IXEMPRA
-
ixabepilone
----------
IXEMPRA™ Kit (ixabepilone) for Injection, for intravenous infusion only
|
||||||||||||||||||||||
FULL PRESCRIBING INFORMATION
WARNING: TOXICITY IN HEPATIC IMPAIRMENT
IXEMPRA in combination with capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN due to increased risk of toxicity and neutropenia-related death [see Contraindications (4) and Warnings and Precautions (5.3)].
1 INDICATIONS AND USAGE
IXEMPRA (ixabepilone) is indicated in combination with capecitabine for the treatment of patients with metastatic or locally advanced breast cancer resistant to treatment with an anthracycline and a taxane, or whose cancer is taxane resistant and for whom further anthracycline therapy is contraindicated. Anthracycline resistance is defined as progression while on therapy or within 6 months in the adjuvant setting or 3 months in the metastatic setting. Taxane resistance is defined as progression while on therapy or within 12 months in the adjuvant setting or 4 months in the metastatic setting.
IXEMPRA is indicated as monotherapy for the treatment of metastatic or locally advanced breast cancer in patients whose tumors are resistant or refractory to anthracyclines, taxanes, and capecitabine.
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
The recommended dosage of IXEMPRA is 40 mg/m2 administered intravenously over 3 hours every 3 weeks. Doses for patients with body surface area (BSA) greater than 2.2 m2 should be calculated based on 2.2 m2.
2.2 Dose Modification
Dose Adjustments During Treatment
Patients should be evaluated during treatment by periodic clinical observation and laboratory tests including complete blood cell counts. If toxicities are present, treatment should be delayed to allow recovery. Dosing adjustment guidelines for monotherapy and combination therapy are shown in Table 1. If toxicities recur, an additional 20% dose reduction should be made.
| IXEMPRA (Monotherapy or Combination Therapy) | IXEMPRA Dose Modification |
|---|---|
| a Toxicities graded in accordance with National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE v3.0). | |
| Nonhematologic: | |
| Grade 2 neuropathy (moderate) lasting ≥7 days | Decrease the dose by 20% |
| Grade 3 neuropathy (severe) lasting <7 days | Decrease the dose by 20% |
| Grade 3 neuropathy (severe) lasting ≥7 days or disabling neuropathy | Discontinue treatment |
| Any grade 3 toxicity (severe) other than neuropathy | Decrease the dose by 20% |
| Transient grade 3 arthralgia/myalgia or fatigue | No change in dose of IXEMPRA |
| Grade 3 hand-foot syndrome (palmar-plantar erythrodysesthesia) | |
| Any grade 4 toxicity (disabling) | Discontinue treatment |
| Hematologic: | |
| Neutrophil <500 cells/mm3 for ≥7 days | Decrease the dose by 20% |
| Febrile neutropenia | Decrease the dose by 20% |
| Platelets <25,000/mm3 or platelets <50,000/mm3 with bleeding | Decrease the dose by 20% |
| CAPECITABINE (when used in combination with IXEMPRA) | Capecitabine Dose Modification |
| Nonhematologic: | Follow Capecitabine Label |
| Hematologic: | |
| Platelets <25,000/mm3or <50,000/mm3 with bleeding | Hold for concurrent diarrhea or stomatitis until platelet count >50,000/mm3, then continue at same dose. |
| Neutrophils <500 cells/mm3 for ≥7 days or febrile neutropenia | Hold for concurrent diarrhea or stomatitis until neutrophil count >1,000 cells/mm3, then continue at same dose. |
Re-treatment Criteria: Dose adjustments at the start of a cycle should be based on nonhematologic toxicity or blood counts from the preceding cycle following the guidelines in Table 1. Patients should not begin a new cycle of treatment unless the neutrophil count is at least 1500 cells/mm3, the platelet count is at least 100,000 cells/mm3, and nonhematologic toxicities have improved to grade 1 (mild) or resolved.
Dose Adjustments in Special Populations - Hepatic Impairment
Combination Therapy:
IXEMPRA in combination with capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN. Patients receiving combination treatment who have AST and ALT ≤2.5 x ULN and bilirubin ≤1 x ULN may receive the standard dose of ixabepilone (40 mg/m2). [See Boxed Warning, Contraindications (4), Warnings and Precautions (5.3), and Use in Specific Populations(8.6).]
Monotherapy:
Patients with hepatic impairment should be dosed with IXEMPRA based on the guidelines in Table 2. Patients with moderate hepatic impairment should be started at 20 mg/m2, the dosage in subsequent cycles may be escalated up to, but not exceeding, 30 mg/m2 if tolerated. Use in patients with AST or ALT >10 x ULN or bilirubin >3 x ULN is not recommended. Limited data are available for patients with baseline AST or ALT >5 x ULN. Caution should be used when treating these patients. [SeeWarnings and Precautions (5.3), and Use in Specific Populations (8.6).]
| Transaminase Levels | Bilirubin Levelsa | IXEMPRAb
(mg/m2) |
||
|---|---|---|---|---|
| a Excluding patients whose total bilirubin is elevated due to Gilbert’s disease. | ||||
| b Dosage recommendations are for first course of therapy; further decreases in subsequent courses should be based on individual tolerance. | ||||
| Mild | AST and ALT ≤2.5 x ULN | and | ≤1 x ULN | 40 |
| AST and ALT ≤10 x ULN | and | ≤1.5 x ULN | 32 | |
| Moderate | AST and ALT ≤10 x ULN | and | >1.5 x ULN - ≤3 x ULN | 20 - 30 |
Strong CYP3A4 Inhibitors
The use of concomitant strong CYP3A4 inhibitors should be avoided (eg, ketoconazole, itraconazole, clarithromycin, atazanavir, nefazodone, saquinavir, telithromycin, ritonavir, amprenavir, indinavir, nelfinavir, delavirdine, or voriconazole). Grapefruit juice may also increase plasma concentrations of IXEMPRA and should be avoided. Based on pharmacokinetic studies, if a strong CYP3A4 inhibitor must be coadministered, a dose reduction to 20 mg/m2 is predicted to adjust the ixabepilone AUC to the range observed without inhibitors and should be considered. If the strong inhibitor is discontinued, a washout period of approximately 1 week should be allowed before the IXEMPRA dose is adjusted upward to the indicated dose. [See Drug Interactions (7.1).]
2.3 Premedication
To minimize the chance of occurrence of a hypersensitivity reaction, all patients must be premedicated approximately 1 hour before the infusion of IXEMPRA with:
- An H1 antagonist (eg, diphenhydramine 50 mg orally or equivalent) and
- An H2 antagonist (eg, ranitidine 150 - 300 mg orally or equivalent).
Patients who experienced a hypersensitivity reaction to IXEMPRA require premedication with corticosteroids (eg, dexamethasone 20 mg intravenously, 30 minutes before infusion or orally, 60 minutes before infusion) in addition to pretreatment with H1 and H2 antagonists.
