weeks in rats, there were findings of myxomatous change in the skin, tongue, and gastrointestinal tract, lymphoid depletion of the bone marrow and thymus, and chronic progressive nephropathy of the kidney at 20 mg/kg/day (approximately 0.6 times the human exposure at the recommended dose of 800 mg). Similar changes occurred in the rat carcinogenicity study along with alterations in the tunica intima of the aorta. Vascular inflammation consistent with polyarteritis nodosa occurred in male rats at 60 mg/kg/day (approximately 1.5 times the human exposure at the recommended dose of 800 mg). There were also dose-dependent findings of minimal to moderate subphyseal or cortical hyperostosis and physeal hypertrophy in the femur that correlated with decreased systemic phosphate levels at doses ≥ 60 mg/kg.
14 CLINICAL STUDIES
14.1 Tenosynovial Giant Cell Tumor
The efficacy of TURALIO was evaluated in ENLIVEN (NCT02371369), a double-blind, randomized (1:1), placebo-controlled, multicenter trial in patients with symptomatic TGCT [also referred to as giant cell tumor of the tendon sheath (GCT-TS) or pigmented villonodular synovitis (PVNS)] for whom surgical removal of the tumor would be associated with worsening functional limitation or severe morbidity. Eligible patients were required to have measurable disease per the Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Patients were randomized to placebo or TURALIO 400 mg in the morning and 600 mg in the evening for 2 weeks followed by 400 mg twice daily. Treatment continued until unacceptable toxicity or disease progression. Randomization was stratified by geographic region (US vs. non-US countries) and disease location (upper extremity vs. lower extremity involvement). Patients who completed treatment in the double-blind, randomized part of the trial were eligible to advance to an open-label extension part in which all patients were given the option to receive pexidartinib.
The major efficacy outcome measure was overall response rate (ORR) as assessed by blinded independent central review (BICR) at Week 25 using RECIST v1.1. Additional efficacy outcome measures were mean change from baseline in range of motion of the affected joint at Week 25 and ORR as assessed by BICR at Week 25 using tumor volume score (TVS). Range of motion was measured as a percent of normal reference range for the affected joint. Range of motion assessments were performed by a third-party clinical assessor using a goniometer. TVS was defined in ENLIVEN as the estimated volume of the maximally distended synovial cavity or tendon sheath involved, measured in 10% increments. Patients in the placebo arm were offered TURALIO at Week 25 beginning with a 400 mg twice daily dose, as permitted by the study protocol.
A total of 120 patients were randomized, 61 to the TURALIO arm and 59 to the placebo arm. The median age was 44 years (range: 18-79); 59% were females; 88% were White; 53% had prior surgery; 88% were diagnosed with diffuse TGCT; and 9% had previously been treated with systemic therapy. Disease locations were knee (61%), ankle (18%), hip (11%), wrist (3%), foot (3%) and other (5%).
ENLIVEN demonstrated a statistically significant improvement in ORR in patients randomized to TURALIO compared with placebo. Efficacy results are summarized in Table 10.
| Efficacy Parameter | TURALIO N=61 | Placebo N=59 |
|---|---|---|
| CI: confidence interval; NA: not applicable; SD: standard deviation; LS: least squares; +: denotes ongoing at last assessment | ||
| Overall Response Rate (ORR)*,† | ||
| ORR (95% CI) | 38% (27%, 50%) | 0 (0, 6%) |
| Complete Response | 15% | 0 |
| Partial Response | 23% | 0 |
| P-value‡ | <0.0001 | |
| Duration of Response (DOR)† | ||
| Range (months) | 6.9+, 24.9+ | NA |
The analysis of mean change from baseline in range of motion at Week 25 demonstrated a statistically significant improvement in patients randomized to TURALIO compared to placebo. Figure 1 shows the change from baseline in range of motion for each patient at Week 25 (TURALIO N=45, placebo N=43). Results were excluded for 1 patient with missing baseline and 31 patients with a missing range of motion assessment at Week 25.
| Figure 1: Change from Baseline in Range of Motion at Week 25 for ENLIVEN |
|
|
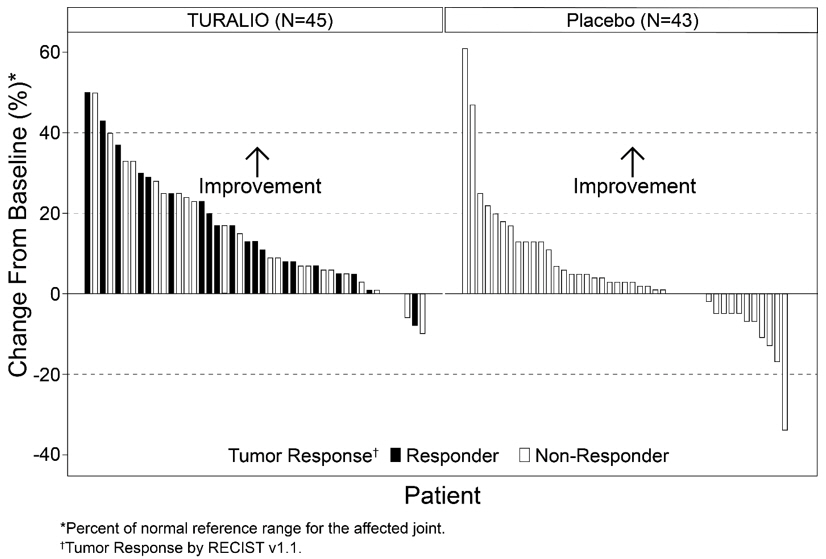
ORR by TVS was 56% (95% CI: 43%, 67%) in patients randomized to the TURALIO arm and 0% in patients randomized to the placebo arm; p < 0.0001.
