MICARDIS
-
telmisartan tablet
Boehringer Ingelheim Pharmaceuticals, Inc.
----------
Micardis®
(telmisartan)
Tablets, 20 mg, 40 mg and 80 mg
Prescribing Information
USE IN PREGNANCY
When used in pregnancy during the second and third trimesters, drugs that act directly on the renin-angiotensin system can cause injury and even death to the developing fetus.
When pregnancy is detected, MICARDIS tablets should be discontinued as soon as possible.
See WARNINGS: Fetal/Neonatal Morbidity and Mortality
DESCRIPTION
MICARDIS tablets is a non-peptide angiotensin II receptor (type AT1) antagonist.
Telmisartan is chemically described as 4'-[(1,4'-dimethyl-2'-propyl [2,6'-bi-1H-benzimidazol]-1'-yl)methyl]-[1,1'-biphenyl]-2-carboxylic acid. Its empirical formula is C33H30N4O2, its molecular weight is 514.63, and its structural formula is:
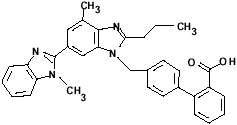
Telmisartan is a white to slightly yellowish solid. It is practically insoluble in water and in the pH range of 3 to 9, sparingly soluble in strong acid (except insoluble in hydrochloric acid), and soluble in strong base.
MICARDIS tablets are available for oral administration, containing 20 mg, 40 mg or 80 mg of telmisartan. The tablets contain the following inactive ingredients: sodium hydroxide, meglumine, povidone, sorbitol, and magnesium stearate. MICARDIS tablets are hygroscopic and require protection from moisture.
CLINICAL PHARMACOLOGY
Mechanism of Action
Angiotensin II is formed from angiotensin I in a reaction catalyzed by angiotensin-converting enzyme (ACE, kininase II). Angiotensin II is the principal pressor agent of the renin-angiotensin system, with effects that include vasoconstriction, stimulation of synthesis and release of aldosterone, cardiac stimulation, and renal reabsorption of sodium. Telmisartan blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the AT1 receptor in many tissues, such as vascular smooth muscle and the adrenal gland. Its action is therefore independent of the pathways for angiotensin II synthesis.
There is also an AT2 receptor found in many tissues, but AT2 is not known to be associated with cardiovascular homeostasis. Telmisartan has much greater affinity (>3,000 fold) for the AT1 receptor than for the AT2 receptor.
Blockade of the renin-angiotensin system with ACE inhibitors, which inhibit the biosynthesis of angiotensin II from angiotensin I, is widely used in the treatment of hypertension. ACE inhibitors also inhibit the degradation of bradykinin, a reaction also catalyzed by ACE. Because telmisartan does not inhibit ACE (kininase II), it does not affect the response to bradykinin. Whether this difference has clinical relevance is not yet known. Telmisartan does not bind to or block other hormone receptors or ion channels known to be important in cardiovascular regulation.
Blockade of the angiotensin II receptor inhibits the negative regulatory feedback of angiotensin II on renin secretion, but the resulting increased plasma renin activity and angiotensin II circulating levels do not overcome the effect of telmisartan on blood pressure.
Pharmacokinetics
General
Following oral administration, peak concentrations (Cmax) of telmisartan are reached in 0.5-1 hour after dosing. Food slightly reduces the bioavailability of telmisartan, with a reduction in the area under the plasma concentration-time curve (AUC) of about 6% with the 40 mg tablet and about 20% after a 160 mg dose. The absolute bioavailability of telmisartan is dose dependent. At 40 and 160 mg the bioavailability was 42% and 58%, respectively. The pharmacokinetics of orally administered telmisartan are nonlinear over the dose range 20-160 mg, with greater than proportional increases of plasma concentrations (Cmax and AUC) with increasing doses. Telmisartan shows bi-exponential decay kinetics with a terminal elimination half life of approximately 24 hours. Trough plasma concentrations of telmisartan with once daily dosing are about 10-25% of peak plasma concentrations. Telmisartan has an accumulation index in plasma of 1.5 to 2.0 upon repeated once daily dosing.
Metabolism and Elimination
Following either intravenous or oral administration of 14C-labeled telmisartan, most of the administered dose (>97%) was eliminated unchanged in feces via biliary excretion; only minute amounts were found in the urine (0.91% and 0.49% of total radioactivity, respectively).
