COGNEX
-
tacrine hydrochloride capsule
Shionogi Inc.
----------
Description
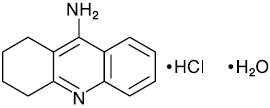
Cognex® (tacrine hydrochloride) is a reversible cholinesterase inhibitor, known chemically as 1,2,3,4-tetrahydro-9-acridinamine monohydrochloride monohydrate. Tacrine hydrochloride is commonly referred to in the clinical and pharmacological literature as THA. It has an empirical formula of C13H14N2• HCl • H2O and a molecular weight of 252.74.
The molecular formula of tacrine hydrochloride is:
Tacrine hydrochloride is a white solid and is freely soluble in distilled water, 0.1N hydrochloric acid, acetate buffer (pH 4.0), phosphate buffer (pH 7.0 to 7.4), methanol, dimethylsulfoxide (DMSO), ethanol, and propylene glycol. The compound is sparingly soluble in linoleic acid and PEG 400.
Each capsule of Cognex® contains tacrine as the hydrochloride. Inactive ingredients are hydrous lactose, magnesium stearate, and microcrystalline cellulose. The hard gelatin capsules contain gelatin, NF; silicon dioxide, NF; sodium lauryl sulfate, NF; and the following dyes: 10 mg: D&C Yellow #10, FD&C Green #3, titanium dioxide; 20 mg: D&C Yellow #10, FD&C Blue #1, titanium dioxide; 30 mg: D&C Yellow #10, FD&C Blue #1, FD&C Red #40, titanium dioxide; 40 mg: D&C Yellow #10, FD&C Blue #1, FD&C Red #40, D&C Red #28, titanium dioxide.
Each 10-, 20-, 30-, and 40-mg Cognex® capsule for oral administration contains 12.75, 25.50, 38.25, and 51.00 mg of tacrine HCl, respectively.
CLINICAL PHARMACOLOGY
Although widespread degeneration of multiple CNS neuronal systems eventually occurs, early pathological changes in Alzheimer's Disease involve, in a relatively selective manner, cholinergic neuronal pathways that project from the basal forebrain to the cerebral cortex and hippocampus. The resulting deficiency of cortical acetylcholine is believed to account for some of the clinical manifestations of mild to moderate dementia. Tacrine, an orally bioavailable, centrally active, reversible cholinesterase inhibitor, presumably acts by elevating acetylcholine concentrations in the cerebral cortex by slowing the degradation of acetylcholine released by still intact cholinergic neurons. If this theoretical mechanism of action is correct, tacrine's effects may lessen as the disease process advances and fewer cholinergic neurons remain functionally intact. There is no evidence that tacrine alters the course of the underlying dementing process.
Clinical Trial Data
The conclusion that Cognex® is an effective treatment for Alzheimer's Disease derives from two adequate and well controlled clinical investigations that evaluated tacrine's effects in patients with probable Alzheimer's disease of mild to moderate severity (NINCDS criteria, Mini-Mental State Examination (MMSE) of Folstein, Folstein and McHugh scores of 10 to 26).
In each study, outcomes during treatment with tacrine and placebo were assessed on two primary measures: (1) the cognitive subscale of the Alzheimer's Disease Assessment Scale (ADAS cog) of Rosen, Mohs, and Davis and (2) a clinician's rated clinical global impression of change.
Study Endpoints
The ADAS cog is a multi-item test battery administered by a psychometrician that examines aspects of memory, attention, praxis, reason, and language. The worst possible score is 70. Elderly, normal adults may score as low as 0 or 1 unit, but individuals judged not to be demented can score higher. The mean score of patients entering each study was approximately 28 units (range 7 to 62). The ADAS cog score is reported to deteriorate at a rate of about 6 to 10 units per year for untreated patients at this stage of dementia.
The clinician's global assessments used in the two studies relied on a clinician's judgment about the overall clinical change observed in patients over the course of the study. Although the conditions for obtaining the clinical assessment differed in each study, the global assessment was rated on a 7-point scale in both studies. A rating of four (4) represents no change; lower ratings indicate improvement from baseline and higher ratings deterioration.
Twelve-Week Study
In one study of 12 weeks duration, patients were randomized to sequences that provided a comparison between placebo, 20, 40, and 80 mg/day by study's end. Statistically significant drug-placebo differences were detected on both primary outcome measures for the group titrated to 80 mg/day. Estimates of the size of the treatment effect varied between 2 and 4 ADAS cog units. The imprecision in these estimates reflects the fact that different analyses, conducted in attempts to account for the effects of the failure of a substantial fraction of the patients randomized to complete the full 12 weeks of the study, yielded different results.
The placebo-80 mg/day comparison also achieved statistical significance on the clinician's global impression of change (CGIC) with a 0.3 to 0.4 unit mean difference. The following diagram illustrates the percentages of patients falling into each global category at trial's end for the patients given placebo or 80 mg/day.
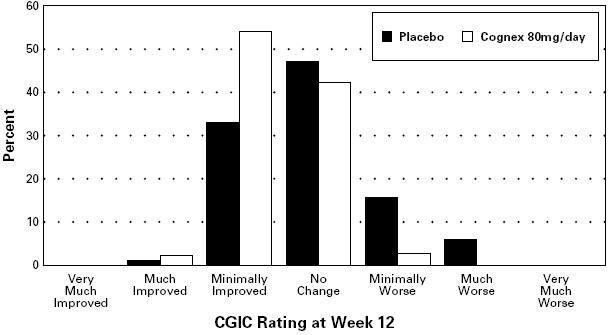
FIGURE 1. Percent of Patients in Each of the Seven Outcome Categories on the Clinician-Rated CGIC for Patients Completing 12 Weeks of Treatment (83% of patients randomized to placebo completed 12 weeks of treatment and are represented above; 56% of those randomized to the 80 mg/day Cognex® sequence completed 12 weeks).
Thirty-Week Study
The second study was 30 weeks long. Six hundred sixty-three patients were randomized to 4 treatment sequences (placebo and 3 drug groups) that called for the daily dose of tacrine to be increased at 6-week intervals, starting with a 40-mg/day dose. By study's end, a comparison between placebo, 80, 120, and 160 mg/day was possible. Patients in the 160 mg group received this dose for the final 12 weeks; the 120 mg group received that dose for 18 weeks.