2.4 Instructions for Preparation and IV Administration
IXEMPRA Kit contains two vials, a vial labeled IXEMPRA (ixabepilone) for injection which contains ixabepilone powder and a vial containing DILUENT for IXEMPRA. Only supplied DILUENT must be used for constituting IXEMPRA (ixabepilone) for injection. IXEMPRA Kit must be stored in a refrigerator at 2° C - 8° C (36° F - 46° F) in the original package to protect from light. Prior to constituting IXEMPRA for injection, the Kit should be removed from the refrigerator and allowed to stand at room temperature for approximately 30 minutes. When the vials are first removed from the refrigerator, a white precipitate may be observed in the DILUENT vial. This precipitate will dissolve to form a clear solution once the DILUENT warms to room temperature. To allow for withdrawal losses, the vial labeled as 15 mg IXEMPRA for injection contains 16 mg of ixabepilone and the vial labeled as 45 mg IXEMPRA for injection contains 47 mg of ixabepilone. The 15-mg IXEMPRA Kit is supplied with a vial providing 8 mL of the DILUENT and the 45-mg IXEMPRA Kit is supplied with a vial providing 23.5 mL of the DILUENT. After constituting with the DILUENT, the concentration of ixabepilone is 2 mg/mL.
Please refer to Preparation and Handling Precautions [see Dosage and Administration (2.5)] before preparation.
A. To constitute:
- With a suitable syringe, aseptically withdraw the DILUENT and slowly inject it into the IXEMPRA for injection vial. The 15-mg IXEMPRA is constituted with 8 mL of DILUENT and the 45-mg IXEMPRA is constituted with 23.5 mL of DILUENT.
- Gently swirl and invert the vial until the powder in IXEMPRA is completely dissolved.
B. To dilute:
Before administration, the constituted solution must be further diluted only with Lactated Ringer's Injection, USP (LRI) supplied in DEHP [di-(2-ethylhexyl) phthalate] free bags. For most doses, a 250 mL bag of Lactated Ringer's Injection is sufficient. However, it is necessary to check the final infusion concentration of each dose based on the volume of Lactated Ringer's Injection to be used. The final concentration for infusion must be between 0.2 mg/mL and 0.6 mg/mL. To calculate the final infusion concentration, use the following formulas:
- Total Infusion Volume = mL of Constituted Solution + mL of LRI
- Final Infusion Concentration = Dose of IXEMPRA (mg)/Total Infusion Volume (mL)
- Aseptically, withdraw the appropriate volume of constituted solution containing 2 mg of ixabepilone per mL.
- Aseptically, transfer to an intravenous (IV) bag containing an appropriate volume of Lactated Ringer's Injection, USP to achieve the final desired concentration of ixabepilone.
- Thoroughly mix the infusion bag by manual rotation.
The infusion solution must be administered through an appropriate in-line filter with a microporous membrane of 0.2 to 1.2 microns. DEHP-free infusion containers and administration sets must be used. Any remaining solution should be discarded according to institutional procedures for antineoplastics.
Stability
After constituting ixabepilone for injection, the constituted solution should be further diluted with Lactated Ringer's Injection as soon as possible, but may be stored in the vial (not the syringe) for a maximum of 1 hour at room temperature and room light. Once diluted with Lactated Ringer's Injection, USP the solution is stable at room temperature and room light for a maximum of 6 hours. Administration of diluted IXEMPRA must be completed within this 6-hour period. Lactated Ringer's Injection, USP is specified because it has a pH range of 6 to 7.5, which is required to maintain IXEMPRA stability. Other diluents should not be used with IXEMPRA.
2.5 Preparation and Handling Precautions
Procedures for proper handling and disposal of antineoplastic drugs [see References (15)] should be followed. To minimize the risk of dermal exposure, impervious gloves should be worn when handling vials containing IXEMPRA, regardless of the setting, including unpacking and inspection, transport within a facility, and dose preparation and administration.
3 DOSAGE FORMS AND STRENGTHS
IXEMPRA for injection, 15 mg supplied with DILUENT for IXEMPRA, 8 mL.
IXEMPRA for injection, 45 mg supplied with DILUENT for IXEMPRA, 23.5 mL.
4 CONTRAINDICATIONS
IXEMPRA is contraindicated in patients with a history of a severe (CTC grade 3/4) hypersensitivity reaction to agents containing Cremophor® EL or its derivatives (eg, polyoxyethylated castor oil) [see Warnings and Precautions (5.4)].
IXEMPRA is contraindicated in patients who have a neutrophil count <1500 cells/mm3 or a platelet count <100,000 cells/mm3 [see Warnings and Precautions (5.2)].
IXEMPRA in combination with capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN [see Boxed Warning and Warnings and Precautions (5.3)].
5 WARNINGS AND PRECAUTIONS
5.1 Peripheral Neuropathy
Peripheral neuropathy was common (see Table 3). Patients treated with IXEMPRA should be monitored for symptoms of neuropathy, such as burning sensation, hyperesthesia, hypoesthesia, paresthesia, discomfort, or neuropathic pain. Neuropathy occurred early during treatment; ~75% of new onset or worsening neuropathy occurred during the first 3 cycles. Patients experiencing new or worsening symptoms may require a reduction or delay in the dose of IXEMPRA [see Dosage and Administration (2.2)]. In clinical studies, peripheral neuropathy was managed through dose reductions, dose delays, and treatment discontinuation. Neuropathy was the most frequent cause of treatment discontinuation due to drug toxicity. In Studies 046 and 081, 80% and 87%, respectively, of patients with peripheral neuropathy who received IXEMPRA had improvement or no worsening of their neuropathy following dose reduction. For patients with grade 3/4 neuropathy in Studies 046 and 081, 76% and 79%, respectively, had documented improvement to baseline or grade 1, twelve weeks after onset.
| IXEMPRA with capecitabine Study 046 | IXEMPRA as monotherapy Study 081 |
|
|---|---|---|
| a Sensory and motor neuropathy combined. | ||
| b 24% and 27% of patients in 046 and 081, respectively, had preexisting neuropathy (grade 1). | ||
| Peripheral neuropathy (all grades)a,b | 67% | 63% |
| Peripheral neuropathy (grade 3/4)a,b | 23% | 14% |
| Discontinuation due to neuropathy | 21% | 6% |
| Median number of cycles to onset of grade 3/4 neuropathy | 4 | 4 |
| Median time to improvement of grade 3/4 neuropathy to baseline or to grade 1 | 6.0 weeks | 4.6 weeks |
A pooled analysis of 945 cancer patients treated with IXEMPRA indicated that patients with diabetes mellitus may be at increased risk of severe neuropathy. The presence of grade 1 neuropathy and prior therapy with neurotoxic chemotherapy agents did not predict either the development or worsening of neuropathy. Patients with moderate to severe neuropathy (grade 2 or greater) were excluded from studies with IXEMPRA. Caution should be used when treating patients with diabetes mellitus or existing moderate to severe neuropathy.
5.2 Myelosuppression
Myelosuppression is dose-dependent and primarily manifested as neutropenia. In clinical studies, grade 4 neutropenia (<500 cells/mm3) occurred in 36% of patients treated with IXEMPRA in combination with capecitabine and 23% of patients treated with IXEMPRA monotherapy. Febrile neutropenia and infection with neutropenia were reported in 5% and 6% of patients treated with IXEMPRA in combination with capecitabine, respectively, and 3% and 5% of patients treated with IXEMPRA as monotherapy, respectively. Neutropenia-related death occurred in 1.9% of 414 patients with normal hepatic function or mild hepatic impairment treated with IXEMPRA in combination with capecitabine. The rate of neutropenia-related deaths was higher (29%, 5 out of 17) in patients with AST or ALT >2.5 x ULN or bilirubin >1.5 x ULN. [See Boxed Warning, Contraindications (4), and Warnings and Precautions (5.3)]. Neutropenia-related death occurred in 0.4% of 240 patients treated with IXEMPRA as monotherapy. No neutropenia-related deaths were reported in 24 patients with AST or ALT >2.5 x ULN or bilirubin >1.5 x ULN treated with IXEMPRA monotherapy. IXEMPRA must not be administered to patients with a neutrophil count <1500 cells/mm3. To monitor for myelosuppression, frequent peripheral blood cell counts are recommended for all patients receiving IXEMPRA. Patients who experience severe neutropenia or thrombocytopenia should have their dose reduced [see Dosage and Administration (2.2)].