Telmisartan is metabolized by conjugation to form a pharmacologically inactive acylglucuronide; the glucuronide of the parent compound is the only metabolite that has been identified in human plasma and urine. After a single dose, the glucuronide represents approximately 11% of the measured radioactivity in plasma. The cytochrome P450 isoenzymes are not involved in the metabolism of telmisartan.
Total plasma clearance of telmisartan is >800 mL/min. Terminal half-life and total clearance appear to be independent of dose.
Distribution
Telmisartan is highly bound to plasma proteins (>99.5%), mainly albumin and α1 - acid glycoprotein. Plasma protein binding is constant over the concentration range achieved with recommended doses. The volume of distribution for telmisartan is approximately 500 liters indicating additional tissue binding.
Special Populations
Pediatric: Telmisartan pharmacokinetics have not been investigated in patients <18 years of age.
Geriatric: The pharmacokinetics of telmisartan do not differ between the elderly and those younger than 65 years (see DOSAGE AND ADMINISTRATION).
Gender: Plasma concentrations of telmisartan are generally 2-3 times higher in females than in males. In clinical trials, however, no significant increases in blood pressure response or in the incidence of orthostatic hypotension were found in women. No dosage adjustment is necessary.
Renal Insufficiency: No dosage adjustment is necessary in patients with decreased renal function. Telmisartan is not removed from blood by hemofiltration (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Hepatic Insufficiency: In patients with hepatic insufficiency, plasma concentrations of telmisartan are increased, and absolute bioavailability approaches 100% (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Drug Interactions: See PRECAUTIONS, Drug Interactions.
Pharmacodynamics
In normal volunteers, a dose of telmisartan 80 mg inhibited the pressor response to an intravenous infusion of angiotensin II by about 90% at peak plasma concentrations with approximately 40% inhibition persisting for 24 hours.
Plasma concentration of angiotensin II and plasma renin activity (PRA) increased in a dose-dependent manner after single administration of telmisartan to healthy subjects and repeated administration to hypertensive patients. The once-daily administration of up to 80 mg telmisartan to healthy subjects did not influence plasma aldosterone concentrations. In multiple dose studies with hypertensive patients, there were no clinically significant changes in electrolytes (serum potassium or sodium), or in metabolic function (including serum levels of cholesterol, triglycerides, HDL, LDL, glucose, or uric acid).
In 30 hypertensive patients with normal renal function treated for 8 weeks with telmisartan 80 mg or telmisartan 80 mg in combination with hydrochlorothiazide 12.5 mg, there were no clinically significant changes from baseline in renal blood flow, glomerular filtration rate, filtration fraction, renovascular resistance, or creatinine clearance.
Clinical Trials
The antihypertensive effects of Micardis® (telmisartan) tablets have been demonstrated in six principal placebo-controlled clinical trials, studying a range of 20-160 mg; one of these examined the antihypertensive effects of telmisartan and hydrochlorothiazide in combination. The studies involved a total of 1773 patients with mild to moderate hypertension (diastolic blood pressure of 95-114 mmHg), 1031 of whom were treated with telmisartan. Following once daily administration of telmisartan, the magnitude of blood pressure reduction from baseline after placebo subtraction was approximately (SBP/DBP) 6-8/6 mmHg for 20 mg, 9-13/6-8 mmHg for 40 mg, and 12-13/7-8 mmHg for 80 mg. Larger doses (up to 160 mg) did not appear to cause a further decrease in blood pressure.
Upon initiation of antihypertensive treatment with telmisartan, blood pressure was reduced after the first dose, with a maximal reduction by about 4 weeks. With cessation of treatment with MICARDIS tablets, blood pressure gradually returned to baseline values over a period of several days to one week. During long term studies (without placebo control) the effect of telmisartan appeared to be maintained for up to at least one year. The antihypertensive effect of telmisartan is not influenced by patient age, gender, weight or body mass index. Blood pressure response in black patients (usually a low-renin population) is noticeably less than that in Caucasian patients. This has been true for most, but not all, angiotensin II antagonists and ACE inhibitors.
In a controlled study, the addition of telmisartan to hydrochlorothiazide produced an additional dose-related reduction in blood pressure that was similar in magnitude to the reduction achieved with telmisartan monotherapy. Hydrochlorothiazide also had an added blood pressure effect when added to telmisartan.
