ZOLINZA
-
vorinostat capsule
Merck & Co., Inc.
----------
ZOLINZA™ (vorinostat) Capsules
|
|||||||||||||||||||||
FULL PRESCRIBING INFORMATION
1. INDICATIONS AND USAGE
ZOLINZA1 is indicated for the treatment of cutaneous manifestations in patients with cutaneous T-cell lymphoma who have progressive, persistent or recurrent disease on or following two systemic therapies.
2. DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The recommended dose is 400 mg orally once daily with food.
Treatment may be continued as long as there is no evidence of progressive disease or unacceptable toxicity.
ZOLINZA capsules should not be opened or crushed [see How Supplied/Storage and Handling (16)].
2.2 Dose Modifications
If a patient is intolerant to therapy, the dose may be reduced to 300 mg orally once daily with food. The dose may be further reduced to 300 mg once daily with food for 5 consecutive days each week, as necessary.
2.3 Dosing in Special Populations
No information is available in patients with renal or hepatic impairment [see Pharmacokinetics (12.3)].
3. DOSAGE FORMS AND STRENGTHS
100 mg white, opaque, hard gelatin capsules with “568” over “100 mg” printed within radial bar in black ink on the capsule body.
4. CONTRAINDICATIONS
None
5. WARNINGS AND PRECAUTIONS
5.1 Thromboembolism
As pulmonary embolism and deep vein thrombosis have been reported as adverse reactions, physicians should be alert to the signs and symptoms of these events, particularly in patients with a prior history of thromboembolic events [see Adverse Reactions (6)].
5.2 Hematologic
Treatment with ZOLINZA can cause dose-related thrombocytopenia and anemia. If platelet counts and/or hemoglobin are reduced during treatment with ZOLINZA, the dose should be modified or therapy discontinued. [See Dosage and Administration (2.2), Warnings and Precautions (5.6) and Adverse Reactions (6).]
5.3 Gastrointestinal
Gastrointestinal disturbances, including nausea, vomiting and diarrhea, have been reported [see Adverse Reactions (6)] and may require the use of antiemetic and antidiarrheal medications. Fluid and electrolytes should be replaced to prevent dehydration [see Adverse Reactions (6.1)]. Pre-existing nausea, vomiting, and diarrhea should be adequately controlled before beginning therapy with ZOLINZA.
5.4 Hyperglycemia
Hyperglycemia has been observed in patients receiving ZOLINZA [see Adverse Reactions (6.1)]. Serum glucose should be monitored, especially in diabetic or potentially diabetic patients. Adjustment of diet and/or therapy for increased glucose may be necessary.
5.5 QTc Prolongation
A definitive study of the effect of vorinostat on QTc has not been conducted. Three of 86 CTCL patients exposed to 400 mg once daily had Grade 1 (>450-470 msec) or 2 (>470-500 msec or increase of >60 msec above baseline) clinical adverse events of QTc prolongation. In a retrospective analysis of three Phase 1 and two Phase 2 studies, 116 patients had a baseline and at least one follow-up ECG. Four patients had Grade 2 (>470-500 msec or increase of >60 msec above baseline) and 1 patient had Grade 3 (>500 msec) QTc prolongation. In 49 non-CTCL patients from 3 clinical trials who had complete evaluation of QT interval, 2 had QTc measurements of >500 msec and 1 had a QTc prolongation of >60 msec.
5.6 Monitoring: Laboratory Tests
Careful monitoring of blood cell counts and chemistry tests, including electrolytes, glucose and serum creatinine, should be performed every 2 weeks during the first 2 months of therapy and monthly thereafter. Electrolyte monitoring should include potassium, magnesium and calcium. Baseline and periodic ECGs should be performed during treatment. ZOLINZA should be administered with particular caution in patients with congenital long QT syndrome, and patients taking anti-arrhythmic medicines or other medicinal products that lead to QT prolongation. Hypokalemia or hypomagnesemia should be corrected prior to administration of ZOLINZA, and consideration should be given to monitoring potassium and magnesium in symptomatic patients (e.g., patients with nausea, vomiting, diarrhea, fluid imbalance or cardiac symptoms). [See Warnings and Precautions (5.5).]
5.7 Other Histone Deacetylase (HDAC) Inhibitors
Severe thrombocytopenia and gastrointestinal bleeding have been reported with concomitant use of ZOLINZA and other HDAC inhibitors (e.g., valproic acid). Monitor platelet count every 2 weeks during the first 2 months. [See Drug Interactions (7.2)].