5.3 Hepatic Impairment
Patients with baseline AST or ALT >2.5 x ULN or bilirubin >1.5 x ULN experienced greater toxicity than patients with baseline AST or ALT ≤2.5 x ULN or bilirubin ≤1.5 x ULN when treated with IXEMPRA at 40 mg/m2 in combination with capecitabine or as monotherapy in breast cancer studies. In combination with capecitabine, the overall frequency of grade 3/4 adverse reactions, febrile neutropenia, serious adverse reactions, and toxicity related deaths was greater [see Warnings and Precautions (5.2)]. With monotherapy, grade 4 neutropenia, febrile neutropenia, and serious adverse reactions were more frequent. The safety and pharmacokinetics of IXEMPRA as monotherapy were evaluated in a dose escalation study in 56 patients with varying degrees of hepatic impairment. Exposure was increased in patients with elevated AST or bilirubin [see Use in Specific Populations (8.6)].
IXEMPRA in combination with capecitabine is contraindicated in patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN due to increased risk of toxicity and neutropenia-related death [see Boxed Warning, Contraindications (4), and Warnings and Precautions (5.2)]. Patients who are treated with IXEMPRA as monotherapy should receive a reduced dose depending on the degree of hepatic impairment [see Dosage and Administration (2.2)]. Use in patients with AST or ALT >10 x ULN or bilirubin >3 x ULN is not recommended. Limited data are available for patients with AST or ALT >5 x ULN. Caution should be used when treating these patients [see Dosage and Administration (2.2)].
5.4 Hypersensitivity Reactions
Patients with a history of a severe hypersensitivity reaction to agents containing Cremophor® EL or its derivatives (eg, polyoxyethylated castor oil) should not be treated with IXEMPRA. All patients should be premedicated with an H1 and an H2 antagonist approximately 1 hour before IXEMPRA infusion and be observed for hypersensitivity reactions (eg, flushing, rash, dyspnea, and bronchospasm). In case of severe hypersensitivity reactions, infusion of IXEMPRA should be stopped and aggressive supportive treatment (eg, epinephrine, corticosteroids) started. Of the 1323 patients treated with IXEMPRA in clinical studies, 9 patients (1%) had experienced severe hypersensitivity reactions (including anaphylaxis). Three of the 9 patients were able to be retreated. Patients who experience a hypersensitivity reaction in one cycle of IXEMPRA must be premedicated in subsequent cycles with a corticosteroid in addition to the H1 and H2 antagonists, and extension of the infusion time should be considered [see Dosage and Administration (2.3) and Contraindications (4)].
5.5 Pregnancy
Pregnancy Category D.
IXEMPRA may cause fetal harm when administered to pregnant women. There are no adequate and well-controlled studies with IXEMPRA in pregnant women. Women should be advised not to become pregnant when taking IXEMPRA. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
Ixabepilone was studied for effects on embryo-fetal development in pregnant rats and rabbits given IV doses of 0.02, 0.08, and 0.3 mg/kg/day and 0.01, 0.03, 0.11 and 0.3 mg/kg/day, respectively. There were no teratogenic effects. In rats, an increase in resorptions and post-implantation loss and a decrease in the number of live fetuses and fetal weight was observed at the maternally toxic dose of 0.3 mg/kg/day (approximately one-tenth the human clinical exposure based on AUC). Abnormalities included a reduced ossification of caudal vertebrae, sternebrae, and metacarpals. In rabbits, ixabepilone caused maternal toxicity (death) and embryo-fetal toxicity (resorptions) at 0.3 mg/kg/day (approximately one-tenth the human clinical dose based on body surface area). No fetuses were available at this dose for evaluation.
5.6 Cardiac Adverse Reactions
The frequency of cardiac adverse reactions (myocardial ischemia and ventricular dysfunction) was higher in the IXEMPRA in combination with capecitabine (1.9%) than in the capecitabine alone (0.3%) treatment group. Supraventricular arrhythmias were observed in the combination arm (0.5%) and not in the capecitabine alone arm. Caution should be exercised in patients with a history of cardiac disease. Discontinuation of IXEMPRA should be considered in patients who develop cardiac ischemia or impaired cardiac function.
5.7 Potential for Cognitive Impairment from Excipients
Since IXEMPRA contains dehydrated alcohol USP, consideration should be given to the possibility of central nervous system and other effects of alcohol [see Description (11)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections.
- Peripheral neuropathy [see Warnings and Precautions (5.1)]
- Myelosuppression [see Warnings and Precautions (5.2)]
- Hypersensitivity reaction [see Warnings and Precautions (5.4)]
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.
Unless otherwise specified, assessment of adverse reactions is based on one randomized study (Study 046) and one single-arm study (Study 081). In Study 046, 369 patients with metastatic breast cancer were treated with IXEMPRA 40 mg/m2 administered intravenously over 3 hours every 21 days, combined with capecitabine 1000 mg/m2 twice daily for 2 weeks followed by a 1-week rest period. Patients treated with capecitabine as monotherapy (n=368) in this study received 1250 mg/m2 twice daily for 2 weeks every 21 days. In Study 081, 126 patients with metastatic or locally advanced breast cancer were treated with IXEMPRA 40 mg/m2 administered intravenously over 3 hours every 3 weeks.
The most common adverse reactions (≥20%) reported by patients receiving IXEMPRA were peripheral sensory neuropathy, fatigue/asthenia, myalgia/arthralgia, alopecia, nausea, vomiting, stomatitis/mucositis, diarrhea, and musculoskeletal pain. The following additional reactions occurred in ≥20% in combination treatment: palmar-plantar erythrodysesthesia (hand-foot) syndrome, anorexia, abdominal pain, nail disorder, and constipation. The most common hematologic abnormalities (>40%) include neutropenia, leukopenia, anemia, and thrombocytopenia.
Table 4 presents nonhematologic adverse reactions reported in 5% or more of patients. Hematologic abnormalities are presented separately in Table 5.
