VECTIBIX
-
panitumumab injection
Amgen Inc.
----------
Vectibix® (panitumumab)
Injection for Intravenous Use
|
||||||||||||||||||||||||||||
FULL PRESCRIBING INFORMATION
WARNING: DERMATOLOGIC TOXICITY and INFUSION REACTIONS
Dermatologic Toxicity: Dermatologic toxicities occurred in 89% of patients and were severe (NCI-CTC grade 3 and higher) in 12% of patients receiving Vectibix monotherapy. [see Dosage and Administration (2.1), Warnings and Precautions (5.1), and Adverse Reactions (6.1)].
Infusion Reactions: Severe infusion reactions occurred in approximately 1% of patients. [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)]. Although not reported with Vectibix, fatal infusion reactions have occurred with other monoclonal antibody products. [see Dosage and Administration (2.1)].
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose and Dose Modifications
The recommended dose of Vectibix is 6 mg/kg, administered as an intravenous infusion over 60 minutes, every 14 days. Doses higher than 1000 mg should be administered over 90 minutes [see Dosage and Adminsitration (2.2)].
Appropriate medical resources for the treatment of severe infusion reactions should be available during Vectibix infusions.
Dose Modifications for Infusion Reactions [see Adverse Reactions (6.1)]
- Reduce infusion rate by 50% in patients experiencing a mild or moderate (grade 1 or 2) infusion reaction for the duration of that infusion.
- Immediately and permanently discontinue Vectibix infusion in patients experiencing severe (grade 3 or 4) infusion reactions.
Dose Modifications for Dermatologic Toxicity [see Adverse Reactions (6.1)]
- Withhold Vectibix for dermatologic toxicities that are grade 3 or higher or are considered intolerable. If toxicity does not improve to ≤ grade 2 within 1 month, permanently discontinue Vectibix.
- If dermatologic toxicity improves to ≤ grade 2, and the patient is symptomatically improved after withholding no more than two doses of Vectibix, treatment may be resumed at 50% of the original dose.
- If toxicities recur, permanently discontinue Vectibix.
- If toxicities do not recur, subsequent doses of Vectibix may be increased by increments of 25% of the original dose until the recommended dose of 6 mg/kg is reached.
2.2 Preparation and Administration
Do not administer Vectibix as an intravenous push or bolus.
Preparation
Prepare the solution for infusion, using aseptic technique, as follows:
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. Although Vectibix should be colorless, the solution may contain a small amount of visible translucent-to-white, amorphous, proteinaceous, panitumumab particulates (which will be removed by filtration; see below). Do not shake. Do not administer Vectibix if discoloration is observed.
- Withdraw the necessary amount of Vectibix for a dose of 6 mg/kg.
- Dilute to a total volume of 100 mL with 0.9% sodium chloride injection, USP. Doses higher than 1000 mg should be diluted to 150 mL with 0.9% sodium chloride injection, USP. Do not exceed a final concentration of 10 mg/mL.
- Mix diluted solution by gentle inversion. Do not shake.
Administration
- Administer using a low-protein-binding 0.2 μm or 0.22 μm in-line filter.
- Vectibix must be administered via infusion pump.
- Flush line before and after Vectibix administration with 0.9% sodium chloride injection, USP, to avoid mixing with other drug products or intravenous solutions. Do not mix Vectibix with, or administer as an infusion with, other medicinal products. Do not add other medications to solutions containing panitumumab.
- Infuse over 60 minutes through a peripheral intravenous line or indwelling intravenous catheter. Doses higher than 1000 mg should be infused over 90 minutes.
Use the diluted infusion solution of Vectibix within 6 hours of preparation if stored at room temperature, or within 24 hours of dilution if stored at 2° to 8°C (36° to 46°F). DO NOT FREEZE.
Discard any unused portion remaining in the vial.
3 DOSAGE FORMS AND STRENGTHS
100 mg of panitumumab in 5 mL (20 mg/mL) single-use vial.
200 mg of panitumumab in 10 mL (20 mg/mL) single-use vial.
400 mg of panitumumab in 20 mL (20 mg/mL) single-use vial.