The study showed statistically significant drug-placebo differences for the 80 and 120 mg/day groups at 18 weeks and for the 120 and 160 mg/day groups at 30 weeks on both a performance-based test of cognitive function (the ADAS cog) and a clinician's assessment of global change (Clinician Interview Based Impression: CIBI). Because many patients failed to complete 30 weeks on treatment, analyses that used each patient's last on-study value or retrieved patients' (see below) 30-week value, even if they were no longer in the study ("intent-to-treat” analysis) were also carried out. All analyses confirmed the effectiveness of tacrine, although the estimated mean treatment effect was different in each analysis.
Effects on ADAS Cog:
The results for the ADAS cog are shown in Figure 2 for the subset of patients actually completing the full 30 weeks of the study. They show that individual patients, whether assigned to tacrine or to placebo, had a wide range of responses. This variability in response is illustrated in the display that follows (Figure 2).
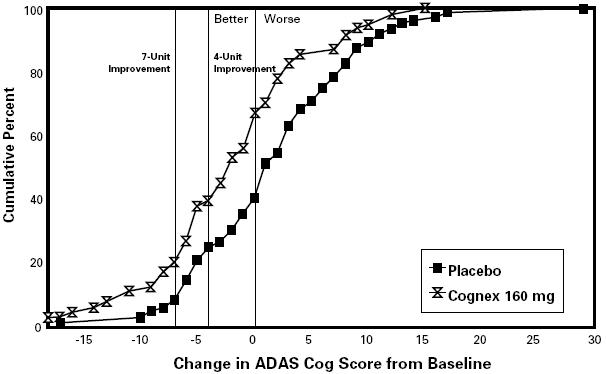
FIGURE 2. Cumulative Percent of Patients Completing 30 Weeks of Treatment Who Attained a Change in ADAS Cog Score From Baseline at Least as Large as the Value on the X Axis. The display is based on scores obtained from a subset of patients (ie, 64% of the 184 randomized to placebo and 27% of the 239 randomized to the 160 mg/daytreatment group).
Figure 2 presents the cumulative percentage (Y axis) of patients assigned to placebo or 160 mg/day who actually completed 30 weeks on treatment and who attained a change in ADAS cog score from baseline at least as large as the ADAS cog change score value given on the X axis. A negative change from baseline represents improvement; a positive change deterioration. Thus, in a display of this type, the curve for an effective treatment is shifted to the left of the curve for placebo. The frequency in each group of any response, e.g., an improvement of 7 ADAS cog units, can be found by plotting the change on the X axis, then reading upward along the Y axis. The variability of response is apparent from the fact that the distribution of responses under both treatment conditions range from large negative to large positive values. Nonetheless, the mean drug-placebo ADAS cog difference for the 30-week 160 mg/day completer patients is 4.8 units, a statistically significant difference.
Effects on CIBI:
The results on the CIBI are shown in Figure 3.
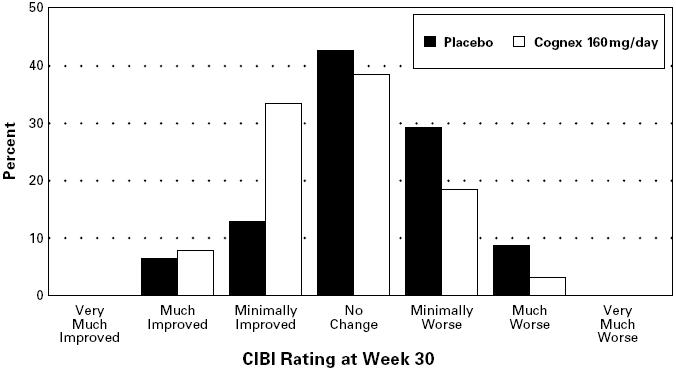
FIGURE 3. Percent of Patients in Each of the Seven Outcome Categories of the CIBI Among Those Completing 30 Weeks. The display is based on scores obtained from the same subset of patients as Figure 2.
Figure 3 is a histogram of the frequency distribution of CIBI scores attained by patients assigned to placebo or to the 160 mg/day tacrine dose group who actually completed the full 30 weeks of the study. The mean tacrine-placebo difference for this group of patients on the CIBI was 0.5 units and was statistically significant.
Expected Responses in Newly Treated Patients:
Although the results described clearly document tacrine's effectiveness, they are based on only a fraction of the patients initially randomized to tacrine, those who could tolerate tacrine and remain on treatment uninterrupted for the full 30 weeks. In considering the expected outcome in a group of patients newly started on tacrine, account must be taken both of the likelihood of staying on therapy and the responses in patients who do so.
Table 1 provides 3 different estimates of the proportion of patients assigned to treatment with tacrine at 160 mg a day or with placebo who attained a particular measure of improvement (ie, a 7 point improvement from baseline in ADAS cog score). The criterion has been chosen entirely for illustrative purposes.
Table 1. Proportion of Patients Attaining 7 Unit Improvement on the
ADAS Cog at the Week 30 Assessment
| Treatment Group N Randomized | I N (%) of Those Randomized | II N (%) of Those Randomized | III N (%) of Those Randomized |
| Placebo (N = 184) | 10/184 ( 5.4) | 10/117 ( 8.5) | 11/1431 ( 7.7) |
| 160 mg/day (N = 239) | 13/239 ( 5.4) | 13/64 (20.3) | 25/1722 (14.5) |
1: 13 of the 143 were receiving tacrine when evaluated.
2: 41 of the 172 were not receiving tacrine when evaluated.
The first column of the table is based on all patients participating in the study. The proportion provides an estimate of the likelihood that a patient entering the study will (1) still be on his or her assigned treatment at week 30 and (2) will improve 7 or more ADAS cognitive points over his or her baseline score. The estimate of response derived in this manner is conservative because the rules under which the 30-week study was conducted required the withdrawal of patients with relatively low (>3 X ULN), asymptomatic, transaminase elevations. In actual clinical practice under the conditions of treatment recommended in the Dosage and Administration Section, a larger fraction of these patients would be able to remain on tacrine and the proportion of those improving 7 or more points on tacrine would be expected, therefore, to be increased (the third column illustrates this).
The second column of the table presents the proportion of 7 unit responders based on the number of patients who (1) were able to complete the full 30 weeks of the study and (2) attained an ADAS cognitive score at week 30 that was 7 or more points better than their baseline score. This analysis provides an optimistic estimate of tacrine's effects because it reflects experience gained only with the minority of patients who were able to remain on treatment to the study's end. The comparison between the proportions of placebo and 160 mg patients attaining a 7 or more point improvement is complicated further by the fact that a larger proportion of tacrine assigned patients withdrew prematurely.