5.8 Pregnancy
Pregnancy Category D
ZOLINZA can cause fetal harm when administered to a pregnant woman. There are no adequate and well-controlled studies of ZOLINZA in pregnant women. Results of animal studies indicate that vorinostat crosses the placenta and is found in fetal plasma at levels up to 50% of maternal concentrations. Doses up to 50 and 150 mg/kg/day were tested in rats and rabbits, respectively (~0.5 times the human exposure based on AUC0-24 hours). Treatment-related developmental effects including decreased mean live fetal weights, incomplete ossifications of the skull, thoracic vertebra, sternebra, and skeletal variations (cervical ribs, supernumerary ribs, vertebral count and sacral arch variations) in rats at the highest dose of vorinostat tested. Reductions in mean live fetal weight and an elevated incidence of incomplete ossification of the metacarpals were seen in rabbits dosed at 150 mg/kg/day. The no observed effect levels (NOELs) for these findings were 15 and 50 mg/kg/day (<0.1 times the human exposure based on AUC) in rats and rabbits, respectively. A dose-related increase in the incidence of malformations of the gall bladder was noted in all drug treatment groups in rabbits versus the concurrent control. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
6. ADVERSE REACTIONS
The most common drug-related adverse reactions can be classified into 4 symptom complexes: gastrointestinal symptoms (diarrhea, nausea, anorexia, weight decrease, vomiting, constipation), constitutional symptoms (fatigue, chills), hematologic abnormalities (thrombocytopenia, anemia), and taste disorders (dysgeusia, dry mouth). The most common serious drug-related adverse reactions were pulmonary embolism and anemia.
6.1 Clinical Trials Experience
The safety of ZOLINZA was evaluated in 107 CTCL patients in two single arm clinical studies in which 86 patients received 400 mg once daily.
The data described below reflect exposure to ZOLINZA 400 mg once daily in the 86 patients for a median number of 97.5 days on therapy (range 2 to 480+ days). Seventeen (19.8%) patients were exposed beyond 24 weeks and 8 (9.3%) patients were exposed beyond 1 year. The population of CTCL patients studied was 37 to 83 years of age, 47.7% female, 52.3% male, and 81.4% white, 16.3% black, and 1.2% Asian or multi-racial.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Common Adverse Reactions
Table 1 summarizes the frequency of CTCL patients with specific adverse events, regardless of causality, using the National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE, version 3.0).
| ZOLINZA 400 mg once daily (N=86) | ||||
| Adverse Events | All Grades | Grades 3-5* | ||
| n | % | n | % | |
|
||||
| Fatigue | 45 | 52.3 | 3 | 3.5 |
| Diarrhea | 45 | 52.3 | 0 | 0.0 |
| Nausea | 35 | 40.7 | 3 | 3.5 |
| Dysgeusia | 24 | 27.9 | 0 | 0.0 |
| Thrombocytopenia | 22 | 25.6 | 5 | 5.8 |
| Anorexia | 21 | 24.4 | 2 | 2.3 |
| Weight Decreased | 18 | 20.9 | 1 | 1.2 |
| Muscle Spasms | 17 | 19.8 | 2 | 2.3 |
| Alopecia | 16 | 18.6 | 0 | 0.0 |
| Dry Mouth | 14 | 16.3 | 0 | 0.0 |
| Blood Creatinine Increased | 14 | 16.3 | 0 | 0.0 |
| Chills | 14 | 16.3 | 1 | 1.2 |
| Vomiting | 13 | 15.1 | 1 | 1.2 |
| Constipation | 13 | 15.1 | 0 | 0.0 |
| Dizziness | 13 | 15.1 | 1 | 1.2 |
| Anemia | 12 | 14.0 | 2 | 2.3 |
| Decreased Appetite | 12 | 14.0 | 1 | 1.2 |
| Peripheral Edema | 11 | 12.8 | 0 | 0.0 |
| Headache | 10 | 11.6 | 0 | 0.0 |
| Pruritus | 10 | 11.6 | 1 | 1.2 |
| Cough | 9 | 10.5 | 0 | 0.0 |
| Upper Respiratory Infection | 9 | 10.5 | 0 | 0.0 |
| Pyrexia | 9 | 10.5 | 1 | 1.2 |
The frequencies of more severe thrombocytopenia, anemia [see Warnings and Precautions (5.2)] and fatigue were increased at doses higher than 400 mg once daily of ZOLINZA.