4 CONTRAINDICATIONS
None.
5 WARNINGS AND PRECAUTIONS
5.1 Dermatologic Toxicity
In Study 1, dermatologic toxicities occurred in 90% of patients and were severe (NCI-CTC grade 3 and higher) in 16% of patients with mCRC receiving Vectibix. The clinical manifestations included, but were not limited to, dermatitis acneiform, pruritus, erythema, rash, skin exfoliation, paronychia, dry skin, and skin fissures. Subsequent to the development of severe dermatologic toxicities, infectious complications, including sepsis, septic death, and abscesses requiring incisions and drainage were reported. Withhold Vectibix for severe or life-threatening dermatologic toxicity. [see Boxed Warning, Adverse Reactions (6.1) and Dosage and Administration (2.1)].
5.2 Infusion Reactions
In Study 1, 4% of patients experienced infusion reactions and in 1% of patients, these reactions were graded as severe (NCI-CTC grade 3–4).
Across all clinical studies, severe infusion reactions occurred with the administration of Vectibix in approximately 1% of patients. Severe infusion reactions included anaphylactic reactions, bronchospasm, and hypotension [see Boxed Warning and Adverse Reactions (6.1)]. Although fatal infusion reactions have not been reported with Vectibix, fatalities have occurred with other monoclonal antibody products. Stop infusion if a severe infusion reaction occurs. Depending on the severity and/or persistence of the reaction, permanently discontinue Vectibix [see Dosage and Administration (2.1)].
5.3 Increased Toxicity With Combination Chemotherapy
Vectibix is not indicated for use in combination with chemotherapy. In an interim analysis of Study 2, the addition of Vectibix to the combination of bevacizumab and chemotherapy resulted in decreased overall survival and increased incidence of NCI-CTC grade 3–5 (87% vs 72%) adverse reactions [see Clinical Studies (14)]. NCI-CTC grade 3–4 adverse drug reactions occurring at a higher rate in Vectibix-treated patients included rash/dermatitis/acneiform (26% vs 1%), diarrhea (23% vs 12%), dehydration (16% vs 5%), primarily occurring in patients with diarrhea, hypokalemia (10% vs 4%), stomatitis/mucositis (4% vs <1%), and hypomagnesemia (4% vs 0). NCI-CTC grade 3-5 pulmonary embolism occurred at a higher rate in Vectibix-treated patients (7% vs 4%) and included fatal events in three (< 1%) Vectibix-treated patients.
As a result of the toxicities experienced, patients randomized to Vectibix, bevacizumab, and chemotherapy received a lower mean relative dose intensity of each chemotherapeutic agent (oxaliplatin, irinotecan, bolus 5-FU, and/or infusional 5-FU) over the first 24 weeks on study, compared with those randomized to bevacizumab and chemotherapy.
In a single-arm study of 19 patients receiving Vectibix in combination with IFL, the incidence of NCI-CTC grade 3–4 diarrhea was 58%; in addition, grade 5 diarrhea occurred in one patient. In a single-arm study of 24 patients receiving Vectibix plus FOLFIRI, the incidence of NCI-CTC grade 3 diarrhea was 25%.
5.4 Pulmonary Fibrosis
Pulmonary fibrosis occurred in less than 1% (2/1467) of patients enrolled in clinical studies of Vectibix. Following the initial fatality, patients with a history of interstitial pneumonitis, pulmonary fibrosis, evidence of interstitial pneumonitis, or pulmonary fibrosis were excluded from clinical studies. Therefore, the estimated risk in a general population that may include such patients is uncertain.
One case occurred in a patient with underlying idiopathic pulmonary fibrosis who received Vectibix in combination with chemotherapy and resulted in death from worsening pulmonary fibrosis after four doses of Vectibix. The second case was characterized by cough and wheezing 8 days following the initial dose, exertional dyspnea on the day of the seventh dose, and persistent symptoms and CT evidence of pulmonary fibrosis following the 11th dose of Vectibix as monotherapy. An additional patient died with bilateral pulmonary infiltrates of uncertain etiology with hypoxia after 23 doses of Vectibix in combination with chemotherapy. Permanently discontinue Vectibix therapy in patients developing interstitial lung disease, pneumonitis, or lung infiltrates.