The third column of the table presents the proportion of patients who had evaluations made at 30 weeks and had a 7-point or greater response. The analysis includes data from patients still on their assigned treatment at week 30 as well as patients who withdrew from the study prior to that time, but were retrieved for a week 30 evaluation. Because patients who withdrew prior to week 30 were permitted to receive tacrine under "open label” conditions, retrieved patients included in this analysis could be receiving either no treatment or treatment with tacrine. In this analysis, patients are considered under the treatment to which they were randomized, regardless of the treatment they were actually receiving at week 30. Thus, some placebo patients could have received tacrine and some tacrine patients could have been receiving no tacrine. Like the analysis based on percent randomized (column I), this analysis, therefore, tends to provide a conservative view of the expected effects of tacrine treatment.
Effects of Cognex® Over Time:
Figure 4 shows for each dose group the time course of change from baseline in ADAS cog scores for patients completing 30 weeks of treatment. There appears to be a persistent difference between groups, but all groups, after initial improvement, deteriorate with time.
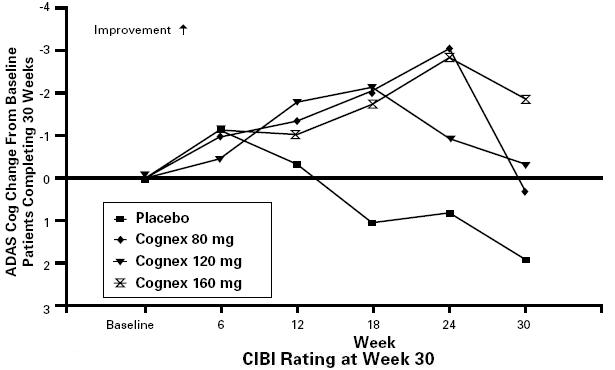
FIGURE 4. ADAS Cog Change From Baseline Over Time for the Subset of Patients Completing 30 Weeks of Treatment. In all tacrine treatment groups dosing was initiated at 40 mg/day and increased in increments of 40 mg every 6 weeks until the target dose was achieved. Patient age, gender, and other baseline patient characteristics were not found to predict clinical outcome.
Clinical Pharmacokinetics (Absorption, Distribution, Metabolism, and Elimination)
Absorption:
Cognex® is rapidly absorbed after oral administration; maximal plasma concentrations occur within 1 to 2 hours. The rate and extent of tacrine absorption following administration of tacrine capsules and solution are virtually indistinguishable. Absolute bioavailability of tacrine is approximately 17 (SD ± 13) %. Food reduces tacrine bioavailability by approximately 30-40%; however, there is no food effect if tacrine is administered at least an hour before meals. The effect of achlorhydria on the absorption of tacrine is unknown.
Distribution:
Mean volume of distribution of tacrine is approximately 349 (SD ± 193) L. Tacrine is about 55% bound to plasma proteins. The extent and degree of tacrine's distribution within various body compartments has not been systematically studied. However, 336 hours after the administration of a single radiolabeled dose, approximately 25% of the radiolabel was not recovered in a mass balance study, suggesting the possibility that tacrine and/or one or more of its metabolites may be retained.
Metabolism:
Tacrine is extensively metabolized by the cytochrome P450 system to multiple metabolites, not all of which have been identified. The vast majority of radiolabeled species present in the plasma following a single dose of 14C radiolabeled tacrine are unidentified (ie, only 5% of radioactivity in plasma has been identified [tacrine and 3-hydroxylated metabolites; 1-, 2-, and 4-hydroxytacrine]).
Studies utilizing human liver preparations demonstrated that cytochrome P450 IA2 is the principal isozyme involved in tacrine metabolism. These findings are consistent with the observation that tacrine and/or one of its metabolites inhibits the metabolism of theophylline in humans (see PRECAUTIONS: Drug-Drug Interactions: theophylline). Results from a study utilizing quinidine to inhibit cytochrome P450 IID6 indicate that tacrine is not metabolized extensively by this enzyme system.
Following aromatic ring hydroxylation, tacrine's metabolites undergo glucuronidation. Whether tacrine and/or its metabolites undergo biliary excretion or entero-hepatic circulation is unknown.
Special Populations:
Age: Based on pooled pharmacokinetic studies (n = 192), there is no clinically relevant influence of age (50 to 84 years) on tacrine clearance. Gender: Average tacrine plasma concentrations are approximately 50% higher in females than in males. This is not explained by differences in body surface area or elimination half-life. The difference is probably due to higher systemic availability after oral dosing and may reflect the known lower activity of cytochrome P450 IA2 in women. Race: The effect of race on tacrine clearance has not been studied. Smoking: Mean plasma tacrine concentrations in current smokers are approximately one third the concentrations in nonsmokers. Cigarette smoking is known to induce cytochrome P450 IA2. Renal disease: Renal disease does not appear to affect the clearance of tacrine. Liver disease: Although studies in patients with liver disease have not been done, it is likely that functional hepatic impairment will reduce the clearance of tacrine and its metabolites.
Presystemic Clearance/Elimination/Excretion:
Tacrine undergoes presystemic clearance (ie, first pass metabolism). The extent of this first pass metabolism depends upon the dose of tacrine administered. Because the enzyme system involved can be saturated at relatively low doses, a larger fraction of a high dose of tacrine will escape first pass elimination than of a smaller dose. Thus, when a 40 mg daily dose is increased by 40 mg, the average plasma concentration will be increased by approximately 6 ng/mL. However, when a daily dose of 80 or 120 mg is increased by 40 mg, the increment in average plasma concentration is approximately 10 ng/mL.
Elimination of tacrine from the plasma, however, is not dose dependent (ie, the half-life is independent of dose or plasma concentration). The elimination half-life is approximately 2 to 4 hours. Following initiation of therapy or a change in daily dose, steady state tacrine plasma concentration should be attained within 24 to 36 hours.
INDICATIONS AND USAGE
Cognex® (tacrine hydrochloride capsules) is indicated for the treatment of mild to moderate dementia of the Alzheimer's type.