Serious Adverse Reactions
The most common serious adverse events, regardless of causality, in the 86 CTCL patients in two clinical studies were pulmonary embolism reported in 4.7% (4/86) of patients, squamous cell carcinoma reported in 3.5% (3/86) of patients and anemia reported in 2.3% (2/86) of patients. There were single events of cholecystitis, death (of unknown cause), deep vein thrombosis, enterococcal infection, exfoliative dermatitis, gastrointestinal hemorrhage, infection, lobar pneumonia, myocardial infarction, ischemic stroke, pelvi-ureteric obstruction, sepsis, spinal cord injury, streptococcal bacteremia, syncope, T-cell lymphoma, thrombocytopenia and ureteric obstruction.
Discontinuations
Of the CTCL patients who received the 400-mg once daily dose, 9.3% (8/86) of patients discontinued ZOLINZA due to adverse events. These adverse events, regardless of causality, included anemia, angioneurotic edema, asthenia, chest pain, exfoliative dermatitis, death, deep vein thrombosis, ischemic stroke, lethargy, pulmonary embolism, and spinal cord injury.
Dose Modifications
Of the CTCL patients who received the 400-mg once daily dose, 10.5% (9/86) of patients required a dose modification of ZOLINZA due to adverse events. These adverse events included increased serum creatinine, decreased appetite, hypokalemia, leukopenia, nausea, neutropenia, thrombocytopenia and vomiting. The median time to the first adverse event resulting in dose reduction was 42 days (range 17 to 263 days).
Laboratory Abnormalities
Laboratory abnormalities were reported in all of the 86 CTCL patients who received the 400-mg once-daily dose.
Increased serum glucose was reported as a laboratory abnormality in 69% (59/86) of CTCL patients who received the 400-mg once daily dose; only 4 of these abnormalities were severe (Grade 3). Increased serum glucose was reported as an adverse event in 8.1% (7/86) of CTCL patients who received the 400-mg once daily dose. [See Warnings and Precautions (5.4).]
Transient increases in serum creatinine were detected in 46.5% (40/86) of CTCL patients who received the 400-mg once daily dose. Of these laboratory abnormalities, 34 were NCI CTCAE Grade 1, 5 were Grade 2, and 1 was Grade 3.
Proteinuria was detected as a laboratory abnormality (51.4%) in 38 of 74 patients tested. The clinical significance of this finding is unknown.
Dehydration
Based on reports of dehydration as a serious drug-related adverse event in clinical trials, patients were instructed to drink at least 2 L/day of fluids for adequate hydration. [See Warnings and Precautions (5.3, 5.6).]
Adverse Reactions in Non-CTCL Patients
The frequencies of individual adverse events were substantially higher in the non-CTCL population. Drug-related serious adverse events reported in the non-CTCL population which were not observed in the CTCL population included single events of blurred vision, asthenia, hyponatremia, tumor hemorrhage, Guillain-Barré syndrome, renal failure, urinary retention, cough, hemoptysis, hypertension, and vasculitis.
7. DRUG INTERACTIONS
7.1 Coumarin-Derivative Anticoagulants
Prolongation of prothrombin time (PT) and International Normalized Ratio (INR) were observed in patients receiving ZOLINZA concomitantly with coumarin-derivative anticoagulants. Physicians should carefully monitor PT and INR in patients concurrently administered ZOLINZA and coumarin derivatives.
7.2 Other HDAC Inhibitors
Severe thrombocytopenia and gastrointestinal bleeding have been reported with concomitant use of ZOLINZA and other HDAC inhibitors (e.g., valproic acid). Monitor platelet count every 2 weeks for the first 2 months. [See Warnings and Precautions (5.7).]
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category D [See Warnings and Precautions (5.8)]
8.3 Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from ZOLINZA, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
The safety and effectiveness of ZOLINZA in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of patients with CTCL in trials (N=107), 46 percent were 65 years of age and over, while 15 percent were 75 years of age and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Use in Patients with Hepatic Impairment
Vorinostat was not evaluated in patients with hepatic impairment. As vorinostat is predominantly eliminated through metabolism, patients with hepatic impairment should be treated with caution. [See Clinical Pharmacology (12.3).]
8.7 Use in Patients with Renal Impairment
Vorinostat was not evaluated in patients with renal impairment. However, renal excretion does not play a role in the elimination of vorinostat. Patients with pre-existing renal impairment should be treated with caution. [See Clinical Pharmacology (12.3).]
10. OVERDOSAGE
No specific information is available on the treatment of overdosage of ZOLINZA.
In the event of overdose, it is reasonable to employ the usual supportive measures, e.g., remove unabsorbed material from the gastrointestinal tract, employ clinical monitoring, and institute supportive therapy, if required. It is not known if vorinostat is dialyzable.