The onset of antihypertensive activity occurs within 3 hours after administration of a single oral dose. At doses of 20, 40, and 80 mg, the antihypertensive effect of once daily administration of telmisartan is maintained for the full 24-hour dose interval. With automated ambulatory blood pressure monitoring and conventional blood pressure measurements, the 24-hour trough-to-peak ratio for 40-80 mg doses of telmisartan was 70-100% for both systolic and diastolic blood pressure. The incidence of symptomatic orthostasis after the first dose in all controlled trials was low (0.04%).
There were no changes in the heart rate of patients treated with telmisartan in controlled trials.
INDICATIONS AND USAGE
Micardis® (telmisartan) tablets are indicated for the treatment of hypertension. It may be used alone or in combination with other antihypertensive agents.
CONTRAINDICATIONS
MICARDIS tablets are contraindicated in patients who are hypersensitive to any component of this product.
WARNINGS
Fetal/Neonatal Morbidity and Mortality
Drugs that act directly on the renin-angiotensin system can cause fetal and neonatal morbidity and death when administered to pregnant women. Several dozen cases have been reported in the world literature in patients who were taking angiotensin converting enzyme inhibitors. When pregnancy is detected, MICARDIS tablets should be discontinued as soon as possible.
The use of drugs that act directly on the renin-angiotensin system during the second and third trimesters of pregnancy has been associated with fetal and neonatal injury, including hypotension, neonatal skull hypoplasia, anuria, reversible or irreversible renal failure, and death. Oligohydramnios has also been reported, presumably resulting from decreased fetal renal function; oligohydramnios in this setting has been associated with fetal limb contractures, craniofacial deformation, and hypoplastic lung development. Prematurity, intrauterine growth retardation, and patent ductus arteriosus have also been reported, although it is not clear whether these occurrences were due to exposure to the drug.
These adverse effects do not appear to have resulted from intrauterine drug exposure that has been limited to the first trimester. Mothers whose embryos and fetuses are exposed to an angiotensin II receptor antagonist only during the first trimester should be so informed. Nonetheless, when patients become pregnant, physicians should have the patient discontinue the use of MICARDIS tablets as soon as possible.
Rarely (probably less often than once in every thousand pregnancies), no alternative to an angiotensin II receptor antagonist will be found. In these rare cases, the mothers should be apprised of the potential hazards to their fetuses, and serial ultrasound examinations should be performed to assess the intra-amniotic environment.
If oligohydramnios is observed, MICARDIS tablets should be discontinued unless they are considered life-saving for the mother. Contraction stress testing (CST), a non-stress test (NST), or biophysical profiling (BPP) may be appropriate, depending upon the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury.
Infants with histories of in utero exposure to an angiotensin II receptor antagonist should be closely observed for hypotension, oliguria, and hyperkalemia. If oliguria occurs, attention should be directed toward support of blood pressure and renal perfusion. Exchange transfusion or dialysis may be required as a means of reversing hypotension and/or substituting for disordered renal function.
There is no clinical experience with the use of MICARDIS tablets in pregnant women. No teratogenic effects were observed when telmisartan was administered to pregnant rats at oral doses of up to 50 mg/kg/day and to pregnant rabbits at oral doses up to 45 mg/kg/day. In rabbits, embryolethality associated with maternal toxicity (reduced body weight gain and food consumption) was observed at 45 mg/kg/day [about 12 times the maximum recommended human dose (MRHD) of 80 mg on a mg/m2 basis]. In rats, maternally toxic (reduction in body weight gain and food consumption) telmisartan doses of 15 mg/kg/day (about 1.9 times the MRHD on a mg/m2 basis), administered during late gestation and lactation, were observed to produce adverse effects in neonates, including reduced viability, low birth weight, delayed maturation, and decreased weight gain. Telmisartan has been shown to be present in rat fetuses during late gestation and in rat milk. The no observed effect doses for developmental toxicity in rats and rabbits, 5 and 15 mg/kg/day, respectively, are about 0.64 and 3.7 times, on a mg/m2 basis, the maximum recommended human dose of telmisartan (80 mg/day).
Hypotension in Volume-Depleted Patients
In patients with an activated renin-angiotensin system, such as volume- and/or salt-depleted patients (e.g., those being treated with high doses of diuretics), symptomatic hypotension may occur after initiation of therapy with Micardis® (telmisartan) tablets. This condition should be corrected prior to administration of MICARDIS tablets, or treatment should start under close medical supervision with a reduced dose.
If hypotension does occur, the patient should be placed in the supine position and, if necessary, given an intravenous infusion of normal saline. A transient hypotensive response is not a contraindication to further treatment, which usually can be continued without difficulty once the blood pressure has stabilized.
