campral (acamprosate calcium) tablet, delayed release
[FOREST PHARMACEUTICALS, Inc. ]
DESCRIPTION
CAMPRAL®(acamprosate calcium) is supplied in an enteric-coated tablet for oral administration. Acamprosate calcium is a synthetic compound with a chemical structure similar to that of the endogenous amino acid homotaurine, which is a structural analogue of the amino acid neurotransmitter γ-aminobutyric acid and the amino acid neuromodulator taurine. Its chemical name is calcium acetylaminopropane sulfonate. Its chemical formula is C10H20N2O8S2Ca and molecular weight is 400.48. Its structural formula is:
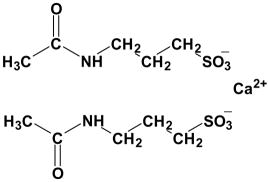
Acamprosate calcium is a white, odorless or nearly odorless powder. It is freely soluble in water, and practically insoluble in absolute ethanol and dichloromethane.
Each CAMPRAL® tablet contains acamprosate calcium 333 mg, equivalent to 300 mg of acamprosate. Inactive ingredients in CAMPRAL® tablets include: crospovidone, microcrystalline cellulose, magnesium silicate, sodium starch glycolate, colloidal anhydrous silica, magnesium stearate, talc, propylene glycol and Eudragit® L 30 D or equivalent. Sulfites were used in the synthesis of the drug substance and traces of residual sulfites may be present in the drug product.
CLINICAL PHARMACOLOGY
Pharmacodynamics
The mechanism of action of acamprosate in maintenance of alcohol abstinence is not completely understood. Chronic alcohol exposure is hypothesized to alter the normal balance between neuronal excitation and inhibition. In vitro and in vivo studies in animals have provided evidence to suggest acamprosate may interact with glutamate and GABA neurotransmitter systems centrally, and has led to the hypothesis that acamprosate restores this balance.
Pharmacodynamic studies have shown that acamprosate calcium reduces alcohol intake in alcohol-dependent animals in a dose-dependent manner and that this effect appears to be specific to alcohol and the mechanisms of alcohol dependence.
Acamprosate calcium has negligible observable central nervous system (CNS) activity in animals outside of its effects on alcohol dependence, exhibiting no anticonvulsant, antidepressant, or anxiolytic activity.
The administration of acamprosate calcium is not associated with the development of tolerance or dependence in animal studies.
CAMPRAL® is not known to cause alcohol aversion and does not cause a disulfiram-like reaction as a result of ethanol ingestion.
Pharmacokinetics
Absorption
The absolute bioavailability of CAMPRAL® after oral administration is about 11%. Steady-state plasma concentrations of acamprosate are reached within 5 days of dosing. Steady-state peak plasma concentrations after CAMPRAL® doses of 2 x 333 mg tablets three times daily average 350 ng/mL and occur at 3-8 hours post-dose. Coadministration of CAMPRAL® with food decreases bioavailability as measured by Cmax and AUC, by approximately 42% and 23%, respectively. The food effect on absorption is not clinically significant and no adjustment of dose is necessary.
Distribution
The volume of distribution for acamprosate following intravenous administration is estimated to be 72-109 liters (approximately 1 L/kg). Plasma protein binding of acamprosate is negligible.
Metabolism
Acamprosate does not undergo metabolism.
Elimination
After oral dosing of 2 x 333 mg of CAMPRAL®, the terminal half-life ranges from approximately 20 - 33 hours. Following oral administration of CAMPRAL®, the major route of excretion is via the kidneys as acamprosate.
Special Populations
Gender: CAMPRAL® does not exhibit any significant pharmacokinetic differences between male and female subjects.
Age: The pharmacokinetics of CAMPRAL® have not been evaluated in a geriatric population. However, since renal function diminishes in elderly patients and acamprosate is excreted unchanged in urine, acamprosate plasma concentrations are likely to be higher in the elderly population compared to younger adults.