11. DESCRIPTION
ZOLINZA contains vorinostat, which is described chemically as N-hydroxy-N'-phenyloctanediamide.
The empirical formula is C14H20N2O3. The molecular weight is 264.32 and the structural formula is:
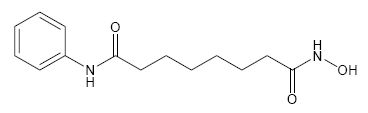
Vorinostat is a white to light orange powder. It is very slightly soluble in water, slightly soluble in ethanol, isopropanol and acetone, freely soluble in dimethyl sulfoxide and insoluble in methylene chloride. It has no chiral centers and is non-hygroscopic. The differential scanning calorimetry ranged from 161.7 (endotherm) to 163.9°C. The pH of saturated water solutions of vorinostat drug substance was 6.6. The pKa of vorinostat was determined to be 9.2.
Each 100 mg ZOLINZA capsule for oral administration contains 100 mg vorinostat and the following inactive ingredients: microcrystalline cellulose, sodium croscarmellose and magnesium stearate. The capsule shell excipients are titanium dioxide, gelatin and sodium lauryl sulfate.
12. CLINICAL PHARMACOLOGY
12.1 Mechanism Of Action
Vorinostat inhibits the enzymatic activity of histone deacetylases HDAC1, HDAC2 and HDAC3 (Class I) and HDAC6 (Class II) at nanomolar concentrations (IC50<86 nM). These enzymes catalyze the removal of acetyl groups from the lysine residues of proteins, including histones and transcription factors. In some cancer cells, there is an overexpression of HDACs, or an aberrant recruitment of HDACs to oncogenic transcription factors causing hypoacetylation of core nucleosomal histones. Hypoacetylation of histones is associated with a condensed chromatin structure and repression of gene transcription. Inhibition of HDAC activity allows for the accumulation of acetyl groups on the histone lysine residues resulting in an open chromatin structure and transcriptional activation. In vitro, vorinostat causes the accumulation of acetylated histones and induces cell cycle arrest and/or apoptosis of some transformed cells. The mechanism of the antineoplastic effect of vorinostat has not been fully characterized.
12.3 Pharmacokinetics
Absorption
The pharmacokinetics of vorinostat were evaluated in 23 patients with relapsed or refractory advanced cancer. After oral administration of a single 400-mg dose of vorinostat with a high-fat meal, the mean ± standard deviation area under the curve (AUC) and peak serum concentration (Cmax) and the median (range) time to maximum concentration (Tmax) were 5.5±1.8 µM●hr, 1.2±0.62 µM and 4 (2-10) hours, respectively.
In the fasted state, oral administration of a single 400-mg dose of vorinostat resulted in a mean AUC and Cmax and median Tmax of 4.2±1.9 µM●hr and 1.2±0.35 µM and 1.5 (0.5-10) hours, respectively. Therefore, oral administration of vorinostat with a high-fat meal resulted in an increase (33%) in the extent of absorption and a modest decrease in the rate of absorption (Tmax delayed 2.5 hours) compared to the fasted state. However, these small effects are not expected to be clinically meaningful. In clinical trials of patients with CTCL, vorinostat was taken with food.
At steady state in the fed-state, oral administration of multiple 400-mg doses of vorinostat resulted in a mean AUC and Cmax and a median Tmax of 6.0±2.0 µM●hr, 1.2±0.53 µM and 4 (0.5-14) hours, respectively.
Distribution
Vorinostat is approximately 71% bound to human plasma proteins over the range of concentrations of 0.5 to 50 µg/mL.
Metabolism
The major pathways of vorinostat metabolism involve glucuronidation and hydrolysis followed by β-oxidation. Human serum levels of two metabolites, O-glucuronide of vorinostat and 4-anilino-4-oxobutanoic acid were measured. Both metabolites are pharmacologically inactive. Compared to vorinostat, the mean steady state serum exposures in humans of the O-glucuronide of vorinostat and 4-anilino-4-oxobutanoic acid were 4-fold and 13-fold higher, respectively.
In vitro studies using human liver microsomes indicate negligible biotransformation by cytochromes P450 (CYP).
Excretion
Vorinostat is eliminated predominantly through metabolism with less than 1% of the dose recovered as unchanged drug in urine, indicating that renal excretion does not play a role in the elimination of vorinostat. The mean urinary recovery of two pharmacologically inactive metabolites at steady state was 16±5.8% of vorinostat dose as the O‑glucuronide of vorinostat, and 36±8.6% of vorinostat dose as 4-anilino-4-oxobutanoic acid. Total urinary recovery of vorinostat and these two metabolites averaged 52±13.3% of vorinostat dose. The mean terminal half-life (t½) was ~2.0 hours for both vorinostat and the O-glucuronide metabolite, while that of the 4-anilino-4-oxobutanoic acid metabolite was 11 hours.