Evidence of Cognex®'s effectiveness in the treatment of dementia of the Alzheimer's type derives from results of two adequate and well-controlled clinical investigations that compared tacrine and placebo on both a performance based measure of cognition and a clinician's global assessment of change. (See CLINICAL PHARMACOLOGY Section: Clinical Trial Data).
CONTRAINDICATIONS
Cognex® is contraindicated in patients with known hypersensitivity to tacrine or acridine derivatives.
Cognex® is contraindicated in patients previously treated with Cognex® who developed treatment-associated jaundice; a serum bilirubin >3 mg/dL; and/or those exhibiting clinical signs or symptoms of hypersensitivity (eg, rash or fever) in association with ALT/SGPT elevations.
WARNINGS
Anesthesia
Cognex®, as a cholinesterase inhibitor, is likely to exaggerate succinylcholine-type muscle relaxation during anesthesia.
Cardiovascular Conditions
Because of its pharmacological action, Cognex® may have vagotonic effects on the sinoatrial and atrioventricular nodes possibly leading to bradycardia and/or heart block. These effects may be particularly harmful to patients with conduction abnormalities, bradyarrhythmias, or a sick sinus syndrome, but may also occur in patients without known preexisting cardiac disease.
Gastrointestinal Disease and Dysfunction
Cognex® is an inhibitor of cholinesterase and may be expected to increase gastric acid secretion due to increased cholinergic activity. Therefore, patients are at increased risk for developing ulcers. Those with a history of ulcer disease or those receiving concurrent nonsteroidal anti-inflammatory drugs (NSAIDs) should be monitored closely for symptoms of active or occult gastrointestinal disease.
Cognex®, also as a predictable consequence of its pharmacological properties, can cause nausea, vomiting, and loose stools at recommended doses.
Liver Injury
Cognex® should be prescribed with care in patients with current evidence or history of abnormal liver function indicated by significant abnormalities in serum transaminase (ALT/SGPT; AST/SGOT), bilirubin, and gamma-glutamyl transpeptidase (GGT) levels (see PRECAUTIONS and DOSAGE AND ADMINISTRATION sections).
The use of tacrine in patients without a prior history of liver disease is commonly associated with serum aminotransferase elevations, some to levels ordinarily considered to indicate clinically important hepatic injury (see Table 2).
Experience gained in more than 12,000 patients who received tacrine in clinical studies and the treatment IND program indicates that if tacrine is promptly withdrawn following detection of these elevations, clinically evident signs and symptoms of liver injury are rare.
Long-term follow up of patients who experience transaminase elevations, however, is limited and it is impossible, therefore, to exclude, with certainty, the possibility of chronic sequelae.
Controlled Clinical Trials, Treatment IND and Post Marketing Experience:
Experience with tacrine in controlled trials and in a large, less closely monitored experience (a treatment IND) is summarized below:
Clinically evident liver toxicity: One of more than 12,000 patients exposed to tacrine in clinical studies and the treatment IND program had documented elevated bilirubin (5.3 X Upper Limit of Normal, ULN) and jaundice with transaminase levels (AST/SGOT) nearly 20 X ULN.
Rare cases of liver toxicity associated with jaundice, raised serum bilirubin, pyrexia, hepatitis and liver failure have been reported in post-marketing experience. Most of these cases have been reversible but some deaths have occurred. Since there was multiple pathology including infection, gallstones and carcinoma it was not possible to clearly establish the relationship to Cognex® treatment.
Blood chemistry signs of liver injury: Experience from the 30-week clinical study (described earlier) provides a representative estimate of the frequency of ALT/SGPT elevations expected for patients whose transaminase levels are monitored weekly and who receive Cognex® according to the recommended regimen for dose introduction and titration (Table 2). A dosing regimen employing a more rapid escalation of the daily dose of tacrine may be associated with more serious clinical events (see Monitoring of Liver function and the Management of the patient who develops transaminase elevations).
Table 2. Cumulative Incidence of ALT/SGPT Elevations
Based on Maximum Values with Weekly Monitoring During the 30-Week Study
[Number and (%) of Patients]
| Maximum ALT | Males N = 229 | Females N = 250 | Total N = 479 |
| Within Normal Limits | 121 (53) | 100 (40) | 221 (46) |
| >ULN | 108 (47) | 150 (60) | 258 (54) |
| >2 times ULN | 77 (34) | 104 (42) | 181 (38) |
| >3 times ULN | 58 (25) | 81 (32) | 139 (29) |
| >10 times ULN | 12 (5) | 19 (8) | 31 (6) |
| >20 times ULN | 3 (1) | 6 (2) | 9 (2) |
Experience in 2446 patients who participated in all clinical trials, including the 30-week study, indicates approximately 50% of patients treated with Cognex® can be expected to have at least 1 ALT/SGPT level above ULN; approximately 25% of patients are likely to develop elevations >3 X ULN, and about 7% of patients may develop elevations >10 X ULN. Data collected from the treatment IND program were consistent with those obtained during clinical studies, and showed 3% of 5665 patients experiencing an ALT/SGPT elevation >10 X ULN.
In clinical trials where transaminases were monitored weekly, the median time to onset of the first ALT/SGPT elevation above ULN was approximately 6 weeks, with maximum ALT/SGPT occurring 1 week later, even in instances when Cognex® treatment was stopped. Under the conditions of forced slow upwards dose titration (increases of 40 mg a day every 6 weeks) employed in clinical studies, 95% of transaminase elevations >3 X ULN occurred within the first 18 weeks of Cognex® therapy, and 99% of the 10-fold elevations occurred by the 12th week and on not more than 80 mg; note, however, that for most patients ALT was monitored weekly and Cognex®was stopped when liver enzymes exceeded 3 X ULN. A total of 276 patients were monitored for ALT/SGPT levels every other week in two double-blind clinical studies, an open-label study, and amended treatment IND. The incidence, severity, time to onset, peak and recovery of ALT/SGPT levels were similar to weekly monitoring. With less frequent monitoring than every other week or the less stringent discontinuation criteria recommended below (see DOSAGE AND ADMINISTRATION), it is possible that marked elevations might be more common. It must also be appreciated that experience with prolonged exposure to the high dose (160 mg/day) is limited. In all cases, transaminase levels returned to within normal limits upon discontinuation of Cognex® treatment or following dosage reduction, usually within 4 to 6 weeks.