Pediatrics: The pharmacokinetics of CAMPRAL® have not been evaluated in a pediatric population.
Renal Impairment: Peak plasma concentrations after administration of a single dose of 2 x 333 mg CAMPRAL® tablets to patients with moderate or severe renal impairment were about 2-fold and 4-fold higher, respectively, compared to healthy subjects. Similarly, elimination half-life was about 1.8-fold and 2.6-fold longer, respectively, compared to healthy subjects. There is a linear relationship between creatinine clearance values and total apparent plasma clearance, renal clearance and plasma half-life of acamprosate. A dose of 1 x 333 mg CAMPRAL®, three times daily, is recommended in patients with moderate renal impairment (creatinine clearance of 30-50 mL/min, see also PRECAUTIONS).
Patients with severe renal impairment (creatinine clearance ≤30 mL/min) should not be given CAMPRAL® (see also CONTRAINDICATIONS).
Hepatic Impairment: Acamprosate is not metabolized by the liver and the pharmacokinetics of CAMPRAL® are not altered in patients with mild to moderate hepatic impairment (groups A and B of the Child-Pugh classification). No adjustment of dosage is recommended in such patients.
Alcohol-dependent subjects: A cross-study comparison of CAMPRAL® at doses of 2 x 333 mg three times daily indicated similar pharmacokinetics between alcohol-dependent subjects and healthy subjects.
Drug-Drug Interactions
Acamprosate had no inducing potential on the cytochrome CYP1A2 and 3A4 systems, and in vitro inhibition studies suggest that acamprosate does not inhibit in vivo metabolism mediated by cytochrome CYP1A2, 2C9, 2C19, 2D6, 2E1, or 3A4. The pharmacokinetics of CAMPRAL® were unaffected when co-administered with alcohol, disulfiram or diazepam. Similarly, the pharmacokinetics of ethanol, diazepam and nordiazepam, imipramine and desipramine, naltrexone and 6-beta naltrexol were unaffected following co-administration with CAMPRAL®. However, co-administration of CAMPRAL® with naltrexone led to a 33% increase in the Cmax and a 25% increase in the AUC of acamprosate. No adjustment of dosage is recommended in such patients.
CLINICAL STUDIES
The efficacy of CAMPRAL® in the maintenance of abstinence was supported by three clinical studies involving a total of 998 patients who were administered at least one dose of CAMPRAL® or placebo as an adjunct to psychosocial therapy. Each study was a double-blind, placebo-controlled trial in alcohol-dependent patients who had undergone inpatient detoxification and were abstinent from alcohol on the day of randomization. Study durations ranged from 90 days to 360 days. CAMPRAL® proved superior to placebo in maintaining abstinence, as indicated by a greater percentage of subjects being assessed as continuously abstinent throughout treatment.
In a fourth study, the efficacy of CAMPRAL® was evaluated in alcoholics, including patients with a history of polysubstance abuse and patients who had not undergone detoxification and were not required to be abstinent at baseline. This study failed to demonstrate superiority of CAMPRAL® over placebo.
INDICATIONS AND USAGE
CAMPRAL® is indicated for the maintenance of abstinence from alcohol in patients with alcohol dependence who are abstinent at treatment initiation. Treatment with CAMPRAL® should be part of a comprehensive management program that includes psychosocial support.
The efficacy of CAMPRAL® in promoting abstinence has not been demonstrated in subjects who have not undergone detoxification and not achieved alcohol abstinence prior to beginning CAMPRAL® treatment. The efficacy of CAMPRAL® in promoting abstinence from alcohol in polysubstance abusers has not been adequately assessed.
CONTRAINDICATIONS
CAMPRAL® is contraindicated in patients who previously have exhibited hypersensitivity to acamprosate calcium or any of its components.
CAMPRAL® is contraindicated in patients with severe renal impairment (creatinine clearance ≤30 mL/min).