Special Populations
Based upon an exploratory analysis of limited data, gender, race and age do not appear to have meaningful effects on the pharmacokinetics of vorinostat.
Pediatric
Vorinostat was not evaluated in patients <18 years of age.
Hepatic Insufficiency
Vorinostat was not evaluated in patients with hepatic impairment. [See Use in Specific Populations (8.6).]
Renal Insufficiency
Vorinostat was not evaluated in patients with renal impairment. However, renal excretion does not play a role in the elimination of vorinostat. [See Use in Specific Populations (8.7).]
Pharmacokinetic effects of vorinostat with other agents
Vorinostat is not an inhibitor of CYP drug metabolizing enzymes in human liver microsomes at steady state Cmax of the 400 mg dose (Cmax of 1.2 µM vs IC50 of >75 µM). Gene expression studies in human hepatocytes detected some potential for suppression of CYP2C9 and CYP3A4 activities by vorinostat at concentrations higher (≥10 µM) than pharmacologically relevant. Thus, vorinostat is not expected to affect the pharmacokinetics of other agents. As vorinostat is not eliminated via the CYP pathways, it is anticipated that vorinostat will not be subject to drug-drug interactions when co-administered with drugs that are known CYP inhibitors or inducers. However, no formal clinical studies have been conducted to evaluate drug interactions with vorinostat.
In vitro studies indicate that vorinostat is not a substrate of human P-glycoprotein (P-gp). In addition, vorinostat has no inhibitory effect on human P-gp-mediated transport of vinblastine (a marker P-gp substrate) at concentrations of up to 100 μM. Thus, vorinostat is not likely to inhibit P-gp at the pharmacologically relevant serum concentration of 2 μM (Cmax) in humans.
13. NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment Of Fertility
Carcinogenicity studies have not been performed with vorinostat.
Vorinostat was mutagenic in vitro in the bacterial reverse mutation assays (Ames test), caused chromosomal aberrations in vitro in Chinese hamster ovary (CHO) cells and increased the incidence of micro-nucleated erythrocytes when administered to mice (Mouse Micronucleus Assay).
Effects on the female reproductive system were identified in the oral fertility study when females were dosed for 14 days prior to mating through gestational day 7. Doses of 15, 50 and 150 mg/kg/day to rats resulted in approximate exposures of 0.15, 0.36 and 0.70 times the expected clinical exposure based on AUC. Dose dependent increases in corpora lutea were noted at ≥15 mg/kg/day, which resulted in increased peri-implantation losses were noted at ≥50 mg/kg/day. At 150 mg/kg/day, there were increases in the incidences of dead fetuses and in resorptions.
No effects on reproductive performance were observed in male rats dosed (20, 50, 150 mg/kg/day; approximate exposures of 0.15, 0.36 and 0.70 times the expected clinical exposure based on AUC), for 70 days prior to mating with untreated females. [See Warnings and Precautions (5.8).]
14. CLINICAL STUDIES
Cutaneous T-cell Lymphoma
In two open-label clinical studies, patients with refractory CTCL have been evaluated to determine their response rate to oral ZOLINZA. One study was a single-arm clinical study and the other assessed several dosing regimens. In both studies, patients were treated until disease progression or intolerable toxicity.
Study 1
In an open-label, single‑arm, multicenter non-randomized study, 74 patients with advanced CTCL were treated with ZOLINZA at a dose of 400 mg once daily. The primary endpoint was response rate to oral ZOLINZA in the treatment of skin disease in patients with advanced CTCL (Stage IIB and higher) who had progressive, persistent, or recurrent disease on or following two systemic therapies. Enrolled patients should have received, been intolerant to or not a candidate for bexarotene. Extent of skin disease was quantitatively assessed by investigators using a modified Severity Weighted Assessment Tool (SWAT). The investigator measured the percentage total body surface area (%TBSA) involvement separately for patches, plaques, and tumors within 12 body regions using the patient’s palm as a “ruler”. The total %TBSA for each lesion type was multiplied by a severity weighting factor (1=patch, 2=plaque and 4=tumor) and summed to derive the SWAT score. Efficacy was measured as either a Complete Clinical Response (CCR) defined as no evidence of disease, or Partial Response (PR) defined as a ≥50% decrease in SWAT skin assessment score compared to baseline. Both CCR and PR had to be maintained for at least 4 weeks.