This relatively benign experience may be the consequence of careful laboratory monitoring that facilitated the discontinuation of patients early on after the onset of their transaminase elevations. Consequently, frequent monitoring of serum transaminase levels is recommended (see DOSAGE AND ADMINISTRATION, WARNINGS: Liver
Injury: Monitoring of Liver Function and the Management of the Patient Who Develops Transaminase Elevations and PRECAUTIONS: Laboratory Tests).
Liver biopsy experience: Liver biopsy results in 7 patients who received tacrine (1 in a Parke-Davis sponsored study and 6 in studies reported in the literature) revealed hepatocellular necrosis in 6 patients, and granulomatous changes in the seventh. In all cases, liver function tests returned to normal with no evidence of persisting hepatic dysfunction.
Experience with the rechallenge of patients with transaminase elevations following recovery: Two hundred and twelve patients among the 866 patients assigned to tacrine in the 12 and 30 week studies were withdrawn because they developed transaminase elevations >3 X ULN. One hundred and forty-five of these patients were subsequently rechallenged with weekly monitoring of ALT/SGPT. During their initial exposure to tacrine, 20 of these 145 had experienced initial elevations >10 times ULN, while the remainder had experienced elevations between 3 and 10 X ULN.
Upon rechallenge with an initial dose of 40 mg/day, only 48 (33%) of the 145 patients developed transaminase elevations greater than 3 X ULN. Of these patients, 44 had elevations that were between 3 and 10 X ULN and 4 had elevations that were > 10 X ULN.
The mean time to onset of elevations occurred earlier on rechallenge than on initial exposure (22 versus 48 days). Of the 145 patients rechallenged, 127 (88%) were able to continue Cognex® treatment, and 91 of these 127 patients titrated to doses higher than those associated with the initial transaminase elevation.
Predictors of the risk of transaminase elevations: The incidence of transaminase elevations is higher among females. There are no other known predictors of the risk of hepatocellular injury.
Monitoring of Liver function and the Management of the patient who develops transaminase elevations. (See also DOSAGE AND ADMINISTRATION and PRECAUTIONS: Laboratory Tests.)
Blood chemistries: Serum transaminase levels (specifically ALT/SGPT) should be monitored every other week from at least week 4 to week 16 following initiation of treatment, after which monitoring may be decreased to every 3 months. For patients who develop ALT/SGPT elevations greater than two times the upper limit of normal, the dose and monitoring regimen should be modified as described in Table 4 (see DOSAGE AND ADMINISTRATION).
A full monitoring sequence should be repeated in the event that a patient suspends treatment with tacrine for more than 4 weeks.
If ALT/SGPT elevations occur, the frequency of monitoring and the dose of Cognex® should be modified according to the table shown below in DOSAGE AND ADMINISTRATION.
Rechallenge: Patients with clinical jaundice confirmed by a significant elevation in total bilirubin (> 3 mg/dL) and/or those exhibiting clinical signs and/or symptoms of hypersensitivity (e.g. rash or fever) in association with ALT/SGPT elevations should immediately and permanently discontinue Cognex® and not be rechallenged.Other patients who are required to discontinue Cognex® treatment because of ALT/SGPT elevations may be rechallenged once ALT/SGPT levels return to within normal limits. (See DOSAGE AND ADMINISTRATION.)
Rechallenge of patients with ALT/SGPT elevations less than 10 X ULN has not resulted in serious liver injury. However, because experience in the rechallenge of patients who had elevations greater than 10 X ULN is limited, the risks associated with the rechallenge of these patients are not well characterized. Careful, frequent (weekly) monitoring of serum ALT/SGPT should be undertaken when rechallenging such patients.
If rechallenged, patients should be given an initial dose of 40 mg/day (10 mg QID) and ALT/SGPT levels monitored weekly. If, after 6 weeks on 40 mg/day, the patient is tolerating the dosage with no unacceptable elevations in ALT/SGPT, recommended dose-titration may be resumed. Weekly monitoring of the ALT/SGPT levels should continue for a total of 16 weeks after which monitoring may be decreased to monthly for 2 months and every 3 months thereafter.
Liver biopsy: Liver biopsy is not indicated in cases of uncomplicated transaminase elevation.
Neurological Conditions
Seizures: Cholinomimetics are believed to have some potential to cause generalized convulsions; seizure activity may, however, also be a manifestation of Alzheimer's disease.
Sudden worsening of the degree of cognitive impairment: Worsening of cognitive function has been reported following abrupt discontinuation of Cognex® or after a large reduction in total daily dose (80 mg/day or more).
PRECAUTIONS
General
Hematology
An absolute neutrophil count (ANC) less than 500/µL occurred in 4 patients who received Cognex® during the course of clinical trials. Three of the 4 patients had concurrent medical conditions commonly associated with a low ANC; 2 of these patients remained on Cognex.® The fourth patient, who had a history of hypersensitivity (penicillin allergy), withdrew from the study as a result of a rash and also developed an ANC <500/µL, which returned to normal; this patient was not rechallenged and, therefore, the role played by Cognex® in this reaction is unknown.
Six patients had an absolute neutrophil count 1500/µL, associated with an elevation of ALT/SGPT.
The total clinical experience in more than 12,000 patients does not indicate a clear association between Cognex® treatment and serious white blood cell abnormalities.
Information for Patients and Caregivers
Patients and caregivers should be advised that the effect of Cognex® (brand of tacrine hydrochloride) therapy is thought to depend upon its administration at regular intervals, as directed.
The caregiver should be advised about the possibility of adverse effects. Two types should be distinguished: (1) those occurring in close temporal association with the initiation of treatment or an increase in dose (eg, nausea, vomiting, loose stools, diarrhea, etc) and (2) those with a delayed onset (eg, rash, jaundice, changes in the color of stool—black, very dark or light [ie, acholic]).
Patients and caregivers should be encouraged to inform the physician about the emergence of new events or any increase in the severity of existing adverse clinical events.
Caregivers should be advised that abrupt discontinuation of Cognex® or a large reduction in total daily dose (80 mg/day or more) may cause a decline in cognitive function and behavioral disturbances. Unsupervised increases in the dose of tacrine may also have serious consequences. Consequently, changes in dose should not be undertaken in the absence of direct instruction of a physician.
Laboratory Tests:(see WARNINGS: Liver Injury and DOSAGE AND ADMINISTRATION)
Serum transaminase levels (specifically ALT/SGPT) should be monitored in patients given Cognex® (see WARNINGS: Liver Injury).