5.5 Electrolyte Depletion/Monitoring
In Study 1, median magnesium levels decreased by 0.1 mmol/L in the panitumumab arm; hypomagnesemia (NCI-CTC grade 3 or 4) requiring oral or intravenous electrolyte repletion occurred in 2% of patients. Hypomagnesemia occurred 6 weeks or longer after the initiation of Vectibix. In some patients, both hypomagnesemia and hypocalcemia occurred. Patients’ electrolytes should be periodically monitored during and for 8 weeks after the completion of Vectibix therapy. Institute appropriate treatment, eg, oral or intravenous electrolyte repletion, as needed.
5.6 Photosensitivity
Exposure to sunlight can exacerbate dermatologic toxicity. Advise patients to wear sunscreen and hats and limit sun exposure while receiving Vectibix.
5.7 EGF Receptor Testing
Detection of EGFR protein expression is necessary for selection of patients appropriate for Vectibix therapy because these are the only patients studied and for whom benefit has been shown [see Indications and Usage (1) and Clinical Studies (14)]. Patients with colorectal cancer enrolled in Study 1 were required to have immunohistochemical evidence of EGFR expression using the Dako EGFR pharmDx® test kit.
Assessment for EGFR expression should be performed by laboratories with demonstrated proficiency in the specific technology being utilized. Improper assay performance, including use of suboptimally fixed tissue, failure to utilize specific reagents, deviation from specific assay instructions, and failure to include appropriate controls for assay validation, can lead to unreliable results. Refer to the package insert for the Dako EGFR pharmDx® test kit, or other test kits approved by FDA, for identification of patients eligible for treatment with Vectibix and for full instructions on assay performance.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Dermatologic Toxicity [see Boxed Warning and Warnings and Precautions (5.1)]
- Infusion Reactions [see Boxed Warning and Warnings and Precautions (5.2)]
- Increased Toxicity With Combination Chemotherapy [see Warnings and Precautions (5.3)]
- Pulmonary Fibrosis [see Warnings and Precautions (5.4)]
- Electrolyte Depletion/Monitoring [see Warnings and Precautions (5.5)]
- Photosensitivity [see Warnings and Precautions (5.6)]
The most common adverse events of Vectibix are skin rash with variable presentations, hypomagnesemia, paronychia, fatigue, abdominal pain, nausea, and diarrhea, including diarrhea resulting in dehydration.
The most serious adverse events of Vectibix are pulmonary fibrosis, pulmonary embolism, severe dermatologic toxicity complicated by infectious sequelae and septic death, infusion reactions, abdominal pain, hypomagnesemia, nausea, vomiting, and constipation. Adverse reactions requiring discontinuation of Vectibix were infusion reactions, severe skin toxicity, paronychia, and pulmonary fibrosis.
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates in the clinical studies of a drug cannot be directly compared to rates in clinical studies of another drug and may not reflect the rates observed in practice. The adverse reaction information from clinical studies does, however, provide a basis for identifying the adverse events that appear to be related to drug use and for approximating rates.
Safety data are available from 15 clinical trials in which 1467 patients received Vectibix; of these, 1293 received Vectibix monotherapy and 174 received Vectibix in combination with chemotherapy [see Warnings and Precautions (5.3)].
The data described in Table 1 and in other sections below, except where noted, reflect exposure to Vectibix administered as a single agent at the recommended dose and schedule (6 mg/kg every 2 weeks) in 229 patients with mCRC enrolled in Study 1, a randomized, controlled trial. The median number of doses was five (range: one to 26 doses), and 71% of patients received eight or fewer doses. The population had a median age of 62 years (range: 27 to 82 years), 63% were male, and 99% were white with < 1% black, < 1% Hispanic, and 0% other.