| Study 046 | Study 081 | |||||
|---|---|---|---|---|---|---|
| IXEMPRA with capecitabine n=369 | Capecitabine n=368 | IXEMPRA monotherapy n=126 |
||||
| System Organ Classa/ Preferred Term | Total (%) | Grade 3/4 (%) | Total (%) | Grade 3/4 (%) | Total (%) | Grade 3/4 (%) |
| a System organ class presented as outlined in Guidelines for Preparing Core Clinical Safety Information on Drugs by the Council for International Organizations of Medical Sciences (CIOMS). | ||||||
| b A composite of multiple MedDRA Preferred Terms. | ||||||
| c NCI CTC grading for febrile neutropenia ranges from Grade 3 to 5. Three patients (1%) experienced Grade 5 (fatal) febrile neutropenia. Other neutropenia-related deaths (9) occurred in the absence of reported febrile neutropenia [see Warnings and Precautions (5.2)]. | ||||||
| d No grade 4 reports. | ||||||
| e Peripheral sensory neuropathy (graded with the NCI CTC scale) was defined as the occurrence of any of the following: areflexia, burning sensation, dysesthesia, hyperesthesia, hypoesthesia, hyporeflexia, neuralgia, neuritis, neuropathy, neuropathy peripheral, neurotoxicity, painful response to normal stimuli, paresthesia, pallanesthesia, peripheral sensory neuropathy, polyneuropathy, polyneuropathy toxic and sensorimotor disorder. Peripheral motor neuropathy was defined as the occurrence of any of the following: multifocal motor neuropathy, neuromuscular toxicity, peripheral motor neuropathy, and peripheral sensorimotor neuropathy. |
||||||
| f Palmar-plantar erythrodysesthesia (hand-foot syndrome) was graded on a 1-3 severity scale in Study 046. | ||||||
| Infections and Infestations | ||||||
| Upper respiratory tract infectionb | 4 | 0 | 3 | 0 | 6 | 0 |
| Blood and Lymphatic System Disorders | ||||||
| Febrile neutropenia | 5 | 4c | 1 | 1d | 3 | 3d |
| Immune System Disorders | ||||||
| Hypersensitivityb | 2 | 1d | 0 | 0 | 5 | 1d |
| Metabolism and Nutrition Disorders | ||||||
| Anorexiab | 34 | 3d | 15 | 1d | 19 | 2d |
| Dehydrationb | 5 | 2 | 2 | <1d | 2 | 1d |
| Psychiatric | ||||||
| Insomniab | 9 | <1d | 2 | 0 | 5 | 0 |
| Nervous System Disorders | ||||||
| Peripheral neuropathy | ||||||
| Sensory neuropathyb,e | 65 | 21 | 16 | 0 | 62 | 14 |
| Motor neuropathyb | 16 | 5d | <1 | 0 | 10 | 1d |
| Headache | 8 | <1d | 3 | 0 | 11 | 0 |
| Taste disorderb | 12 | 0 | 4 | 0 | 6 | 0 |
| Dizziness | 8 | 1d | 5 | 1d | 7 | 0 |
| Eye Disorders | ||||||
| Lacrimation increased | 5 | 0 | 4 | <1d | 4 | 0 |
| Vascular Disorders | ||||||
| Hot flushb | 5 | 0 | 2 | 0 | 6 | 0 |
| Respiratory, Thoracic, and Mediastinal Disorders | ||||||
| Dyspneab | 7 | 1 | 4 | 1 | 9 | 1d |
| Coughb | 6 | 0 | 2 | 0 | 2 | 0 |
| Gastrointestinal Disorders | ||||||
| Nausea | 53 | 3d | 40 | 2d | 42 | 2d |
| Vomitingb | 39 | 4d | 24 | 2 | 29 | 1d |
| Stomatitis/mucositisb | 31 | 4 | 20 | 3d | 29 | 6 |
| Diarrheab | 44 | 6d | 39 | 9 | 22 | 1d |
| Constipation | 22 | 0 | 6 | <1d | 16 | 2d |
| Abdominal painb | 24 | 2d | 14 | 1d | 13 | 2d |
| Gastroesophageal reflux diseaseb | 7 | 1d | 8 | 0 | 6 | 0 |
| Skin and Subcutaneous Tissue Disorders | ||||||
| Alopeciab | 31 | 0 | 3 | 0 | 48 | 0 |
| Skin rashb | 17 | 1d | 7 | 0 | 9 | 2d |
| Nail disorderb | 24 | 2d | 10 | <1d | 9 | 0 |
| Palmar-plantar erythrodysesthesia syndromeb,f | 64 | 18d | 63 | 17d | 8 | 2d |
| Pruritus | 5 | 0 | 2 | 0 | 6 | 1d |
| Skin exfoliationb | 5 | <1d | 3 | 0 | 2 | 0 |
| Skin hyperpigmentationb | 11 | 0 | 14 | 0 | 2 | 0 |
| Musculoskeletal, Connective Tissue, and Bone Disorders | ||||||
| Myalgia/arthralgiab | 39 | 8d | 5 | <1d | 49 | 8d |
| Musculoskeletal painb | 23 | 2d | 5 | 0 | 20 | 3d |
| General Disorders and Administrative Site Conditions | ||||||
| Fatigue/astheniab | 60 | 16 | 29 | 4 | 56 | 13 |
| Edemab | 8 | 0 | 5 | <1d | 9 | 1d |
| Pyrexia | 10 | 1d | 4 | 0 | 8 | 1d |
| Painb | 9 | 1d | 2 | 0 | 8 | 3d |
| Chest painb | 4 | 1d | <1 | 0 | 5 | 1d |
| Investigations | ||||||
| Weight decreased | 11 | 0 | 3 | 0 | 6 | 0 |
| Study 046 | Study 081 | |||||
|---|---|---|---|---|---|---|
| IXEMPRA with capecitabine n=369 | Capecitabine n=368 | IXEMPRA monotherapy n=126 |
||||
| Hematology Parameter | Grade 3 (%) | Grade 4 (%) | Grade 3 (%) | Grade 4 (%) | Grade 3 (%) | Grade 4 (%) |
| a G-CSF (granulocyte colony stimulating factor) or GM-CSF (granulocyte macrophage stimulating factor) was used in 20% and 17% of patients who received IXEMPRA in Study 046 and Study 081, respectively. | ||||||
| Neutropeniaa | 32 | 36 | 9 | 2 | 31 | 23 |
| Leukopenia (WBC) | 41 | 16 | 5 | 1 | 36 | 13 |
| Anemia (Hgb) | 8 | 2 | 4 | 1 | 6 | 2 |
| Thrombocytopenia | 5 | 3 | 2 | 2 | 5 | 2 |
The following serious adverse reactions were also reported in 1323 patients treated with IXEMPRA as monotherapy or in combination with other therapies in Phase 2 and 3 studies.
Infections and Infestations: sepsis, pneumonia, infection, neutropenic infection, urinary tract infection, bacterial infection, enterocolitis, laryngitis, lower respiratory tract infection
Blood and Lymphatic System Disorders: coagulopathy, lymphopenia
Metabolism and Nutrition Disorders: hyponatremia, metabolic acidosis, hypokalemia, hypovolemia
Nervous System Disorders: cognitive disorder, syncope, cerebral hemorrhage, abnormal coordination, lethargy
Cardiac Disorders: myocardial infarction, supraventricular arrhythmia, left ventricular dysfunction, angina pectoris, atrial flutter, cardiomyopathy, myocardial ischemia
Vascular Disorders: hypotension, thrombosis, embolism, hemorrhage, hypovolemic shock, vasculitis
Respiratory, Thoracic, and Mediastinal Disorders: pneumonitis, hypoxia, respiratory failure, acute pulmonary edema, dysphonia, pharyngolaryngeal pain
Gastrointestinal Disorders: ileus, colitis, impaired gastric emptying, esophagitis, dysphagia, gastritis, gastrointestinal hemorrhage
Hepatobiliary Disorders: acute hepatic failure, jaundice
Skin and Subcutaneous Tissue Disorders: erythema multiforme
Musculoskeletal, Connective Tissue Disorders, and Bone Disorders: muscular weakness, muscle spasms, trismus
Renal and Urinary Disorders: nephrolithiasis, renal failure
General Disorders and Administration Site Conditions: chills
Investigations: increased transaminases, increased blood alkaline phosphatase, increased gamma-glutamyltransferase
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on Ixabepilone
Drugs That May Increase Ixabepilone Plasma Concentrations
CYP3A4 Inhibitors: Co-administration of ixabepilone with ketoconazole, a potent CYP3A4 inhibitor, increased ixabepilone AUC by 79% compared to ixabepilone treatment alone. If alternative treatment cannot be administered, a dose adjustment should be considered. The effect of mild or moderate inhibitors (eg, erythromycin, fluconazole, or verapamil) on exposure to ixabepilone has not been studied. Therefore, caution should be used when administering mild or moderate CYP3A4 inhibitors during treatment with IXEMPRA, and alternative therapeutic agents that do not inhibit CYP3A4 should be considered. Patients receiving CYP3A4 inhibitors during treatment with IXEMPRA should be monitored closely for acute toxicities (eg, frequent monitoring of peripheral blood counts between cycles of IXEMPRA). [See Dosage and Administration (2.2).]