PRECAUTIONS
Use of CAMPRAL® does not eliminate or diminish withdrawal symptoms.
General
Renal Impairment: Treatment with CAMPRAL® in patients with moderate renal impairment (creatinine clearance of 30-50 mL/min) requires a dose reduction. Patients with severe renal impairment (creatinine clearance of ≤30 mL/min) should not be given CAMPRAL® (see also CONTRAINDICATIONS).
Suicidality: In controlled clinical trials of CAMPRAL®, adverse events of a suicidal nature (suicidal ideation, suicide attempts, completed suicides) were infrequent overall, but were more common in CAMPRAL®-treated patients than in patients treated with placebo (1.4% vs. 0.5% in studies of 6 months or less; 2.4% vs. 0.8% in year-long studies). Completed suicides occurred in 3 of 2272 (0.13%) patients in the pooled acamprosate group from all controlled studies and 2 of 1962 patients (0.10%) in the placebo group. Adverse events coded as "depression" were reported at similar rates in CAMPRAL®-treated and placebo-treated patients. Although many of these events occurred in the context of alcohol relapse, no consistent pattern of relationship between the clinical course of recovery from alcoholism and the emergence of suicidality was identified. The interrelationship between alcohol dependence, depression and suicidality is well-recognized and complex. Alcohol-dependent patients, including those patients being treated with CAMPRAL® should be monitored for the development of symptoms of depression or suicidal thinking. Families and caregivers of patients being treated with CAMPRAL® should be alerted to the need to monitor patients for the emergence of symptoms of depression or suicidality, and to report such symptoms to the patient's health care provider.
Information for Patients
Physicians are advised to discuss the following issues with patients for whom they prescribe CAMPRAL®.
Any psychoactive drug may impair judgment, thinking, or motor skills. Patients should be cautioned about operating hazardous machinery, including automobiles, until they are reasonably certain that CAMPRAL® therapy does not affect their ability to engage in such activities.
Patients should be advised to notify their physician if they become pregnant or intend to become pregnant during therapy.
Patients should be advised to notify their physician if they are breast-feeding.
Patients should be advised to continue CAMPRAL® therapy as directed, even in the event of relapse and should be reminded to discuss any renewed drinking with their physician.
Patients should be advised that CAMPRAL® has been shown to help maintain abstinence only when used as a part of a treatment program that includes counseling and support.
Drug Interactions
The concomitant intake of alcohol and CAMPRAL® does not affect the pharmacokinetics of either alcohol or acamprosate.
Pharmacokinetic studies indicate that administration of disulfiram or diazepam does not affect the pharmacokinetics of acamprosate. Co-administration of naltrexone with CAMPRAL® produced a 25% increase in AUC and a 33% increase in the Cmax of acamprosate. No adjustment of dosage is recommended in such patients.
The pharmacokinetics of naltrexone and its major metabolite 6-beta-naltrexol were unaffected following co-administration with CAMPRAL®.
Other concomitant therapies: In clinical trials, the safety profile in subjects treated with CAMPRAL® concomitantly with anxiolytics, hypnotics and sedatives (including benzodiazepines), or non-opioid analgesics was similar to that of subjects taking placebo with these concomitant medications. Patients taking CAMPRAL® concomitantly with antidepressants more commonly reported both weight gain and weight loss, compared with patients taking either medication alone.
Carcinogenicity, Mutagenicity and Impairment of Fertility
A carcinogenicity study was conducted in which Sprague-Dawley rats received acamprosate calcium in their diet at doses of 25, 100 or 400 mg/kg/day (0.2, 0.7 or 2.5-fold the maximum recommended human dose based on an AUC comparison). There was no evidence of an increased incidence of tumors in this carcinogenicity study in the rat. An adequate carcinogenicity study in the mouse has not been conducted.