|
||||
| Patients Treated With Vectibix Plus BSC (n = 229) |
Best Supportive Care Alone (n = 234) |
|||
| Grade* | ||||
| Body System |
All Grades % |
Grade 3–4 % |
All Grades % |
Grade 3–4 % |
| Body as a Whole | ||||
| Fatigue | 26 | 4 | 15 | 3 |
| General Deterioration | 11 | 8 | 4 | 3 |
| Digestive | ||||
| Abdominal Pain | 25 | 7 | 17 | 5 |
| Nausea | 23 | 1 | 16 | < 1 |
| Diarrhea | 21 | 2 | 11 | 0 |
| Constipation | 21 | 3 | 9 | 1 |
| Vomiting | 19 | 2 | 12 | 1 |
| Stomatitis | 7 | 0 | 1 | 0 |
| Mucosal Inflammation | 6 | < 1 | 1 | 0 |
| Metabolic/Nutritional | ||||
| Hypomagnesemia (Lab) | 38 | 4 | 2 | 0 |
| Peripheral Edema | 12 | 1 | 6 | < 1 |
| Respiratory | ||||
| Cough | 14 | < 1 | 7 | 0 |
| Skin/Appendages | ||||
| All Skin/Integument Toxicity | 90 | 16 | 9 | 0 |
| Skin | 90 | 14 | 6 | 0 |
| Erythema | 65 | 5 | 1 | 0 |
| Acneiform Dermatitis | 57 | 7 | 1 | 0 |
| Pruritus | 57 | 2 | 2 | 0 |
| Nail | 29 | 2 | 0 | 0 |
| Paronychia | 25 | 2 | 0 | 0 |
| Skin Exfoliation | 25 | 2 | 0 | 0 |
| Rash | 22 | 1 | 1 | 0 |
| Skin Fissures | 20 | 1 | < 1 | 0 |
| Eye | 15 | < 1 | 2 | 0 |
| Acne | 13 | 1 | 0 | 0 |
| Dry Skin | 10 | 0 | 0 | 0 |
| Other Nail Disorder | 9 | 0 | 0 | 0 |
| Hair | 9 | 0 | 1 | 0 |
| Growth of Eyelashes | 6 | 0 | 0 | 0 |
Dermatologic, Mucosal, and Ocular Toxicity
In Study 1, dermatologic toxicities occurred in 90% of patients receiving Vectibix. Skin toxicity was severe (NCI-CTC grade 3 and higher) in 16% of patients. Ocular toxicities occurred in 15% of patients and included, but were not limited to: conjunctivitis (4%), ocular hyperemia (3%), increased lacrimation (2%), and eye/eyelid irritation (1%). Stomatitis (7%) and oral mucositis (6%) were reported. One patient experienced a NCI-CTC grade 3 event of mucosal inflammation. The incidence of paronychia was 25% and was severe in 2% of patients. Nail disorders occurred in 9% of patients [see Warnings and Precautions (5.1)].
Median time to the development of dermatologic, nail, or ocular toxicity was 14 days; the time to most severe skin/ocular toxicity was 15 days after the first dose of Vectibix; and the median time to resolution after the last dose of Vectibix was 84 days. Severe toxicity necessitated dose interruption in 11% of Vectibix-treated patients [see Dosage and Administration (2.1)].
Subsequent to the development of severe dermatologic toxicities, infectious complications, including sepsis, septic death, and abscesses requiring incisions and drainage, were reported.
Infusion Reactions
Infusional toxicity was defined as any event described at any time during the clinical study as allergic reaction or anaphylactoid reaction, or any event occurring on the first day of dosing described as allergic reaction, anaphylactoid reaction, fever, chills, or dyspnea. Vital signs and temperature were measured within 30 minutes prior to initiation and upon completion of the Vectibix infusion. The use of premedication was not standardized in the clinical trials. Thus, the utility of premedication in preventing the first or subsequent episodes of infusional toxicity is unknown. Across several clinical trials of Vectibix monotherapy, 3% (43/1336) experienced infusion reactions of which approximately 1% (6/1336) were severe (NCI-CTC grade 3–4). In one patient, Vectibix was permanently discontinued for a serious infusion reaction [see Dosage and Administration (2.1)].