Drugs That May Decrease Ixabepilone Plasma Concentrations
CYP3A4 Inducers: IXEMPRA is a CYP3A4 substrate. Strong CYP3A4 inducers (eg, dexamethasone, phenytoin, carbamazepine, rifampin, rifampicin, rifabutin, and phenobarbital) may decrease ixabepilone concentrations leading to subtherapeutic levels. Therefore, therapeutic agents with low enzyme induction potential should be considered for coadministration with IXEMPRA. St. John's Wort may decrease ixabepilone plasma concentrations unpredictably and should be avoided.
7.2 Effect of Ixabepilone on Other Drugs
Ixabepilone does not inhibit CYP enzymes at relevant clinical concentrations and is not expected to alter the plasma concentrations of other drugs [see Clinical Pharmacology (12.3)].
7.3 Capecitabine
In patients with cancer who received ixabepilone (40 mg/m2) in combination with capecitabine (1000 mg/m2), ixabepilone Cmax decreased by 19%, capecitabine Cmax decreased by 27%, and 5-fluorouracil AUC increased by 14%, as compared to ixabepilone or capecitabine administered separately. The interaction is not clinically significant given that the combination treatment is supported by efficacy data.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category D [See Warnings and Precautions (5.5)]
8.3 Nursing Mothers
It is not known whether ixabepilone is excreted into human milk. Following intravenous administration of radiolabeled ixabepilone to rats on days 7 to 9 postpartum, concentrations of radioactivity in milk were comparable with those in plasma and declined in parallel with the plasma concentrations. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from ixabepilone, a decision must be made whether to discontinue nursing or to discontinue IXEMPRA taking into account the importance of the drug to the mother.
8.4 Pediatric Use
The safety and effectiveness of IXEMPRA in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of IXEMPRA did not include sufficient numbers of subjects aged sixty-five and over to determine whether they respond differently from younger subjects.
Forty-five of 431 patients treated with IXEMPRA in combination with capecitabine were ≥65 years of age and 3 patients were ≥75. Overall, the incidence of grade 3/4 adverse reactions were higher in patients ≥65 years of age versus those <65 years of age (82% versus 68%) including grade 3/4 stomatitis (9% versus 1%), diarrhea (9% versus 6%), palmar-plantar erythrodysesthesia syndrome (27% versus 20%), peripheral neuropathy (24% versus 22%), febrile neutropenia (9% versus 3%), fatigue (16% versus 12%), and asthenia (11% versus 6%). Toxicity-related deaths occurred in 2 (4.7%) of 43 patients ≥65 years with normal baseline hepatic function or mild impairment.
Thirty-two of 240 breast cancer patients treated with IXEMPRA as monotherapy were ≥65 years of age and 6 patients were ≥75. No overall differences in safety were observed in these patients compared to those <65 years of age.
8.6 Hepatic Impairment
IXEMPRA was evaluated in 56 patients with mild to severe hepatic impairment defined by bilirubin levels and AST levels. Compared to patients with normal hepatic function (n=17), the area under the curve (AUC0‑infinity) of ixabepilone increased by:
- 22% in patients with a) bilirubin >1 – 1.5 x ULN or b) AST >ULN but bilirubin <1.5 x ULN;
- 30% in patients with bilirubin >1.5 – 3 x ULN and any AST level; and
- 81% in patients with bilirubin >3 x ULN and any AST level.
Doses of 10 and 20 mg/m2 as monotherapy were tolerated in 17 paitents with severe hepatic impairment (bilirubin >3 x ULN).
IXEMPRA in combination with capecitabine must not be given to patients with AST or ALT >2.5 x ULN or bilirubin >1 x ULN [see Boxed Warning, Contraindications (4), and Warnings and Precautions (5.3)]. Dose reduction is recommended when administering IXEMPRA as monotherapy to patients with hepatic impairment [see Dosage and Administration (2.3)]. Because there is a need for dosage adjustment based upon hepatic function, assessment of hepatic function is recommended before initiation of IXEMPRA and periodically thereafter.
8.7 Renal Impairment
Ixabepilone is minimally excreted via the kidney. No controlled pharmacokinetic studies were conducted with IXEMPRA in patients with renal impairment. IXEMPRA in combination with capecitabine has not been evaluated in patients with calculated creatinine clearance of <50 mL/min. IXEMPRA as monotherapy has not been evaluated in patients with creatinine >1.5 times ULN. In a population pharmacokinetic analysis of IXEMPRA as monotherapy, there was no meaningful effect of mild and moderate renal insufficiency (CrCL >30 mL/min) on the pharmacokinetics of ixabepilone.
10 OVERDOSAGE
One case of overdose of IXEMPRA has been reported. The patient mistakenly received 100 mg/m2 (total dose 185 mg) and was admitted to the hospital for observation. The patient experienced myalgia (grade 1) and fatigue (grade 1) one day after infusion and was treated with a centrally acting analgesic. The patient recovered and was discharged without incident.
There is no known antidote for overdosage of IXEMPRA. In case of overdosage, the patient should be closely monitored and supportive treatment should be administered. Management of overdose should include supportive medical interventions to treat the presenting clinical manifestations.
11 DESCRIPTION
IXEMPRA (ixabepilone) is a microtubule inhibitor belonging to a class of antineoplastic agents, the epothilones and their analogs. The epothilones are isolated from the myxobacterium Sorangium cellulosum. Ixabepilone is a semisynthetic analog of epothilone B, a 16-membered polyketide macrolide, with a chemically modified lactam substitution for the naturally existing lactone.
The chemical name for ixabepilone is (1S,3S,7S,10R,11S,12S,16R)-7,11-dihydroxy-8,8,10,12,16-pentamethyl-3-[(1E)-1-methyl-2-(2-methyl-4-thiazolyl)ethenyl]-17-oxa-4-azabicyclo[14.1.0] heptadecane-5,9-dione, and it has a molecular weight of 506.7. Ixabepilone has the following structural formula:
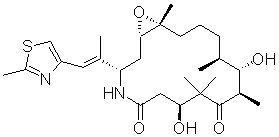
IXEMPRA (ixabepilone) for Injection is intended for intravenous infusion only after constitution with the supplied DILUENT and after further dilution with Lactated Ringer's Injection, USP. IXEMPRA (ixabepilone) for injection is supplied as a sterile, non-pyrogenic single-use vial containing 15 mg or 45 mg ixabepilone as lyophilized white powder. The DILUENT for IXEMPRA is a sterile, non-pyrogenic solution of 52.8% (w/v) purified polyoxyethylated castor oil and 39.8% (w/v) dehydrated alcohol, USP. The IXEMPRA (ixabepilone) for injection and the DILUENT for IXEMPRA are co-packaged and supplied as IXEMPRA Kit.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ixabepilone is a semi-synthetic analog of epothilone B. Ixabepilone binds directly to β-tubulin subunits on microtubules, leading to suppression of microtubule dynamics. Ixabepilone suppresses the dynamic instability of αβ−II and αβ−III microtubules. Ixabepilone possesses low in vitro susceptibility to multiple tumor resistance mechanisms including efflux transporters, such as MRP-1 and P-glycoprotein (P-gp). Ixabepilone blocks cells in the mitotic phase of the cell division cycle, leading to cell death.