Secondary efficacy endpoints included response duration, time to progression, and time to objective response.
The population had been exposed to a median of three prior therapies (range 1 to 12).
Table 2 summarizes the demographic and disease characteristics of the Study 1 population.
| Vorinostat | |
| Characteristics | (N=74) |
| Age (year) | |
| Mean (SD) | 61.2 (11.3) |
| Median (Range) | 60.0 (39.0, 83.0) |
| Gender, n (%) | |
| Male | 38 (51.4%) |
| Female | 36 (48.6%) |
| CTCL stage, n (%) | |
| IB | 11 (14.9%) |
| IIA | 2 (2.7%) |
| IIB | 19 (25.7%) |
| III | 22 (29.7%) |
| IVA | 16 (21.6%) |
| IVB | 4 (5.4%) |
| Racial Origin, n (%) | |
| Asian | 1 (1.4%) |
| Black | 11 (14.9%) |
| Other | 1 (1.4%) |
| White | 61 (82.4%) |
| Time from Initial CTCL Diagnosis (year) | |
| Median (Range) | 2.6 (0.0, 27.3) |
| Clinical Characteristics | |
| Number of prior systemic treatments, median (range) | 3.0 (1.0, 12.0) |
The overall objective response rate was 29.7% (22/74, 95% CI [19.7 to 41.5%]) in all patients treated with ZOLINZA. In patients with Stage IIB and higher CTCL, the overall objective response rate was 29.5% (18/61). One patient with Stage IIB CTCL achieved a CCR. Median times to response were 55 and 56 days (range 28 to 171 days), respectively in the overall population and in patients with Stage IIB and higher CTCL. However, in rare cases it took up to 6 months for patients to achieve an objective response to ZOLINZA.
The median response duration was not reached since the majority of responses continued at the time of analysis, but was estimated to exceed 6 months for both the overall population and in patients with Stage IIB and higher CTCL. When end of response was defined as a 50% increase in SWAT score from the nadir, the estimated median response duration was 168 days and the median time to tumor progression was 202 days.
Using a 25% increase in SWAT score from the nadir as criterion for tumor progression, the estimated median time-to-progression was 148 days for the overall population and 169 days in the 61 patients with Stage IIB and higher CTCL.
Response to any previous systemic therapy does not appear to be predictive of response to ZOLINZA.
Study 2
In an open-label, non-randomized study, ZOLINZA was evaluated to determine the response rate for patients with CTCL who were refractory or intolerant to at least one treatment. In this study, 33 patients were assigned to one of 3 cohorts: Cohort 1, 400 mg once daily; Cohort 2, 300 mg twice daily 3 days/week; or Cohort 3, 300 mg twice daily for 14 days followed by a 7-day rest (induction). In Cohort 3, if at least a partial response was not observed then patients were dosed with a maintenance regimen of 200 mg twice daily. The primary efficacy endpoint, objective response, was measured by the 7‑point Physician’s Global Assessment (PGA) scale. The investigator assessed improvement or worsening in overall disease compared to baseline based on overall clinical impression. Index and non-index cutaneous lesions as well as cutaneous tumors, lymph nodes and all other disease manifestations were also assessed and included in the overall clinical impression. CCR required 100% clearing of all findings, and PR required at least 50% improvement in disease findings.
The median age was 67.0 years (range 26.0 to 82.0). Fifty-five percent of patients were male, and 45% of patients were female. Fifteen percent of patients had Stage IA, IB, or IIA CTCL and 85% of patients had Stage IIB, III, IVA, or IVB CTCL. The median number of prior systemic therapies was 4 (range 0.0 to 11.0).
In all patients treated, the objective response was 24.2% (8/33) in the overall population, 25% (7/28) in patients with Stage IIB or higher disease and 36.4% (4/11) in patients with Sezary syndrome. The overall response rates were 30.8%, 9.1% and 33.3% in Cohort 1, Cohort 2 and Cohort 3, respectively. The 300 mg twice daily regimen had higher toxicity with no additional clinical benefit over the 400 mg once daily regimen. No CCR was observed.
Among the 8 patients who responded to study treatment, the median time to response was 83.5 days (range 25 to 153 days). The median response duration was 106 days (range 66 to 136 days). Median time to progression was 211.5 days (range 94 to 255 days).
15. REFERENCES
- NIOSH Alert: Preventing occupational exposures to antineoplastic and other hazardous drugs in healthcare settings. 2004. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2004-165.
- OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999.http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html
- NIH [2002]. 1999 recommendations for the safe handling of cytotoxic drugs. U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, NIH Publication No. 92-2621.