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The immunogenicity of Vectibix has been evaluated using two different screening immunoassays for the detection of anti-panitumumab antibodies: an acid dissociation bridging enzyme-linked immunosorbent assay (ELISA) (detecting high-affinity antibodies) and a Biacore® biosensor immunoassay (detecting both high- and low-affinity antibodies). The incidence of binding antibodies to panitumumab (excluding predose and transient positive patients), as detected by the acid dissociation ELISA, was 3/613 (< 1%) and as detected by the Biacore® assay was 28/613 (4.6%).
For patients whose sera tested positive in screening immunoassays, an in vitro biological assay was performed to detect neutralizing antibodies. Excluding predose and transient positive patients, 10 of the 613 patients (1.6%) with postdose samples and 3/356 (0.8%) of the patients with follow-up samples tested positive for neutralizing antibodies.
No evidence of altered pharmacokinetic profile or toxicity profile was found between patients who developed antibodies to panitumumab as detected by screening immunoassays and those who did not.
The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to panitumumab with the incidence of antibodies to other products may be misleading.
7 DRUG INTERACTIONS
No formal drug-drug interaction studies have been conducted with Vectibix.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C. There are no studies of Vectibix in pregnant women. Reproduction studies in cynomolgus monkeys treated with 1.25 to 5 times the recommended human dose of panitumumab resulted in significant embryolethality and abortions; however, no other evidence of teratogenesis was noted in offspring [see Reproductive and Developmental Toxicology (13.3)]. Vectibix should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Based on animal models, EGFR is involved in prenatal development and may be essential for normal organogenesis, proliferation, and differentiation in the developing embryo. Human IgG is known to cross the placental barrier; therefore, panitumumab may be transmitted from the mother to the developing fetus, and has the potential to cause fetal harm when administered to pregnant women.
Women who become pregnant during Vectibix treatment are encouraged to enroll in Amgen’s Pregnancy Surveillance Program. Patients or their physicians should call 1-800-772-6436 (1-800-77-AMGEN) to enroll.
8.3 Nursing Mothers
It is not known whether panitumumab is excreted into human milk; however human IgG is excreted into human milk. Published data suggest that breast milk antibodies do not enter the neonatal and infant circulation in substantial amounts. Because many drugs are excreted into human milk and because of the potential for serious adverse reactions in nursing infants from Vectibix, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. If nursing is interrupted, based on the mean half-life of panitumumab, nursing should not be resumed earlier than 2 months following the last dose of Vectibix [see Clinical Pharmacology (12.3)].
8.4 Pediatric Use
The safety and effectiveness of Vectibix have not been established in pediatric patients. The pharmacokinetic profile of Vectibix has not been studied in pediatric patients.
8.5 Geriatric Use
Of 229 patients with mCRC who received Vectibix in Study 1, 96 (42%) were ≥ age 65. Although the clinical study did not include a sufficient number of geriatric patients to determine whether they respond differently from younger patients, there were no apparent differences in safety and effectiveness of Vectibix between these patients and younger patients.
10 OVERDOSAGE
Doses up to approximately twice the recommended therapeutic dose (12 mg/kg) resulted in adverse reactions of skin toxicity, diarrhea, dehydration, and fatigue.
11 DESCRIPTION
Vectibix (panitumumab) is a recombinant, human IgG2 kappa monoclonal antibody that binds specifically to the human epidermal growth factor receptor (EGFR). Panitumumab has an approximate molecular weight of 147 kDa. Panitumumab is produced in genetically engineered mammalian (Chinese Hamster Ovary) cells.
Vectibix is a sterile, colorless, pH 5.6 to 6.0 liquid for intravenous (IV) infusion, which may contain a small amount of visible translucent-to-white, amorphous, proteinaceous, panitumumab particulates. Each single-use 5 mL vial contains 100 mg of panitumumab, 29 mg sodium chloride, 34 mg sodium acetate, and Water for Injection, USP. Each single-use 10 mL vial contains 200 mg of panitumumab, 58 mg sodium chloride, 68 mg sodium acetate, and Water for Injection, USP. Each single-use 20 mL vial contains 400 mg of panitumumab, 117 mg sodium chloride, 136 mg sodium acetate, and Water for Injection, USP.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The EGFR is a transmembrane glycoprotein that is a member of a subfamily of type I receptor tyrosine kinases, including EGFR, HER2, HER3, and HER4. EGFR is constitutively expressed in normal epithelial tissues, including the skin and hair follicle. EGFR is over-expressed in certain human cancers, including colon and rectum cancers. Interaction of EGFR with its normal ligands (eg, EGF, transforming growth factor-alpha) leads to phosphorylation and activation of a series of intracellular proteins, which in turn regulate transcription of genes involved with cellular growth and survival, motility, and proliferation.