At completion of the open-label extension part of the study in which all patients received TURALIO, the ORR using RECIST v1.1 was 61% (95% CI: 48%, 72%) in the 61 patients originally randomized to the TURALIO arm. The median duration of response was not reached (range: 4.6+, 63.4+ months) in the 37 responders.
16 HOW SUPPLIED/STORAGE AND HANDLING
TURALIO 200 mg capsules are supplied as size 0 with white opaque body and dark green opaque cap with white print "T10", available in:
| • 28 count bottle | NDC#: 65597-402-28 |
| • 120 count bottle | NDC#: 65597-402-20 |
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Hepatotoxicity
Advise patients of the risk of hepatotoxicity that could be fatal and that they will need to undergo monitoring for liver injury and to report immediately any signs or symptoms of severe liver injury to their healthcare provider [see Warnings and Precautions (5.1)].
TURALIO REMS Program
- TURALIO is available only through a restricted program called TURALIO REMS Program and patients are required to be part of the patient registry [see Warnings and Precautions (5.2)].
- TURALIO is available only from certified pharmacies participating in the program. Therefore, provide patients with the telephone number and website for information on how to obtain the product.
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.3), Use in Specific Populations (8.1, 8.3)].
- Advise females of reproductive potential to use effective non-hormonal contraception during treatment with TURALIO and for 1 month after the final dose [see Drug Interactions (7.3), Use in Specific Populations (8.3)].
- Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 1 week after the final dose [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Lactation
Advise females not to breastfeed during treatment with TURALIO and for 1 week after the final dose [see Use in Specific Populations (8.2)].
Infertility
Advise females and males of reproductive potential that TURALIO may impair fertility [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Drug Interactions
Advise patients to inform their healthcare providers of all concomitant products, including over-the-counter products and supplements [see Dosage and Administration (2), Drug Interactions (7)].
Administration
Instruct patients to take TURALIO on an empty stomach (at least 1 hour before or 2 hours after a meal or snack). Instruct patients to swallow capsules whole (do not open, break, or chew) [see Dosage and Administration (2.1, 2.2)].
Manufactured for: Daiichi Sankyo, Inc.
Basking Ridge, NJ 07920
TURALIO® is a registered trademark of Daiichi Sankyo Company, Limited.
©2022, Daiichi Sankyo, Inc.
USPI-TUR-C6-1022-r005
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 10/2021 |
| Medication Guide
TURALIO® (tur-a-lee-oh) (pexidartinib) Capsules |
|
| What is the most important information I should know about TURALIO? TURALIO can cause serious side effects, including: Serious Liver Problems which may be severe and can lead to death. Your healthcare provider will do blood tests to check for liver problems:
Stop taking TURALIO and call your healthcare provider right away if you develop:
|
|
|
|
| TURALIO Risk Evaluation and Mitigation Strategy (REMS): Because of the risk of serious liver problems, TURALIO is available only through a restricted program called the TURALIO REMS Program. Your healthcare provider must be enrolled in the program in order for you to be prescribed TURALIO. There is a registry that collects information about the effects of taking TURALIO over time. You must complete and sign an enrollment form for the TURALIO REMS Program and the registry. Ask your healthcare provider for more information. | |
| What is TURALIO?
TURALIO is a prescription medicine used to treat certain adults who have tenosynovial giant cell tumor (TGCT) that is not likely to improve with surgery. TGCT is also known as giant cell tumor of the tendon sheath (GCT-TS) or pigmented villonodular synovitis (PVNS). It is not known if TURALIO is safe and effective in children. |
|
Before taking TURALIO, tell your healthcare provider about all of your medical conditions, including if you:
|
|
How should I take TURALIO?
|
|
What should I avoid while taking TURALIO?
|
|
| What are the possible side effects of TURALIO? TURALIO can cause serious side effects. See "What is the most important information I should know about TURALIO?" The most common side effects of TURALIO include: |
|
|
|
| TURALIO may affect fertility in females and males, which may affect your ability to have children. Talk to your healthcare provider if you have concerns about fertility. These are not all of the possible side effects of TURALIO. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store TURALIO?
|
|
| General information about the safe and effective use of TURALIO
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use TURALIO for a condition for which it was not prescribed. Do not give TURALIO to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TURALIO that is written for health professionals. |
|
| What are the ingredients in TURALIO? Active Ingredient: pexidartinib Inactive Ingredients: poloxamer 407, mannitol, crospovidone, and magnesium stearate. Capsule shell: hypromellose, titanium dioxide, black iron oxide and yellow iron oxide Manufactured for: Daiichi Sankyo, Inc., Basking Ridge, NJ 07920 TURALIO® is a registered trademark of Daiichi Sankyo Company, Limited. ©2020, Daiichi Sankyo, Inc. USMG-TUR-C4-1021-r003 For more information, call 1-877-437-7763 or go to https://www.turalio.com/. |
|


| TURALIO
pexidartinib capsule |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Daiichi Sankyo Inc. (068605067) |