Drug-Drug Interactions
Possible metabolic basis for interactions.
Tacrine is primarily eliminated by hepatic metabolism via cytochrome P450 drug metabolizing enzymes. Drug-drug interactions may occur when Cognex® is given concurrently with agents such as theophylline that undergo extensive metabolism via cytochrome P450 IA2.
Theophylline.
Coadministration of tacrine with theophylline increased theophylline elimination half-life and average plasma theophylline concentrations by approximately 2-fold. Therefore, monitoring of plasma theophylline concentrations and appropriate reduction of theophylline dose are recommended in patients receiving tacrine and theophylline concurrently. The effect of theophylline on tacrine pharmacokinetics has not been assessed.
Cimetidine.
Cimetidine increased the Cmax and AUC of tacrine by approximately 54% and 64%, respectively.
Anticholinergics.
Because of its mechanism of action, Cognex® has the potential to interfere with the activity of anticholinergic medications.
Cholinomimetics and Cholinesterase Inhibitors.
A synergistic effect is expected when Cognex® is given concurrently with succinylcholine (see WARNINGS), cholinesterase inhibitors, or cholinergic agonists such as bethanechol.
Fluvoxamine.
In a study of 13 healthy, male volunteers, a single 40 mg dose of tacrine added to fluvoxamine 100 mg/day administered at steady-state was associated with five- and eight-fold increases in tacrine Cmax and AUC, respectively, compared to the administration of tacrine alone. Five subjects experienced nausea, vomiting, sweating, and diarrhea following coadministration, consistent with the cholinergic effects of tacrine.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Tacrine was mutagenic to bacteria in the Ames test. Unscheduled DNA synthesis was induced in rat and mouse hepatocytes in vitro. Results of cytogenetic (chromosomal aberration) studies were equivocal. Tacrine was not mutagenic in an in vitro mammalian mutation test. Overall, the results of these tests, along with the fact that tacrine belongs to a chemical class (acridines) containing some members which are animal carcinogens, suggest that tacrine may be carcinogenic.
Studies of the effects of tacrine on fertility have not been performed.
ADVERSE REACTIONS
Common Adverse Events Leading to Discontinuation
In clinical trials, approximately 17% of the 2706 patients who received Cognex® and 5% of the 1886 patients who received placebo withdrew permanently because of adverse events. It should be noted that some of the placebo-treated patients were exposed to Cognex® prior to receiving placebo due to the variety of study designs used, including crossover studies. Transaminase elevations were the most common reason for withdrawals during Cognex® treatment (8% of all Cognex®-treated patients, or 212 of 456 patients withdrawn). The controlled clinical trial protocols required that any patient with an ALT/SGPT elevation >3 X ULN be withdrawn, because of concern about potential hepatotoxicity. Apart from withdrawals due to transaminase elevations, 244 patients (9%) withdrew for adverse events while receiving Cognex®.
Other adverse events that most frequently led to the withdrawal of tacrine-treated patients in clinical trials were nausea and/or vomiting (1.5%), agitation (0.9%), rash (0.7%), anorexia (0.7%), and confusion (0.5%). These adverse events also most frequently led to the withdrawal of placebo-treated patients, although at lower frequencies (0.1% to 0.2%).
Most Frequent Adverse Clinical Events Seen in Association With the Use of Tacrine
The events identified here are those that occurred at an absolute incidence of at least 5% of patients treated with Cognex®, and at a rate at least 2-fold higher in patients treated with Cognex®than placebo.
The most common adverse events associated with the use of Cognex® were elevated transaminases, nausea and/or vomiting, diarrhea, dyspepsia, myalgia, anorexia, and ataxia. Of these events, nausea and/or vomiting, diarrhea, dyspepsia, and anorexia appeared to be dose-dependent.
Adverse Events Reported in Controlled Trials
Adverse Events Reported in Controlled Trials
The events cited in the tables below reflect experience gained under closely monitored conditions of clinical trials with a highly selected patient population. In actual clinical practice or in other clinical trials, these frequency estimates may not apply, as the conditions of use, reporting behavior, and the kinds of patients treated may differ.
Table 3 lists treatment-emergent signs and symptoms that occurred in at least 2% of patients with Alzheimer's disease in placebo-controlled trials and who received the recommended regimen for dose introduction and titration of Cognex® (see DOSAGE AND ADMINISTRATION).
Table 3. Adverse Events Occurring in at Least 2% of Patients Receiving Cognex®
at a Starting Dose of 40 mg/day with Titration in 40 mg/day Increments
Every 6 Weeks in Controlled Clinical Trials
[Number (%) of Patients]
| BODY SYSTEM/Adverse Events | Cognex®N = 634 | PlaceboN = 342 |
| LABORATORY DEVIATIONS | ||
| Elevated Transaminasea | 184 (29) | 5 (2) |
| BODY AS A WHOLE | ||
| Headache | 67 (11) | 52 (15) |
| Fatigue | 26 (4) | 9 (3) |
| Chest Pain | 24 (4) | 18 (5) |
| Weight Decrease | 21 (3) | 4 (1) |
| Back Pain | 15 (2) | 14 (4) |
| Asthenia | 15 (2) | 7 (2) |
| DIGESTIVE SYSTEM | ||
| Nausea and/or Vomiting | 178 (28) | 29 (9) |
| Diarrhea | 99 (16) | 18 (5) |
| Dyspepsia | 57 (9) | 22 (6) |
| Anorexia | 54 (9) | 11 (3) |
| Abdominal Pain | 48 (8) | 24 (7) |
| Flatulence | 22 (4) | 5 (2) |
| Constipation | 24 (4) | 8 (2) |
| HEMIC AND LYMPHATIC SYSTEM | ||
| Purpura | 15 (2) | 8 (2) |
| MUSCULOSKELETAL SYSTEM | ||
| Myalgia | 54 (9) | 18 (5) |
| NERVOUS SYSTEM | ||
| Dizziness | 73 (12) | 39 (11) |
| Confusion | 42 (7) | 24 (7) |
| Ataxia | 36 (6) | 12 (4) |
| Insomnia | 37 (6) | 18 (5) |
| Somnolence | 22 (4) | 11 (3) |
| Tremor | 14 (2) | 2 (<1) |
| PSYCHOBIOLOGIC FUNCTION | ||
| Agitation | 43 (7) | 30 (9) |
| Depression | 22 (4) | 14 (4) |
| Thinking Abnormal | 17 (3) | 14 (4) |
| Anxiety | 16 (3) | 7 (2) |
| Hallucination | 15 (2) | 12 (4) |
| Hostility | 15 (2) | 5 (2) |
| RESPIRATORY SYSTEM | ||
| Rhinitis | 51 (8) | 22 (6) |
| Upper Respiratory Infection | 18 (3) | 11 (3) |
| Coughing | 17 (3) | 18 (5) |
| SKIN AND APPENDAGES | ||
| Rashb | 46 (7) | 18 (5) |
| Facial Flushing, Skin Flushing | 16 (3) | 3 (<1) |
| UROGENITAL SYSTEM | ||
| Urination Frequency | 21 (3) | 12 (4) |
| Urinary Tract Infection | 21 (3) | 20 (6) |
| Urinary Incontinence | 16 (3) | 9 (3) |
a ALT or AST value of approximately 3 X ULN or greater or that resulted in a change in patient management. Patients were monitored weekly.