Panitumumab binds specifically to EGFR on both normal and tumor cells, and competitively inhibits the binding of ligands for EGFR. Nonclinical studies show that binding of panitumumab to the EGFR prevents ligand-induced receptor autophosphorylation and activation of receptor-associated kinases, resulting in inhibition of cell growth, induction of apoptosis, decreased proinflammatory cytokine and vascular growth factor production, and internalization of the EGFR. In vitro assays and in vivo animal studies demonstrate that panitumumab inhibits the growth and survival of selected human tumor cell lines expressing EGFR.
12.3 Pharmacokinetics
Panitumumab administered as a single agent exhibits nonlinear pharmacokinetics.
Following single-dose administrations of panitumumab as 1-hour infusions, the area under the concentration-time curve (AUC) increased in a greater than dose-proportional manner, and clearance (CL) of panitumumab decreased from 30.6 to 4.6 mL/day/kg as the dose increased from 0.75 to 9 mg/kg. However, at doses above 2 mg/kg, the AUC of panitumumab increased in an approximately dose-proportional manner.
Following the recommended dose regimen (6 mg/kg given once every 2 weeks as a 1-hour infusion), panitumumab concentrations reached steady-state levels by the third infusion with mean (± SD) peak and trough concentrations of 213 ± 59 and 39 ± 14 mcg/mL, respectively. The mean (± SD) AUC0-tau and CL were 1306 ± 374 mcg•day/mL and 4.9 ± 1.4 mL/kg/day, respectively. The elimination half-life was approximately 7.5 days (range: 3.6 to 10.9 days).
A population pharmacokinetic analysis was performed to explore the potential effects of selected covariates on panitumumab pharmacokinetics. Results suggest that age (21–88 years), gender, race (15% non-white), mild-to-moderate renal dysfunction, mild-to-moderate hepatic dysfunction, and EGFR membrane-staining intensity (1+, 2+, 3+) in tumor cells had no apparent impact on the pharmacokinetics of panitumumab.
No formal pharmacokinetic studies of panitumumab have been conducted in patients with renal or hepatic impairment.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or mutagenicity studies of panitumumab have been conducted. It is not known if panitumumab can impair fertility in humans. Prolonged menstrual cycles and/or amenorrhea occurred in normally cycling, female cynomolgus monkeys treated weekly with 1.25 to 5 times the recommended human dose of panitumumab (based on body weight). Menstrual cycle irregularities in panitumumab-treated female monkeys were accompanied by both a decrease and delay in peak progesterone and 17β-estradiol levels. Normal menstrual cycling resumed in most animals after discontinuation of panitumumab treatment. A no-effect level for menstrual cycle irregularities and serum hormone levels was not identified. The effects of panitumumab on male fertility have not been studied. However, no adverse effects were observed microscopically in reproductive organs from male cynomolgus monkeys treated for 26 weeks with panitumumab at doses of up to approximately 5-fold the recommended human dose (based on body weight).
13.2 Animal Toxicology and/or Pharmacology
Weekly administration of panitumumab to cynomolgus monkeys for 4 to 26 weeks resulted in dermatologic findings, including dermatitis, pustule formation and exfoliative rash, and deaths secondary to bacterial infection and sepsis at doses of 1.25 to 5-fold higher (based on body weight) than the recommended human dose.
13.3 Reproductive and Developmental Toxicology
Pregnant cynomolgus monkeys were treated weekly with panitumumab during the period of organogenesis (gestation day [GD] 20–50). While no panitumumab was detected in serum of neonates from panitumumab-treated dams, anti-panitumumab antibody titers were present in 14 of 27 offspring delivered at GD 100. There were no fetal malformations or other evidence of teratogenesis noted in the offspring. However, significant increases in embryolethality and abortions occurred at doses of approximately 1.25 to 5 times the recommended human dose (based on body weight).