PRECAUTIONS
General
Impaired Hepatic Function: As the majority of telmisartan is eliminated by biliary excretion, patients with biliary obstructive disorders or hepatic insufficiency can be expected to have reduced clearance. MICARDIS tablets should be used with caution in these patients.
Impaired Renal Function: As a consequence of inhibiting the renin-angiotensin-aldosterone system, changes in renal function may be anticipated in susceptible individuals. In patients whose renal function may depend on the activity of the renin-angiotensin-aldosterone system (e.g., patients with severe congestive heart failure), treatment with angiotensin-converting enzyme inhibitors and angiotensin receptor antagonists has been associated with oliguria and/or progressive azotemia and (rarely) with acute renal failure and/or death. Similar results may be anticipated in patients treated with MICARDIS tablets.
In studies of ACE inhibitors in patients with unilateral or bilateral renal artery stenosis, increases in serum creatinine or blood urea nitrogen were observed. There has been no long term use of MICARDIS tablets in patients with unilateral or bilateral renal artery stenosis but an effect similar to that seen with ACE inhibitors should be anticipated.
Dual Blockade of the Renin-angiotensin-aldosterone System: As a consequence of inhibiting the renin-angiotensin-aldosterone system, changes in renal function (including acute renal failure) have been reported. Dual blockade of the renin-angiotensin-aldosterone system (e.g., by adding an ACE-inhibitor to an angiotensin II receptor antagonist) should be used with caution and should include close monitoring of renal function.
Information for Patients
Pregnancy: Female patients of childbearing age should be told about the consequences of second- and third-trimester exposure to drugs that act on the renin-angiotensin system, and they should also be told that these consequences do not appear to have resulted from intrauterine drug exposure that has been limited to the first trimester. These patients should be asked to report pregnancies to their physicians as soon as possible.
Drug Interactions
Digoxin: When telmisartan was co-administered with digoxin, median increases in digoxin peak plasma concentration (49%) and in trough concentration (20%) were observed. It is, therefore, recommended that digoxin levels be monitored when initiating, adjusting, and discontinuing telmisartan to avoid possible over- or under-digitalization.
Warfarin: Telmisartan administered for 10 days slightly decreased the mean warfarin trough plasma concentration; this decrease did not result in a change in International Normalized Ratio (INR).
Other Drugs: Co-administration of telmisartan did not result in a clinically significant interaction with acetaminophen, amlodipine, glibenclamide, simvastatin, hydrochlorothiazide or ibuprofen. Telmisartan is not metabolized by the cytochrome P450 system and had no effects in vitro on cytochrome P450 enzymes, except for some inhibition of CYP2C19. Telmisartan is not expected to interact with drugs that inhibit cytochrome P450 enzymes; it is also not expected to interact with drugs metabolized by cytochrome P450 enzymes, except for possible inhibition of the metabolism of drugs metabolized by CYP2C19.
Carcinogenesis, Mutagenesis, Impairment of Fertility
There was no evidence of carcinogenicity when telmisartan was administered in the diet to mice and rats for up to 2 years. The highest doses administered to mice (1000 mg/kg/day) and rats (100 mg/kg/day) are, on a mg/m2 basis, about 59 and 13 times, respectively, the maximum recommended human dose (MRHD) of telmisartan. These same doses have been shown to provide average systemic exposures to telmisartan >100 times and >25 times, respectively, the systemic exposure in humans receiving the MRHD (80 mg/day).
Genotoxicity assays did not reveal any telmisartan-related effects at either the gene or chromosome level. These assays included bacterial mutagenicity tests with Salmonella and E. coli (Ames), a gene mutation test with Chinese hamster V79 cells, a cytogenetic test with human lymphocytes, and a mouse micronucleus test.
No drug-related effects on the reproductive performance of male and female rats were noted at 100 mg/kg/day (the highest dose administered), about 13 times, on a mg/m2 basis, the MRHD of telmisartan. This dose in the rat resulted in an average systemic exposure (telmisartan AUC as determined on day 6 of pregnancy) at least 50 times the average systemic exposure in humans at the MRHD (80 mg/day).
Pregnancy
Pregnancy Categories C (first trimester) and D (second and third trimesters). See WARNINGS, Fetal/Neonatal Morbidity and Mortality.