b Includes COSTART terms: rash, rash-erythematous, rash-maculopapular, urticaria, petechial rash, rash-vesiculobullous, and pruritus.
Other Adverse Events Observed During All Clinical Trials
Cognex® has been administered to 2706 individuals during clinical trials. A total of 1471 patients were treated for at least 3 months, 1137 for at least 6 months, and 773 for at least 1 year. Any untoward reactions that occurred during these trials were recorded as adverse events by the clinical investigators using terminology of their own choosing. To provide a meaningful estimate of the proportion of individuals having similar types of events, the events were grouped into a smaller number of standardized categories using a modified COSTART dictionary. These categories are used in the listing below. The frequencies represent the proportion of the 2706 individuals exposed to Cognex® who experienced that event while receiving Cognex®. All adverse events are included except those already listed on the previous table and those COSTART terms too general to be informative. Events are further classified by body system categories and listed using the following definitions: frequent adverse events are defined as those occurring in at least 1/100 patients; infrequent adverse events are those occurring in 1/100 to 1/1000 patients; and rare adverse events are those occurring in less than 1/1000 patients. These adverse events are not necessarily related to Cognex® treatment. Only rare adverse events deemed to be potentially important are included.
Body As a Whole:
Frequent: Chill, fever, malaise, peripheral edema. Infrequent: Face edema, dehydration, weight increase, cachexia, edema (generalized), lipoma. Rare: Heat exhaustion, sepsis, cholingeric crisis, death.
Cardiovascular System:
Frequent: Hypotension, hypertension. Infrequent: Heart failure, myocardial infarction, angina pectoris, cerebrovascular accident, transient ischemic attack, phlebitis, venous insufficiency, abdominal aortic aneurysm, atrial fibrillation or flutter, palpitation, tachycardia, bradycardia, pulmonary embolus, migraine, hypercholesterolemia. Rare: Heart arrest, premature atrial contractions, A-V block, bundle branch block.
Digestive System:
Infrequent: Glossitis, gingivitis, mouth or throat dry, stomatitis, increased salivation, dysphagia, esophagitis, gastritis, gastroenteritis, GI hemorrhage, stomach ulcer, hiatal hernia, hemorrhoids, stools bloody, diverticulitis, fecal impaction, fecal incontinence, hemorrhage (rectum), cholelithiasis, cholecystitis, increased appetite. Rare: Duodenal ulcer, bowel obstruction.
Hemic and Lymphatic:
Infrequent: Anemia, lymphadenopathy. Rare: Leukopenia, thrombocytopenia, hemolysis, pancytopenia.
Musculoskeletal:
Frequent: Fracture, arthralgia, arthritis, hypertonia. Infrequent: Osteoporosis, tendinitis, bursitis, gout. Rare: Myopathy.
Nervous System:
Frequent: Convulsions, vertigo, syncope, hyperkinesia, paresthesia. Infrequent: Dreaming abnormal, dysarthria, aphasia, amnesia, wandering, twitching, hypesthesia, delirium, paralysis, bradykinesia, movement disorder, cogwheel rigidity, paresis, neuritis, hemiplegia, Parkinson's disease, neuropathy, extrapyramidal syndrome, reflexes decreased/absent. Rare: Tardive dyskinesia, dysesthesia, dystonia, encephalitis, coma, apraxia, oculogyric crisis, akathisia, oral facial dyskinesia, Bell's palsy, exacerbation of Parkinson's disease.
Psychobiologic Function:
Frequent: Nervousness. Infrequent: Apathy, increased libido, paranoia, neurosis. Rare: Suicidal, psychosis, hysteria.
Respiratory System:
Frequent: Pharyngitis, sinusitis, bronchitis, pneumonia, dyspnea. Infrequent: Epistaxis, chest congestion, asthma, hyperventilation, lower respiratory infection. Rare: Hemoptysis, lung edema, lung cancer, acute epiglottitis.
Skin and Appendages:
Frequent: Sweating increased. Infrequent: Acne, alopecia, dermatitis, eczema, skin dry, herpes zoster, psoriasis, cellulitis, cyst, furunculosis, herpes simplex, hyperkeratosis, basal cell carcinoma, skin cancer. Rare: Desquamation, seborrhea, squamous cell carcinoma, ulcer (skin), skin necrosis, melanoma.
Urogenital System:
Infrequent: Hematuria, renal stone, kidney infection, glycosuria, dysuria, polyuria, nocturia, pyuria, cystitis, urinary retention, urination urgency, vaginal hemorrhage, pruritus (genital), breast pain, impotence, prostate cancer. Rare: Bladder tumor, renal tumor, renal failure, urinary obstruction, breast cancer, epididymitis, carcinoma (ovary).
Special Senses:
Frequent: Conjunctivitis. Infrequent: Cataract, eyes dry, eye pain, visual field defect, diplopia, amblyopia, glaucoma, hordeolum, deafness, earache, tinnitus, inner ear infection, otitis media, unusual taste. Rare: Vision loss, ptosis, blepharitis, labyrinthitis, inner ear disturbance.
Postintroduction Reports
Voluntary reports of adverse events temporally associated with Cognex® that have been received since market introduction, that are not listed above, and that may have no causal relationship with the drug include the following: pancreatitis, perforated peptic ulcer, and falling.