12.2 Pharmacodynamics
In cancer patients, ixabepilone has a plasma concentration-dependent effect on tubulin dynamics in peripheral blood mononuclear cells that is observed as the formation of microtubule bundles. Ixabepilone has antitumor activity in vivo against multiple human tumor xenografts, including drug-resistant types that overexpress P-gp, MRP-1, and βIII tubulin isoforms, or harbor tubulin mutations. Ixabepilone is active in xenografts that are resistant to multiple agents including taxanes, anthracyclines, and vinca alkaloids. Ixabepilone demonstrated synergistic antitumor activity in combination with capecitabine in vivo. In addition to direct antitumor activity, ixabepilone has antiangiogenic activity.
12.3 Pharmacokinetics
Absorption
Following administration of a single 40 mg/m2 dose of IXEMPRA in patients with cancer, the mean Cmax was 252 ng/mL (coefficient of variation, CV 56%) and the mean AUC was 2143 ng•hr/mL (CV 48%). Typically Cmax occurred at the end of the 3 hour infusion. In cancer patients, the pharmacokinetics of ixabepilone were linear at doses of 15 to 57 mg/m2.
Distribution
The mean volume of distribution of 40 mg/m2 ixabepilone at steady-state was in excess of 1000 L. In vitro, the binding of ixabepilone to human serum proteins ranged from 67 to 77%, and the blood-to-plasma concentration ratios in human blood ranged from 0.65 to 0.85 over a concentration range of 50 to 5000 ng/mL.
Metabolism
Ixabepilone is extensively metabolized in the liver. In vitro studies indicated that the main route of oxidative metabolism of ixabepilone is via CYP3A4. More than 30 metabolites of ixabepilone are excreted into human urine and feces. No single metabolite accounted for more than 6% of the administered dose. The biotransformation products generated from ixabepilone by human liver microsomes were not active when tested for in vitro cytotoxicity against a human tumor cell line.
In vitro studies using human liver microsomes indicate that clinically relevant concentrations of ixabepilone do not inhibit CYP3A4, CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6. Ixabepilone does not induce the activity or the corresponding mRNA levels of CYP1A2, CYP2B6, CYP2C9, or CYP3A4 in cultured human hepatocytes at clinically relevant concentrations. Therefore, it is unlikely that ixabepilone will affect the plasma levels of drugs that are substrates of CYP enzymes.
Elimination
Ixabepilone is eliminated primarily as metabolized drug. After an intravenous 14[C]-ixabepilone dose to patients, approximately 86% of the dose was eliminated within 7 days in feces (65% of the dose) and in urine (21% of the dose). Unchanged ixabepilone accounted for approximately 1.6% and 5.6% of the dose in feces and urine, respectively. Ixabepilone has a terminal elimination half-life of approximately 52 hours. No accumulation in plasma is expected for ixabepilone administered every 3 weeks.
Effects of Age, Gender, and Race
Based upon a population pharmacokinetic analysis in 676 cancer patients, gender, race, and age do not have meaningful effects on the pharmacokinetics of ixabepilone.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with ixabepilone have not been conducted. Ixabepilone did not induce mutations in the microbial mutagenesis (Ames) assay and was not clastogenic in an in vitro cytogenetic assay using primary human lymphocytes. Ixabepilone was clastogenic (induction of micronuclei) in the in vivo rat micronucleus assay at doses ≥0.625 mg/kg/day.
There were no effects on male or female rat mating or fertility at doses up to 0.2 mg/kg/day in both males and females (approximately one-fifteenth the expected human clinical exposure based on AUC). The effect of ixabepilone on human fertility is unknown. However, when rats were given an IV infusion of ixabepilone during breeding and through the first 7 days of gestation, a significant increase in resorptions and pre- and post-implantation loss and a decrease in the number of corpora lutea was observed at 0.2 mg/kg/day. Testicular atrophy or degeneration was observed in 6-month rat and 9-month dog studies when ixabepilone was given every 21 days at intravenous doses of 6.7 mg/kg (40 mg/m2) in rats (approximately 2.1 times the expected clinical exposure based on AUC) and 0.5 and 0.75 mg/kg (10 and 15 mg/m2) in dogs (approximately 0.2 and 0.4 times the expected clinical exposure based on AUC).
13.2 Animal Toxicology
Overdose
In rats, single intravenous doses of ixabepilone from 60 to 180 mg/m2 (mean AUC values ≥8156 ng•h/mL) were associated with mortality occurring between 5 and 14 days after dosing, and toxicity was principally manifested in the gastrointestinal, hematopoietic (bone-marrow), lymphatic, peripheral-nervous, and male-reproductive systems. In dogs, a single intravenous dose of 100 mg/m2 (mean AUC value of 6925 ng•h/mL) was markedly toxic, inducing severe gastrointestinal toxicity and death 3 days after dosing.
14 CLINICAL STUDIES
Combination Therapy
In an open-label, multicenter, multinational, randomized trial of 752 patients with metastatic or locally advanced breast cancer, the efficacy and safety of IXEMPRA (40 mg/m2 every 3 weeks) in combination with capecitabine (at 1000 mg/m2 twice daily for 2 weeks followed by 1 week rest) were assessed in comparison with capecitabine as monotherapy (at 1250 mg/m2 twice daily for 2 weeks followed by 1 week rest). Patients were previously treated with anthracyclines and taxanes. Patients were required to have demonstrated tumor progression or resistance to taxanes and anthracyclines as follows:
- tumor progression within 3 months of the last anthracycline dose in the metastatic setting or recurrence within 6 months in the adjuvant or neoadjuvant setting, and
- tumor progression within 4 months of the last taxane dose in the metastatic setting or recurrence within 12 months in the adjuvant or neoadjuvant setting.
For anthracyclines, patients who received a minimum cumulative dose of 240 mg/m2 of doxorubicin or 360 mg/m2 of epirubicin were also eligible.
Sixty-seven percent of patients were White, 23% were Asian, and 3% were Black. Both arms were evenly matched with regards to race, age (median 53 years), baseline performance status (Karnofsky 70-100%), and receipt of prior adjuvant or neo-adjuvant chemotherapy (75%). Tumors were ER-positive in 47% of patients, ER-negative in 43%, HER2-positive in 15%, HER2-negative in 61%, and ER-negative, PR-negative, HER2-negative in 25%. The baseline disease characteristics and previous therapies for all patients (n=752) are shown in Table 6.
| a For IXEMPRA plus capecitabine versus capecitabine only, prior treatment in the metastatic setting included cyclophosphamide (25% vs. 23%), fluorouracil (22% vs. 16%), vinorelbine (11% vs. 12%), gemcitabine (9% each arm), carboplatin (9% vs. 7%), liposomal doxorubicin (3% each arm), and cisplatin (2% vs. 3%). | ||
| b Tumor progression within 3 months in the metastatic setting or recurrence within 6 months in the adjuvant or neoadjuvant setting. | ||
| c 24% and 21% of patients had received 2 or more taxane-containing regimens in the combination and single agent treatment groups, respectively. | ||
| IXEMPRA with capecitabine n=375 | Capecitabine n=377 |
|
|---|---|---|
| Site of disease | ||
| Visceral disease (liver or lung) | 316 (84%) | 315 (84%) |
| Liver | 245 (65%) | 228 (61%) |
| Lung | 180 (48%) | 174 (46%) |
| Lymph node | 250 (67%) | 249 (66%) |
| Bone | 168 (45%) | 162 (43%) |
| Skin/soft tissue | 60 (16%) | 62 (16%) |
| Number of prior chemotherapy regimens in metastatic settinga | ||
| 0 | 27 (7%) | 33 (9%) |
| 1 | 179 (48%) | 184 (49%) |
| 2 | 152 (41%) | 138 (37%) |
| ≥3 | 17 (5%) | 22 (6%) |
| Anthracycline resistanceb | 164 (44%) | 165 (44%) |
| Taxane Resistancec | ||
| Neoadjuvant/adjuvant setting | 40 (11%) | 44 (12%) |
| Metastatic setting | 327 (87%) | 319 (85%) |
The patients in the combination treatment group received a median of 5 cycles of treatment and patients in the capecitabine monotherapy treatment group received a median of 4 cycles of treatment.