Acamprosate calcium was negative in all genetic toxicology studies conducted. Acamprosate calcium demonstrated no evidence of genotoxicity in an in vitro bacterial reverse point mutation assay (Ames assay) or an in vitro mammalian cell gene mutation test using Chinese Hamster Lung V79 cells. No clastogenicity was observed in an in vitro chromosomal aberration assay in human lymphocytes and no chromosomal damage detected in an in vivo mouse micronucleus assay.
Acamprosate calcium had no effect on fertility after treatment for 70 days prior to mating in male rats and for 14 days prior to mating, throughout mating, gestation and lactation in female rats at doses up to 1000 mg/kg/day (approximately 4 times the maximum recommended human daily oral dose on a mg/m2 basis). In mice, acamprosate calcium administered orally for 60 days prior to mating and throughout gestation in females at doses up to 2400 mg/kg/day (approximately 5 times the maximum recommended human daily oral dose on a mg/m2 basis) had no effect on fertility.
Pregnancy Category C
Teratogenic effects: Acamprosate calcium has been shown to be teratogenic in rats when given in doses that are approximately equal to the human dose (on a mg/m2 basis) and in rabbits when given in doses that are approximately 3 times the human dose (on a mg/m2 basis). Acamprosate calcium produced a dose-related increase in the number of fetuses with malformations in rats at oral doses of 300 mg/kg/day or greater (approximately equal to the maximum recommended human daily oral dose on a mg/m2 basis). The malformations included hydronephrosis, malformed iris, retinal dysplasia, and retroesophageal subclavian artery. No findings were observed at an oral dose of 50 mg/kg/day (approximately one-fifth the maximum recommended human daily oral dose on a mg/m2 basis). An increased incidence of hydronephrosis was also noted in Burgundy Tawny rabbits at oral doses of 400 mg/kg/day or greater (approximately 3 times the maximum recommended human daily oral dose on a mg/m2 basis). No developmental effects were observed in New Zealand white rabbits at oral doses up to 1000 mg/kg/day (approximately 8 times the maximum recommended human daily oral dose on a mg/m2 basis). The findings in animals should be considered in relation to known adverse developmental effects of ethyl alcohol, which include the characteristics of fetal alcohol syndrome (craniofacial dysmorphism, intrauterine and postnatal growth retardation, retarded psychomotor and intellectual development) and milder forms of neurological and behavioral disorders in humans. There are no adequate and well controlled studies in pregnant women. CAMPRAL® should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nonteratogenic effects: A study conducted in pregnant mice that were administered acamprosate calcium by the oral route starting on Day 15 of gestation through the end of lactation on postnatal day 28 demonstrated an increased incidence of still-born fetuses at doses of 960 mg/kg/day or greater (approximately 2 times the maximum recommended human daily oral dose on a mg/m2 basis). No effects were observed at a dose of 320 mg/kg/day (approximately one-half the maximum recommended human daily dose on a mg/m2 basis).
Labor and Delivery
The potential for CAMPRAL® to affect the duration of labor and delivery is unknown.
Nursing Mothers
In animal studies, acamprosate was excreted in the milk of lactating rats dosed orally with acamprosate calcium. The concentration of acamprosate in milk compared to blood was 1.3:1. It is not known whether acamprosate is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when CAMPRAL® is administered to a nursing woman.
Pediatric Use
The safety and efficacy of CAMPRAL® have not been established in the pediatric population.
Geriatric Use
Forty-one of the 4234 patients in double-blind, placebo-controlled, clinical trials of CAMPRAL® were 65 years of age or older, while none were 75 years of age or over. There were too few patients in the ≥65 age group to evaluate any differences in safety or effectiveness for geriatric patients compared to younger patients.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function (See Clinical Pharmacology, Adverse Reactions, and Dosage and Administration).
ADVERSE REACTIONS
The adverse event data described below reflect the safety experience in over 7000 patients exposed to CAMPRAL® for up to one year, including over 2000 CAMPRAL®-exposed patients who participated in placebo-controlled trials.