OVERDOSAGE
As in any case of overdose, general supportive measures should be utilized. Overdosage with cholinesterase inhibitors can cause a cholinergic crisis characterized by severe nausea/vomiting, salivation, sweating, bradycardia, hypotension, collapse, and convulsions. Increasing muscle weakness is a possibility and may result in death if respiratory muscles are involved.
Tertiary anticholinergics such as atropine may be used as an antidote for Cognex® overdosage. Intravenous atropine sulfate titrated to effect is recommended: in adults, initial dose of 1.0 to 2.0 mg IV with subsequent doses based on clinical response. In children, the usual IM or IV dose is 0.05 mg/kg, repeated every 10-30 minutes until muscarinic signs and symptoms subside and repeated if they reappear. Atypical increases in blood pressure and heart rate have been reported with other cholinomimetics when coadministered with quaternary anticholinergics such as glycopyrrolate.
It is not known whether Cognex® or its metabolites can be eliminated by dialysis (hemodialysis, peritoneal dialysis, or hemofiltration).
The estimated median lethal dose of tacrine following a single oral dose in rats is 40 mg/kg, or approximately 12 times the maximum recommended human dose of 160 mg/day. Dose-related signs of cholinergic stimulation were observed in animals and included vomiting, diarrhea, salivation, lacrimation, ataxia, convulsions, tremor, and stereotypic head and body movements.
DOSAGE AND ADMINISTRATION
The recommendations for dose titration are based on experience from clinical trials. The rate of dose escalation may be slowed if a patient is intolerant to the titration schedule recommended below. It is not advisable, however, to accelerate the dose incrementation plan.
Following initiation of therapy, or any dosage increase, patients should be observed carefully for adverse effects. Cognex® should be taken between meals whenever possible; however, if minor GI upset occurs, Cognex®may be taken with meals to improve tolerability. Taking Cognex® with meals can be expected to reduce plasma levels approximately 30% to 40%.
Initiation of Treatment
The initial dose of Cognex® brand of tacrine hydrochloride is 40 mg/day (10 mg QID). This dose should be maintained for a minimum of 4 weeks with every other week monitoring of transaminase levels beginning 4 weeks after initiation of treatment. It is important that the dose not be increased during this period because of the potential for delayed onset of transaminase elevations.
Dose Titration
Following 4 weeks of treatment at 40 mg/day (10 mg QID), the dose of Cognex® should then be increased to 80 mg/day (20 mg QID), providing there are no significant transaminase elevations and the patient is tolerating treatment. Patients should be titrated to higher doses (120 and 160 mg/day, in divided doses on a QID schedule) at 4- week intervals on the basis of tolerance.
Dose Adjustment
Serum ALT/SGPT should be monitored every other week from at least week 4 to week 16 following initiation of treatment, after which monitoring may be decreased to every 3 months. For patients who develop ALT/SGPT elevations greater than two times the upper limit of normal, the dose and monitoring regimen should be modified as described in Table 4.
A full monitoring and dose titration sequence must be repeated in the event that a patient suspends treatment with tacrine for more than 4 weeks.
Table 4. Recommended Dose and Monitoring Regimen
Modification in Response to ALT/SGPT Elevations
| ALT/SGPT Level | Treatment and Monitoring Regimen |
| 2 X ULN | Continue treatment according to recommended titration and monitoring schedule. |
| > 2 to3 X ULN | Continue treatment according to recommended titration. Monitor ALT/SGPT levels weekly until levels return to normal limits. |
| > 3 to5 X ULN | Reduce the daily dose of Cognex® by 40 mg/day. Monitor ALT/SGPT levels weekly. Resume dose titration and every other week monitoring when the levels of the ALT/SGPT return to normal limits. |
| > 5 X ULN | Stop Cognex® treatment. Monitor the patient closely for signs and symptoms associated with hepatitis and follow ALT/SGPT levels until within normal limits. See Rechallenge section below. |
| | Experience is limited in patients with ALT/SGPT >10 X ULN. The risk of rechallenge must be considered against demonstrated clinical benefit. |
| | Patients with clinical jaundice confirmed by a significant elevation in total bilirubin (> 3 mg/dL) and/or those exhibiting clinical signs and/or symptoms of hypersensitivity (e.g. rash or fever) in association with ALT/SGPT elevations should immediately and permanently discontinue Cognex® and not be rechallenged.
|
Rechallenge
Patients who are required to discontinue Cognex® treatment because of ALT/SGPT elevations may be rechallenged once ALT/SGPT levels return to normal limits.
Rechallenge of patients exposed to ALT/SGPT elevations less than 10 X ULN has not resulted in serious liver injury. However, because experience in the rechallenge of patients who had elevations greater than 10 X ULN is limited, the risks associated with the rechallenge of these patients are not well characterized. Careful, frequent (weekly) monitoring of serum ALT/SGPT should be undertaken when rechallenging such patients.
If rechallenged, patients should be given an initial dose of 40 mg/day (10 mg QID) and ALT/SGPT levels monitored weekly. If, after 6 weeks on 40 mg/day, the patient is tolerating the dosage with no unacceptable elevations in ALT/SGPT, the recommended dose-titration may be resumed. Weekly monitoring of the ALT/SGPT levels should continue for a total of 16 weeks after which monitoring may be decreased to monthly for 2 months and every 3 months thereafter.
HOW SUPPLIED
Cognex® is supplied as capsules of tacrine hydrochloride containing 10, 20, 30, and 40 mg of tacrine. The capsule logo is "Cognex®” with the strength (eg, 10, 20, 30, or 40) printed underneath
| 10 mg (yellow/dark green) | Bottles of 120 (NDC 59630-190-12) |
| 20 mg (yellow/light blue) | Bottles of 120 (NDC 59630-191-12) |
| 30 mg (yellow/swedish orange) | Bottles of 120 (NDC 59630-192-12) |
| 40 mg (yellow/lavender) | Bottles of 120 (NDC 59630-193-12) |





| COGNEX
tacrine hydrochloride capsule |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020070 | 09/09/1993 | 09/30/2010 |
| COGNEX
tacrine hydrochloride capsule |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020070 | 09/09/1993 | 09/30/2010 |
| COGNEX
tacrine hydrochloride capsule |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020070 | 09/09/1993 | 09/30/2010 |
| COGNEX
tacrine hydrochloride capsule |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020070 | 09/09/1993 | 09/30/2010 |
| Labeler - Shionogi Inc. (949127786) |
Revised: 09/2012 Shionogi Inc.