The primary endpoint of the study was progression-free survival (PFS) defined as time from randomization to radiologic progression as determined by Independent Radiologic Review (IRR), clinical progression of measurable skin lesions or death from any cause. Other study endpoints included objective tumor response based on Response Evaluation Criteria in Solid Tumors (RECIST), time to response, response duration, and overall survival. The data for overall survival analysis are not mature.
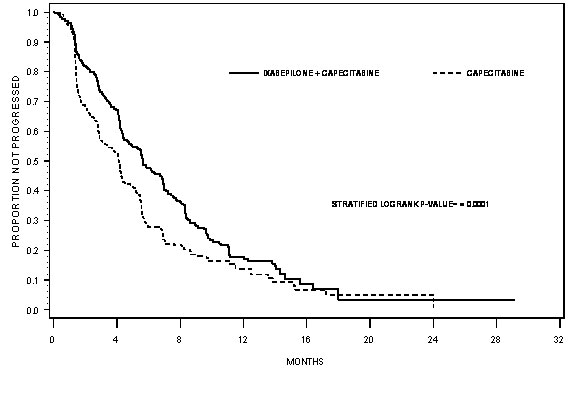
IXEMPRA in combination with capecitabine resulted in a statistically significant improvement in PFS compared to capecitabine. The results of the study are presented in Table 7 and Figure 1.
| a Patients were censored for PFS at the last date of tumor assessment prior to the start of subsequent therapy. In patients where independent review was not available PFS was censored at the randomization date. | ||||
| b For the hazard ratio, a value less than 1.00 favors combination treatment, CI adjusted for interim analysis. | ||||
| c Stratified by visceral metastasis in liver/lung, prior chemotherapy in metastatic setting, and anthracycline resistance. | ||||
| d Cochran-Mantel-Haenszel test | ||||
| Efficacy Parameter | IXEMPRA with Capecitabine n=375 | Capecitabine n=377 |
||
|---|---|---|---|---|
| PFS | ||||
| Number of eventsa | 242 | 256 | ||
| Median (95% CI) | 5.7 months (4.8 - 6.7) | 4.1 months (3.1 - 4.3) |
||
| Hazard Ratiob (95% CI) | 0.69 (0.58 - 0.83) | |||
| p-valuec (Log rank) | <0.0001 | |||
| Objective Tumor Response Rate (95% CI) | 34.7% (29.9 - 39.7) | 14.3% (10.9 - 18.3) |
||
| p-valuec (CMH)d | <0.0001 | |||
| Duration of Response, Median (95% CI) | 6.4 months (5.6 - 7.1) | 5.6 months (4.2 - 7.5) |
||
Figure 1: Progression-free Survival Kaplan Meier Curves
Monotherapy
IXEMPRA was evaluated as a single agent in a multicenter single-arm study in 126 women with metastatic or locally advanced breast cancer. The study enrolled patients whose tumors had recurred or had progressed following two or more chemotherapy regimens including an anthracycline, a taxane, and capecitabine. Patients who had received a minimum cumulative dose of 240 mg/m2 of doxorubicin or 360 mg/m2 of epirubicin were also eligible. Tumor progression or recurrence were prospectively defined as follows:
- Disease progression while on therapy in the metastatic setting (defined as progression while on treatment or within 8 weeks of last dose),
- Recurrence within 6 months of the last dose in the adjuvant or neoadjuvant setting (only for anthracycline and taxane),
- HER2 positive patients must also have progressed during or after discontinuation of trastuzumab.
In this study, the median age was 51 years (range, 30-78), and 79% were White, 5% Black, and 2% Asian, Karnofsky performance status was 70-100%, 88% had received two or more prior chemotherapy regimens for metastatic disease, and 86% had liver and/or lung metastases. Tumors were ER-positive in 48% of patients, ER-negative in 44%, HER2-positive in 7%, HER2-negative in 72%, and ER-negative, PR-negative, HER2-negative in 33%.
IXEMPRA was administered at a dose of 40 mg/m2 intravenously over 3 hours every 3 weeks. Patients received a median of 4 cycles (range 1 to 18) of IXEMPRA therapy.
Objective tumor response was determined by independent radiologic and investigator review using RECIST. Efficacy results are presented in Table 8.
| a All responses were partial. | |
| b As assessed by IRR. | |
| Endpoint | Result |
| Objective tumor response rate (95% CI) IRR Assessmenta (n=113) Investigator Assessment (n=126) |
12.4% (6.9 - 19.9) 18.3% (11.9 - 26.1) |
| Time to responseb (n=14) | |
| Median, weeks (min - max) | 6.1 (5 - 54.4) |
| Duration of responseb (n=14) | |
| Median, months (95% CI) | 6.0 (5.0 - 7.6) |
15 REFERENCES
- Preventing Occupational Exposures to Antineoplastic and Other Hazardous Drugs in Health Care Settings. NIOSH Alert 2004-165.
- OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999. http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html
- American Society of Health-System Pharmacists. ASHP guidelines on handling hazardous drugs. Am J Health-Syst Pharm. 2006;63:1172-1193.
- Polovich, M., White, J.M., & Kelleher, L.O. (eds.) 2005. Chemotherapy and biotherapy guidelines and recommendations for practice (2nd. ed.) Pittsburgh, PA: Oncology Nursing Society.
16 HOW SUPPLIED/STORAGE AND HANDLING
IXEMPRA is supplied as a Kit containing one vial of IXEMPRA™ (ixabepilone) for injection and one vial of DILUENT for IXEMPRA.
| NDC 0015-1910-12 | IXEMPRA™Kit containing one vial of IXEMPRA™ (ixabepilone) for injection, 15 mg and one vial of DILUENT for IXEMPRA, 8 mL |
| NDC 0015-1911-13 | IXEMPRA™Kit containing one vial of IXEMPRA™ (ixabepilone) for injection, 45 mg and one vial of DILUENT for IXEMPRA, 23.5 mL |
IXEMPRA Kit must be stored in a refrigerator at 2° C to 8° C (36° F to 46° F). Retain in original package until time of use to protect from light.
Procedures for proper handling and disposal of antineoplastic drugs [see References(15)] should be followed. To minimize the risk of dermal exposure, impervious gloves should be worn when handling vials containing IXEMPRA, regardless of the setting, including unpacking and inspection, transport within a facility, and dose preparation and administration.
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Patient Labeling (17.6)
17.1 Peripheral Neuropathy
Patients should be advised to report to their physician any numbness and tingling of the hands or feet [see Warnings and Precautions (5.1)].
17.2 Fever/Neutropenia
Patients should be instructed to call their physician if a fever of 100.5° F or greater or other evidence of potential infection such as chills, cough, or burning or pain on urination develops [see Warnings and Precautions (5.2)].