Adverse Events Leading to Discontinuation
In placebo-controlled trials of 6 months or less, 8% of CAMPRAL®-treated patients discontinued treatment due to an adverse event, as compared to 6% of patients treated with placebo. In studies longer than 6 months, the discontinuation rate due to adverse events was 7% in both the placebo-treated and the CAMPRAL®-treated patients. Only diarrhea was associated with the discontinuation of more than 1% of patients (2% of CAMPRAL®-treated vs. 0.7% of placebo-treated patients). Other events, including nausea, depression, and anxiety, while accounting for discontinuation in less than 1% of patients, were nevertheless more commonly cited in association with discontinuation in CAMPRAL®-treated patients than in placebo-treated patients.
Common Adverse Events Reported in Controlled Trials
Common, non-serious adverse events were collected spontaneously in some controlled studies and using a checklist in other studies. The overall profile of adverse events was similar using either method. Table 1 shows those events that occurred in any CAMPRAL® treatment group at a rate of 3% or greater and greater than the placebo group in controlled clinical trials with spontaneously reported adverse events. The reported frequencies of adverse events represent the proportion of individuals who experienced, at least once, a treatment-emergent adverse event of the type listed, without regard to the causal relationship of the events to the drug.
| Body System/Preferred Term | Number of Patients (%) with Events | |||
|---|---|---|---|---|
|
CAMPRAL® 1332 mg/day | CAMPRAL®
1998 mg/day1 | CAMPRAL®
Pooled2 | Placebo | |
|
*includes events coded as “fracture” by sponsor; **includes events coded as “nervousness” by sponsor |
||||
|
1 includes 258 patients treated with acamprosate calcium 2000 mg/day, using a different dosage strength and regimen. |
||||
|
2 includes all patients in the first two columns as well as 83 patients treated with acamprosate calcium 3000 mg/day, using a different dosage strength and regimen. |
||||
| Number of patients in Treatment Group | 397 | 1539 | 2019 | 1706 |
| Number (%) of patients with an AE | 248 (62%) | 910 (59%) | 1231 (61%) | 955 (56%) |
| Body as a Whole | 121 (30%) | 513 (33%) | 685 (34%) | 517 (30%) |
| Accidental Injury* | 17 ( 4%) | 44 ( 3%) | 70 ( 3%) | 52 ( 3%) |
| Asthenia | 29 ( 7%) | 79 ( 5%) | 114 ( 6%) | 93 ( 5%) |
| Pain | 6 ( 2%) | 56 ( 4%) | 65 ( 3%) | 55 ( 3%) |
| Digestive System | 85 (21%) | 440 (29%) | 574 (28%) | 344 (20%) |
| Anorexia | 20 ( 5%) | 35 ( 2%) | 57 ( 3%) | 44 ( 3%) |
| Diarrhea | 39 (10%) | 257 (17%) | 329 (16%) | 166 (10%) |
| Flatulence | 4 ( 1%) | 55 ( 4%) | 63 ( 3%) | 28 ( 2%) |
| Nausea | 11 ( 3%) | 69 ( 4%) | 87 ( 4%) | 58 ( 3%) |
| Nervous System | 150 (38%) | 417 (27%) | 598 (30%) | 500 (29%) |
| Anxiety** | 32 ( 8%) | 80 ( 5%) | 118 ( 6%) | 98 ( 6%) |
| Depression | 33 ( 8%) | 63 ( 4%) | 102 ( 5%) | 87 ( 5%) |
| Dizziness | 15 ( 4%) | 49 ( 3%) | 67 ( 3%) | 44 ( 3%) |
| Dry mouth | 13 ( 3%) | 23 ( 1%) | 36 ( 2%) | 28 ( 2%) |
| Insomnia | 34 ( 9%) | 94 ( 6%) | 137 ( 7%) | 121 ( 7%) |
| Paresthesia | 11 ( 3%) | 29 ( 2%) | 40 ( 2%) | 34 ( 2%) |
| Skin and Appendages | 26 ( 7%) | 150 (10%) | 187 ( 9%) | 169 (10%) |
| Pruritus | 12 ( 3%) | 68 ( 4%) | 82 ( 4%) | 58 ( 3%) |
| Sweating | 11 ( 3%) | 27 ( 2%) | 40 ( 2%) | 39 ( 2%) |
Other Events Observed During the Premarketing Evaluation of CAMPRAL®
Following is a list of terms that reflect treatment-emergent adverse events reported by patients treated with CAMPRAL® in 20 clinical trials (4461 patients treated with CAMPRAL®, 3526 of whom received the maximum recommended dose of 1998 mg/day for up to one year in duration). This listing does not include those events already listed above; events for which a drug cause was considered remote; event terms which were so general as to be uninformative; and events reported only once which were not likely to be acutely life-threatening.