- American Society of Health-System Pharmacists. (2006) ASHP Guidelines on Handling Hazardous Drugs.
- Polovich, M., White, J. M., & Kelleher, L.O. (eds.) 2005. Chemotherapy and biotherapy guidelines and recommendations for practice (2nd. ed.) Pittsburgh, PA: Oncology Nursing Society.
16. HOW SUPPLIED/STORAGE AND HANDLING
ZOLINZA capsules, 100 mg, are white, opaque hard gelatin capsules with “568” over “100 mg” printed within the radial bar in black ink on the capsule body. They are supplied as follows:
NDC 0006‑0568-40.
Each bottle contains 120 capsules.
Storage and Handling
Store at 20-25°C (68-77°F), excursions permitted between 15-30°C (59-86°F). [See USP Controlled Room Temperature.]
Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published.1-5 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
ZOLINZA (vorinostat) capsules should not be opened or crushed. Direct contact of the powder in ZOLINZA capsules with the skin or mucous membranes should be avoided. If such contact occurs, wash thoroughly as outlined in the references. Personnel should avoid exposure to crushed and/or broken capsules [see Nonclinical Toxicology (13.1)].
17. PATIENT COUNSELING INFORMATION
[See FDA-Approved Patient Labeling (17.2)]
17.1 Instructions
Patients should be instructed to drink at least 2 L/day of fluid to prevent dehydration and should promptly report excessive vomiting or diarrhea to their physician. Patients should be instructed about the signs of deep vein thrombosis and should consult their physician should any evidence of deep vein thrombosis develop. Patients receiving ZOLINZA should seek immediate medical attention if unusual bleeding occurs. ZOLINZA capsules should not be opened or crushed.
Patients should be instructed to read the patient insert carefully.
Manufactured for:
MERCK & CO., INC., Whitehouse Station, NJ 08889, USA
Manufactured by:
Patheon, Inc.
Mississauga, Ontario, Canada L5N 7K9
Printed in USA
9762601
U.S. Patent Nos. RE 38,506 E, 6,087,367
1Trademark of MERCK & CO., Inc., Whitehouse Station, New Jersey 08889 USA
COPYRIGHT © 2006, 2008 MERCK & CO., Inc.
All rights reserved
17.2 FDA-Approved Patient Labeling
Patient Information
ZOLINZA™ (zo LINZ ah)
(vorinostat)
Capsules
Read the patient information that comes with ZOLINZA1 before you start taking it and each time you get a refill. There may be new information. This leaflet is a summary of the information for patients. Your doctor or pharmacist can give you additional information. This leaflet does not take the place of talking with your doctor about your medical condition or your treatment.
What is ZOLINZA?
ZOLINZA is a prescription medicine used to treat a type of cancer called cutaneous T-cell lymphoma (CTCL) in patients when the CTCL gets worse, does not go away, or comes back after treatment with other medicines.
ZOLINZA has not been studied in children under the age of 18.
What should I tell my doctor before taking ZOLINZA?
Tell your doctor about all of your medical conditions, including if you:
- Have any allergies
- Have had a blood clot in your lung (pulmonary embolus)
- Have had a blood clot in a vein (a blood vessel) anywhere in your body (deep vein thrombosis)
- Have nausea, vomiting, or diarrhea
- Have high blood sugar or diabetes
- Have heart problems
- Are pregnant or plan to become pregnant. ZOLINZA may harm your unborn baby. ZOLINZA has not been studied in pregnant women. If you use ZOLINZA during pregnancy, tell your doctor immediately.
- Are breast-feeding or plan to breast-feed. It is not known if ZOLINZA will pass into your breast milk. Talk to your doctor about the best way to feed your baby while you are taking ZOLINZA.
Tell your doctor about all of the medicines you take, including prescription and non-prescription medicines, vitamins and herbal supplements. Some medicines may affect how ZOLINZA works, or ZOLINZA may affect how your other medicines work. Especially tell your doctor if you take:
- Valproic acid: a medicine used to treat seizures. Your doctor will decide if you should continue to take valproic acid and may want to test your blood more frequently.
- COUMADIN®: (warfarin) or any other blood thinner. Ask your doctor if you are not sure if you are taking a blood thinner. Your doctor may want to test your blood more frequently.
Know the medicines you take. Keep a list of your medicines and show it to your doctor and pharmacist when you get a new medicine.
How should I take ZOLINZA?
- Take ZOLINZA exactly as your doctor tells you to.
- Your doctor will tell you how many ZOLINZA capsules to take and when to take them.