17.3 Hypersensitivity Reactions
Patients should be advised to call their physician if they experience urticaria, pruritus, rash, flushing, swelling, dyspnea, chest tightness or other hypersensitivity related symptoms following an infusion of IXEMPRA [see Warnings and Precautions (5.4)].
17.4 Pregnancy
Patients should be advised to use effective contraceptive measures to prevent pregnancy and to avoid nursing during treatment with IXEMPRA [see Warnings and Precautions (5.5) and Use in Specific Populations (8.1, 8.3)].
17.5 Cardiac Adverse Reactions
Patients should be advised to report to their physician chest pain, difficulty breathing, palpitations or unusual weight gain [see Warnings and Precautions (5.6)].
17.6 FDA-Approved Patient Labeling
Patient Information
IXEMPRA™Kit (pronounced as ĭk-'sĕm-pră)
(ixabepilone)
for Injection, for intravenous infusion only
Read the Patient Information that comes with IXEMPRA before you start receiving it and before each injection. There may be new information. This leaflet does not take the place of talking with your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about IXEMPRA?
Your healthcare provider should do blood tests to check your liver function:
- before you begin receiving IXEMPRA
- as needed while you are receiving IXEMPRA
If blood tests show that you have liver problems, do not receive injections of IXEMPRA along with the medicine capecitabine. Taking these two medicines together if you have liver problems increases your chance of serious problems. These include: serious infection and death due to a very low white blood cell count (neutropenia).
What is IXEMPRA?
IXEMPRA is a cancer medicine. IXEMPRA is used alone or with another cancer medicine called capecitabine. IXEMPRA is used to treat breast cancer, when certain other medicines have not worked or no longer work.
Who should not take IXEMPRA?
Do not receive injections of IXEMPRA if you:
- are allergic to a medicine, such as TAXOL®, that contains Cremophor® EL or polyoxyethylated castor oil.
- have low white blood cell or platelet counts. Your healthcare provider will check your blood counts.
- are also taking a cancer medicine called capecitabine and you have liver problems. See “What is the most important information I should know about IXEMPRA?”
What should I tell my healthcare provider before receiving IXEMPRA?
IXEMPRA may not be right for you. Before you receive IXEMPRA, tell your healthcare provider about all of your medical conditions, including if you:
- have liver problems
- have heart problems or a history of heart problems
- have had an allergic reaction to IXEMPRA. You will receive medicines before each injection of IXEMPRA to decrease the chance of an allergic reaction. See “How will I receive IXEMPRA?”
- are pregnant or plan to become pregnant. You should not receive IXEMPRA during pregnancy because it may harm your unborn baby. Talk with your healthcare provider about how to prevent pregnancy while receiving IXEMPRA. Tell your healthcare provider right away if you become pregnant or think you are pregnant while receiving IXEMPRA.
- are breast-feeding. It is not known if IXEMPRA passes into breast milk. You and your healthcare provider should decide if you will take IXEMPRA or breast-feed. You should not do both.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
IXEMPRA and certain other medicines may affect each other causing side effects. IXEMPRA may affect the way other medicines work, and other medicines may affect how IXEMPRA works. Know the medicines you take. Keep a list of your medicines with you to show your healthcare provider.
How will I receive IXEMPRA?
IXEMPRA is given by an injection directly into your vein (intravenous infusion). IXEMPRA is usually given once every three weeks. Each treatment with IXEMPRA will take about 3 hours.
Your healthcare provider will decide how much IXEMPRA you will receive and how often you will receive it.
To lower the chance of allergic reaction, you will receive other medicines about 1 hour before each treatment with IXEMPRA. See “What are the possible side effects of IXEMPRA?”
If you have an allergic reaction to IXEMPRA, you will receive a steroid medicine before future doses of IXEMPRA. You may also need to receive your doses of IXEMPRA more slowly.
What should I avoid while receiving IXEMPRA?
IXEMPRA contains alcohol. If you are dizzy or drowsy, avoid activities that may be dangerous, such as driving or operating machinery.
Do not drink grapefruit juice while receiving IXEMPRA. Drinking grapefruit juice may cause you to have too much IXEMPRA in your blood and lead to side effects.
What are the possible side effects of IXEMPRA?
IXEMPRA may cause serious side effects including:
- Numbness, tingling, or burning in the hands or feet can occur while taking IXEMPRA (neuropathy). These symptoms may be new or get worse while you are receiving IXEMPRA. These symptoms often occur early during treatment with IXEMPRA. Tell your healthcare provider if you have any of these symptoms. Your dose of IXEMPRA may need to be decreased, stopped until your symptoms get better, or totally stopped.
- Low white blood cell count (neutropenia). White blood cells help protect the body from infections caused by bacteria. If you get a fever or infection when your white blood cells are very low, you can become seriously ill and die. You may need treatment in the hospital with antibiotic medicines. Your healthcare provider will monitor your white blood cell count often with blood tests. Tell your healthcare provider right away or go to the nearest hospital emergency room if you have a fever (temperature above 100.5° F) or other sign of infection, such as chills, cough, burning or pain when you urinate, any time between treatments with IXEMPRA.
- Allergic Reactions. Severe allergic reactions can occur with IXEMPRA and may cause death in rare cases. Allergic reactions are most likely to occur while IXEMPRA is being injected into your vein. Tell your healthcare provider right away if you get any of the following signs and symptoms of an allergic reaction:
• itching, hives (raised itchy welts), rash
• flushed face
• sudden swelling of face, throat or tongue
• chest tightness, trouble breathing
• feel dizzy or faint
• feel your heart beating (palpitations)
• Harm to an unborn child. See “What should I tell my healthcare provider before taking IXEMPRA?”
• Heart problems. IXEMPRA might cause decreased blood flow to the heart, problems with heart function, and abnormal heart beat. This is seen more often in patients who also take capecitabine. Tell your healthcare provider right away if you have any of the following symptoms:
• chest pain,
• difficulty breathing,
• feel your heart beating (palpitations), or
• unusual weight gain.
The most common side effects with IXEMPRA used alone or with capecitabine may include:
- tiredness
- loss of appetite
- disorders of toenails and fingernails
- hair loss
- fever
- decreased red blood cells (anemia)
- joint and muscle pain
- headache
- decreased platelets (thrombocytopenia)
- nausea, vomiting, diarrhea, constipation, and abdominal pain
- sores on the lip, in the mouth and esophagus
- tender, red palms and soles of feet (hand-foot syndrome) that looks like a sunburn; the skin may become dry and peel. There may also be numbness and tingling.
Tell your healthcare provider about any side effect that bothers you or that does not go away.
These are not all the side effects of IXEMPRA. Ask your healthcare provider or pharmacist for more information if you have questions or concerns.
General information about IXEMPRA?
This patient information leaflet summarizes the most important information about IXEMPRA. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information Leaflet. If you would like more information about IXEMPRA, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about IXEMPRA that is written for health professionals. For more information about IXEMPRA, call 1–888–IXEMPRA.
IXEMPRA™ (ixabepilone) for injection Manufactured by: Baxter Oncology GmbH, 33790 Halle/Westfalen, Germany
DILUENT for IXEMPRA Manufactured by: Baxter Oncology GmbH, 33790 Halle/Westfalen, Germany
Distributed by Bristol-Myers Squibb Company, Princeton, NJ 08543 USA
1236925A1
5645-0001
Rev November 2007
| IXEMPRA
ixabepilone kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| IXEMPRA
ixabepilone kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
Revised: 12/2008