14 CLINICAL STUDIES
Vectibix Monotherapy
The safety and efficacy of Vectibix were studied in Study 1, an open-label, multinational, randomized, controlled trial of 463 patients with EGFR-expressing, metastatic carcinoma of the colon or rectum (mCRC). Patients were required to have progressed on or following treatment with a regimen(s) containing a fluoropyrimidine, oxaliplatin, and irinotecan; progression was confirmed by an independent review committee (IRC) for 75% of the patients. All patients were required to have EGFR expression defined as at least 1+ membrane staining in ≥ 1% of tumor cells by the Dako EGFR pharmDx® test kit. Patients were randomized 1:1 to receive panitumumab at a dose of 6 mg/kg given once every 2 weeks plus best supportive care (BSC) (n = 231) or BSC alone (n = 232) until investigator-determined disease progression. Randomization was stratified based on ECOG performance status (0–1 vs 2) and geographic region (western Europe, eastern/central Europe, or other). Upon investigator-determined disease progression, patients in the BSC-alone arm were eligible to receive panitumumab and were followed until disease progression was confirmed by the IRC. The analyses of progression-free survival (PFS), objective response, and response duration were based on events confirmed by the IRC that was masked to treatment assignment.
Among the 463 patients, 63% were male, the median age was 62 years, 40% were 65 years or older, 99% were white, 86% had a baseline ECOG performance status of 0 or 1, and 67% had colon cancer. The median number of prior therapies for metastatic disease was 2.4. The membrane-staining intensity for EGFR was 3+ in 19%, 2+ in 51%, and 1+ in 30% of patients’ tumors. The percentage of tumor cells with EGFR membrane staining in the following categories of > 35%, > 20%–35%, 10%–20%, and 1%–< 10% was 38%, 8%, 31%, and 22%, respectively.
Based upon IRC determination of disease progression, a statistically significant prolongation in PFS was observed in patients receiving Vectibix compared to those receiving BSC alone. The mean PFS was 96 days in the Vectibix arm and 60 days in the BSC-alone arm. Results are presented in Figure 1 below.
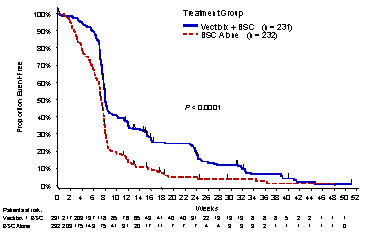
Figure 1. Kaplan-Meier Plot of Progression-Free Survival Time as Determined by the IRC
In a series of sensitivity analyses, including one adjusting for potential ascertainment bias, ie, assessment for progressive disease at a nonstudy specified time point, PFS was still significantly prolonged among patients receiving Vectibix as compared to patients receiving BSC alone.
Of the 232 patients randomized to BSC alone, 75% of patients crossed over to receive Vectibix following investigator determination of disease progression; the median time to cross over was 8.4 weeks (0.3–26.4 weeks).
Partial responses were identified by the IRC in 19 patients randomized to Vectibix, for an overall response of 8% (95% CI: 5.0%, 12.6%). No patient in the control arm had an objective response identified by the IRC. The median duration of response was 17 weeks (95% CI: 16 weeks, 25 weeks). There was no difference in overall survival between the study arms.
Vectibix in Combination With Bevacizumab and Chemotherapy
Vectibix shortened PFS, decreased survival time, and increased toxicity when given in combination with bevacizumab and chemotherapy in Study 2, a randomized, open-label, multicenter trial in the first-line treatment of metastatic colorectal cancer. Patients (n = 1053) were randomized 1:1 to Vectibix at a dose of 6 mg/kg given once every 2 weeks, in combination with bevacizumab and an oxaliplatin- or irinotecan-based 5-fluorouracil-containing chemotherapy regimen, or to bevacizumab and chemotherapy alone. Randomization was stratified by type of regimen (oxaliplatin- or irinotecan-based); 86% of patients received an oxaliplatin-based regimen and 14% received an irinotecan-based regimen.