- Swallow each capsule whole. Do not chew or break open the capsule. If you can’t swallow ZOLINZA capsules whole, tell your doctor. You may need a different medicine.
- Take ZOLINZA with food.
- If ZOLINZA capsules are accidentally opened or crushed, do not touch the capsules or the powder contents of the capsules. If the powder from an open or crushed capsule gets on your skin or in your eyes, wash the contacted area well with plenty of plain water. Call your doctor.
- Drink at least eight 8-ounce glasses of liquids every day while taking ZOLINZA. Drinking enough fluids may help to decrease the chances of losing too much fluid from your body (dehydration) especially if you are having symptoms such as nausea, vomiting or diarrhea while taking ZOLINZA.
- If you miss a dose, take it as soon as you remember. If you do not remember until it is almost time for your next dose, just skip the missed dose. Just take the next dose at your regular time. Do not take two doses of ZOLINZA at the same time.
- If you take too much ZOLINZA, call your doctor, local emergency room, or poison control center right away.
- Your doctor will check your blood cell counts, blood sugar, and other chemistries every two weeks for the first two months of your treatment with ZOLINZA and then monthly. Your doctor may decide to do other tests to check your health as needed.
- If you have high blood sugar (hyperglycemia) or diabetes, continue to monitor your blood sugar as your doctor tells you to. Your doctor may need to change your diet or medicine to help control your blood sugar while you take ZOLINZA. Be sure to tell your doctor if you are unable to eat or drink normally due to nausea, vomiting or diarrhea.
What are the possible side effects of ZOLINZA?
ZOLINZA may cause serious side effects. Tell your doctor right away if you have any of the following symptoms:
-
Blood clots in the legs (deep vein thrombosis)
- sudden swelling in a leg
- pain or tenderness in the leg. The pain may only be felt when standing or walking.
- increased warmth in the area where the swelling is.
- skin redness or change in skin color
-
Blood clots that travel to the lungs (pulmonary embolus)
- sudden sharp chest pain
- shortness of breath
- cough with bloddy secretions
- sweating
- rapid pulse
- fainting
- feeling anxious
- Dehydration (loss of too much fluid from the body). This can happen if you are having nausea, vomiting or diarrhea and can not drink fluids well.
-
Low blood cell counts: Your doctor will periodically do blood tests to check your blood counts.
- Low red blood cells. Low red blood cells may make you feel tired and get tired easily. You may look pale, and feel short of breath.
- Low platelets. Low platelets can cause unusual bleeding or bruising under the skin. Talk to your doctor right away if this happens.
- High blood sugar (blood glucose). If you have high blood sugar or diabetes, monitor your blood sugar frequently as directed by your doctor. Tell your doctor right away if your blood sugar is higher than normal.
- Electrocardiogram abnormality. An electrocardiogram, or EKG, is a test that records the electrical activity of your heart. Your doctor will check your blood electrolytes and electrocardiogram periodically.
In addition, the most common side effects with ZOLINZA include:
- Stomach and intestinal problems, including diarrhea, nausea, vomiting, loss of appetite, constipation and weight loss
- Tiredness
- Dizziness
- Headache
- Changes in the way things taste and dry mouth
- Muscle aches
- Hair loss
- Chills
- Fever
- Upper respiratory infection
- Cough
- Increase in blood creatinine
- Swelling in the foot, ankle and leg
- Itching
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of ZOLINZA. For more information, ask your doctor or pharmacist.
General information about ZOLINZA
Medicines are sometimes prescribed for conditions that are not mentioned in patient information leaflets. Do not use ZOLINZA for a condition for which it was not prescribed. Do not give ZOLINZA to other people, even if they have the same symptoms you have. It may harm them.
Keep ZOLINZA and all medicines out of the reach of children.
This leaflet summarizes the most important information about ZOLINZA. If you would like to know more information, talk to your doctor. You can ask your doctor or pharmacist for information about ZOLINZA that is written for health professionals.
What are the ingredients in ZOLINZA?
Active ingredient: vorinostat
Inactive ingredients: microcrystalline cellulose, sodium croscarmellose and magnesium stearate. The inactive ingredients in the capsule shell are titanium dioxide, gelatin, and sodium lauryl sulfate.
How should I store ZOLINZA?
Store ZOLINZA at room temperature, 68° F to 77° F (20° C to 25° C).
Issued: July 2008
MERCK & CO., INC.
Whitehouse Station, NJ 08889, USA
9762601
| ZOLINZA
vorinostat capsule |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
Revised: 10/2008Merck & Co., Inc.