Nursing Mothers
It is not known whether telmisartan is excreted in human milk, but telmisartan was shown to be present in the milk of lactating rats. Because of the potential for adverse effects on the nursing infant, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Geriatric Use
Of the total number of patients receiving Micardis® (telmisartan) tablets in clinical studies, 551 (18.6%) were 65 to 74 years of age and 130 (4.4%) were 75 years or older. No overall differences in effectiveness and safety were observed in these patients compared to younger patients and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
ADVERSE REACTIONS
MICARDIS tablets have been evaluated for safety in more than 3700 patients, including 1900 treated for over six months and more than 1300 for over one year. Adverse experiences have generally been mild and transient in nature and have only infrequently required discontinuation of therapy.
In placebo-controlled trials involving 1041 patients treated with various doses of telmisartan (20-160 mg) monotherapy for up to 12 weeks, an overall incidence of adverse events similar to that of placebo was observed.
Adverse events occurring at an incidence of 1% or more in patients treated with telmisartan and at a greater rate than in patients treated with placebo, irrespective of their causal association, are presented in the following table.
| Telmisartan | Placebo | |
| n = 1455 | n = 380 | |
| % | % | |
| Upper respiratory tract infection | 7 | 6 |
| Back pain | 3 | 1 |
| Sinusitis | 3 | 2 |
| Diarrhea | 3 | 2 |
| Pharyngitis | 1 | 0 |
In addition to the adverse events in the table, the following events occurred at a rate of 1% but were at least as frequent in the placebo group: influenza-like symptoms, dyspepsia, myalgia, urinary tract infection, abdominal pain, headache, dizziness, pain, fatigue, coughing, hypertension, chest pain, nausea and peripheral edema. Discontinuation of therapy due to adverse events was required in 2.8% of 1455 patients treated with Micardis® (telmisartan) tablets and 6.1% of 380 placebo patients in placebo-controlled clinical trials.
The incidence of adverse events was not dose-related and did not correlate with gender, age, or race of patients.
The incidence of cough occurring with telmisartan in six placebo-controlled trials was identical to that noted for placebo-treated patients (1.6%).
In addition to those listed above, adverse events that occurred in more than 0.3% of 3500 patients treated with MICARDIS tablets monotherapy in controlled or open trials are listed below. It cannot be determined whether these events were causally related to MICARDIS tablets:
Autonomic Nervous System: impotence, increased sweating, flushing; Body as a Whole: allergy, fever, leg pain, malaise; Cardiovascular: palpitation, dependent edema, angina pectoris, tachycardia, leg edema, abnormal ECG; CNS: insomnia, somnolence, migraine, vertigo, paresthesia, involuntary muscle contractions, hypoaesthesia; Gastrointestinal: flatulence, constipation, gastritis, vomiting, dry mouth, hemorrhoids, gastroenteritis, enteritis, gastroesophageal reflux, toothache, non-specific gastrointestinal disorders; Metabolic: gout, hypercholesterolemia, diabetes mellitus; Musculoskeletal: arthritis, arthralgia, leg cramps; Psychiatric: anxiety, depression, nervousness; Resistance Mechanism: infection, fungal infection, abscess, otitis media; Respiratory: asthma, bronchitis, rhinitis, dyspnea, epistaxis; Skin: dermatitis, rash, eczema, pruritus; Urinary: micturition frequency, cystitis; Vascular: cerebrovascular disorder; and Special Senses: abnormal vision, conjunctivitis, tinnitus, earache.
During initial clinical studies, a single case of angioedema was reported (among a total of 3781 patients treated).
Clinical Laboratory Findings
In placebo-controlled clinical trials, clinically relevant changes in standard laboratory test parameters were rarely associated with administration of MICARDIS tablets.
Hemoglobin: A greater than 2 g/dL decrease in hemoglobin was observed in 0.8% telmisartan patients compared with 0.3% placebo patients. No patients discontinued therapy due to anemia.
Creatinine: A 0.5 mg/dL rise or greater in creatinine was observed in 0.4% telmisartan patients compared with 0.3% placebo patients. One telmisartan-treated patient discontinued therapy due to increases in creatinine and blood urea nitrogen.
Liver Enzymes: Occasional elevations of liver chemistries occurred in patients treated with telmisartan; all marked elevations occurred at a higher frequency with placebo. No telmisartan-treated patients discontinued therapy due to abnormal hepatic function.
Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of Micardis® (telmisartan) tablets. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate reliably their frequency or establish a causal relationship to drug exposure. Decisions to include these reactions in labeling are typically based on one or more of the following factors: (1) seriousness of the reaction, (2) frequency of reporting, or (3) strength of causal connection to MICARDIS tablets. The most frequently spontaneously reported events include: headache, dizziness, asthenia, coughing, nausea, fatigue, weakness, edema, face edema, lower limb edema, angioneurotic edema, urticaria, hypersensitivity, sweating increased, erythema, chest pain, atrial fibrillation, congestive heart failure, myocardial infarction, blood pressure increased, hypertension aggravated, hypotension (including postural hypotension), hyperkalemia, syncope, dyspepsia, diarrhea, pain, urinary tract infection, erectile dysfunction, back pain, abdominal pain, muscle cramps (including leg cramps), myalgia, bradycardia, eosinophilia, thrombocytopenia, uric acid increased, abnormal hepatic function/liver disorder, renal impairment including acute renal failure, anemia, increased CPK, anaphylactic reaction, and tendon pain (including tendonitis, tenosynovitis).
Rare cases of rhabdomyolysis have been reported in patients receiving angiotensin II receptor blockers, including MICARDIS tablets.
OVERDOSAGE
Limited data are available with regard to overdosage in humans. The most likely manifestation of overdosage with MICARDIS tablets would be hypotension, dizziness and tachycardia; bradycardia could occur from parasympathetic (vagal) stimulation. If symptomatic hypotension should occur, supportive treatment should be instituted. Telmisartan is not removed by hemodialysis.
DOSAGE AND ADMINISTRATION
Dosage must be individualized. The usual starting dose of MICARDIS tablets is 40 mg once a day. Blood pressure response is dose related over the range of 20-80 mg (see CLINICAL PHARMACOLOGY, Clinical Trials).
-
Special Populations: Patients with depletion of intravascular volume should have the condition corrected or MICARDIS tablets should be initiated under close medical supervision (see WARNINGS, Hypotension in Volume-Depleted Patients). Patients with biliary obstructive disorders or hepatic insufficiency should have treatment started under close medical supervision (see PRECAUTIONS, General, Impaired Hepatic Function and Impaired Renal Function).
Most of the antihypertensive effect is apparent within two weeks and maximal reduction is generally attained after four weeks. When additional blood pressure reduction beyond that achieved with 80 mg MICARDIS tablets is required, a diuretic may be added.
No initial dosing adjustment is necessary for elderly patients or patients with renal impairment, including those on hemodialysis. Patients on dialysis may develop orthostatic hypotension; their blood pressure should be closely monitored.
MICARDIS tablets may be administered with other antihypertensive agents.
MICARDIS tablets may be administered with or without food.
HOW SUPPLIED
MICARDIS tablets are available as white or off-white, uncoated tablets containing telmisartan 20 mg, 40 mg or 80 mg. Tablets are marked with the BOEHRINGER INGELHEIM logo on one side, and on the other side, with either 50H, 51H or 52H for the 20 mg, 40 mg, and 80 mg strengths, respectively. Tablets are provided as follows:
Micardis® (telmisartan) tablets 20 mg are round and individually blister-sealed in cartons of 30 tablets as 3 x 10 cards (NDC 0597-0039-37).
MICARDIS tablets 40 mg are oblong shaped and individually blister-sealed in cartons of 30 tablets as 3 x 10 cards (NDC 0597-0040-37).
MICARDIS tablets 80 mg are oblong shaped and individually blister-sealed in cartons of 30 tablets as 3 x 10 cards (NDC 0597-0041-37).
Storage
Store at 25°C (77°F); excursions permitted to 15°-30°C (59°-86°F) [see USP Controlled Room Temperature]. Tablets should not be removed from blisters until immediately before administration.
Distributed by:
Boehringer Ingelheim Pharmaceuticals, Inc.
Ridgefield, CT 06877 USA
Licensed from: Boehringer Ingelheim International GmbH, Ingelheim, Germany
©Copyright 2008, Boehringer Ingelheim International GmbH, ALL RIGHTS RESERVED
MICARDIS tablets are covered by U.S. Patent 5,591,762
Rev: September 2008
OT1200DI1008
340194/4
IT12004A
10005385/01
| MICARDIS
telmisartan tablet |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| MICARDIS
telmisartan tablet |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| MICARDIS
telmisartan tablet |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
Revised: 09/2008Boehringer Ingelheim Pharmaceuticals, Inc.