The major study objective was comparison of PFS in the oxaliplatin stratum as determined by an independent central review. An interim analysis based on 257 PFS events in the oxaliplatin stratum demonstrated shorter PFS in patients receiving Vectibix, bevacizumab, and chemotherapy compared to those receiving bevacizumab and chemotherapy alone (median PFS were 8.8 months and 10.5 months; hazard ratio 1.44 [95% CI: 1.12, 1.85], p-value = 0.0024, Cox model with randomization factors as covariates). An unplanned analysis of overall survival after 155 deaths (both strata combined), conducted at the time of the interim analysis of PFS, yielded an adjusted hazard ratio of 1.55 [95% CI: 1.12, 2.14], comparing patients receiving Vectibix, bevacizumab, and chemotherapy (92 deaths) to those receiving bevacizumab and chemotherapy alone (63 deaths) [see Warnings and Precautions (5.3)].
16 HOW SUPPLIED/STORAGE AND HANDLING
Vectibix is supplied as a sterile, colorless, preservative-free solution containing 20 mg/mL Vectibix (panitumumab) in a single-use vial.
Vectibix is provided as one vial per carton.
Each 5 mL single-use vial contains 100 mg of panitumumab in 5 mL (20 mg/mL) (NDC 55513-954-01).
Each 10 mL single-use vial contains 200 mg of panitumumab in 10 mL (20 mg/mL) (NDC 55513-955-01).
Each 20 mL single-use vial contains 400 mg of panitumumab in 20 mL (20 mg/mL) (NDC 55513-956-01).
Store vials in the original carton under refrigeration at 2° to 8°C (36° to 46°F) until time of use. Protect from direct sunlight. DO NOT FREEZE. Since Vectibix does not contain preservatives, any unused portion remaining in the vial must be discarded.
The diluted infusion solution of Vectibix should be used within 6 hours of preparation if stored at room temperature, or within 24 hours of dilution if stored at 2° to 8°C (36° to 46°F). DO NOT FREEZE.
17 PATIENT COUNSELING INFORMATION
Advise patients to contact a healthcare professional for any of the following:
- Skin and ocular/visual changes [see Boxed Warning and Warnings and Precautions (5.1)],
- Signs and symptoms of infusion reactions including fever, chills, or breathing problems [see Boxed Warning and Warnings and Precautions (5.2)],
- Persistent or recurrent coughing, wheezing, or dyspnea [see Warnings and Precautions (5.4)],
- Pregnancy or nursing [see Use in Specific Populations (8.1, 8.3)].
Advise patients of the need for:
- Periodic monitoring of electrolytes [see Warnings and Precautions (5.5)],
- Limitation of sun exposure (use sunscreen, wear hats) while receiving Vectibix and for 2 months after the last dose of Vectibix therapy. [see Warnings and Precautions (5.6)],
- Adequate contraception in both males and females while receiving Vectibix and for 6 months after the last dose of Vectibix therapy [see Use in Specific Populations (8.1, 8.3)].
[Amgen logo]
Vectibix® (panitumumab) Injection for Intravenous Use
Manufactured by:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, CA 91320-1799
USA
This product, its production, and/or its use may be covered by one or more US Patents, including US Patent No. 6,235,883, as well as other patents or patents pending.
© 2006-2008 Amgen Inc. All rights reserved.
3xxxxxx - v4
| VECTIBIX
panitumumab injection |
|||||||||||||||||||
|
|||||||||||||||||||
|
|||||||||||||||||||
|
|||||||||||||||||||
|
|||||||||||||||||||
| VECTIBIX
panitumumab injection |
|||||||||||||||||||
|
|||||||||||||||||||
|
|||||||||||||||||||
|
|||||||||||||||||||
|
|||||||||||||||||||
| VECTIBIX
panitumumab injection |
|||||||||||||||||||
|
|||||||||||||||||||
|
|||||||||||||||||||
|
|||||||||||||||||||
|
|||||||||||||||||||
Revised: 07/2008Amgen Inc.