Events are further categorized by body system and listed in order of decreasing frequency according to the following definitions: frequent adverse events are those occurring in at least 1/100 patients (only those not already listed in the summary of adverse events in controlled trials appear in this listing); infrequent adverse events are those occurring in 1/100 to 1/1000 patients; rare events are those occurring in fewer than 1/1000 patients.
Body as a Whole –Frequent: headache, abdominal pain, back pain, infection, flu syndrome, chest pain, chills, suicide attempt; Infrequent: fever, intentional overdose, malaise, allergic reaction, abscess, neck pain, hernia, intentional injury; Rare: ascites, face edema, photosensitivity reaction, abdomen enlarged, sudden death.
Cardiovascular System –Frequent: palpitation, syncope; Infrequent: hypotension, tachycardia, hemorrhage, angina pectoris, migraine, varicose vein, myocardial infarct, phlebitis, postural hypotension; Rare: heart failure, mesenteric arterial occlusion, cardiomyopathy, deep thrombophlebitis, shock.
Digestive System –Frequent: vomiting, dyspepsia, constipation, increased appetite; Infrequent: liver function tests abnormal, gastroenteritis, gastritis, dysphagia, eructation, gastrointestinal hemorrhage, pancreatitis, rectal hemorrhage, liver cirrhosis, esophagitis, hematemesis, nausea and vomiting, hepatitis; Rare: melena, stomach ulcer, cholecystitis, colitis, duodenal ulcer, mouth ulceration, carcinoma of liver.
Endocrine System –Rare: goiter, hypothyroidism.
Hemic and Lymphatic System –Infrequent: anemia, ecchymosis, eosinophilia, lymphocytosis, thrombocytopenia; Rare: leukopenia, lymphadenopathy, monocytosis.
Metabolic and Nutritional Disorders –Frequent – peripheral edema, weight gain; Infrequent: weight loss, hyperglycemia, SGOT increased, SGPT increased, gout, thirst, hyperuricemia, diabetes mellitus, avitaminosis, bilirubinemia; Rare: alkaline phosphatase increased, creatinine increased, hyponatremia, lactic dehydrogenase increased.
Musculoskeletal System –Frequent – myalgia, arthralgia; Infrequent: leg cramps; Rare: rheumatoid arthritis, myopathy.
Nervous System –Frequent –somnolence, libido decreased, amnesia, thinking abnormal, tremor, vasodilatation, hypertension; Infrequent: convulsion, confusion, libido increased, vertigo, withdrawal syndrome, apathy, suicidal ideation, neuralgia, hostility, agitation, neurosis, abnormal dreams, hallucinations, hypesthesia; Rare: alcohol craving, psychosis, hyperkinesia, twitching, depersonalization, increased salivation, paranoid reaction, torticollis, encephalopathy, manic reaction.
Respiratory System –Frequent: rhinitis, cough increased, dyspnea, pharyngitis, bronchitis; Infrequent: asthma, epistaxis, pneumonia; Rare: laryngismus, pulmonary embolus.
Skin and Appendages –Frequent: rash; Infrequent: acne, eczema, alopecia, maculopapular rash, dry skin, urticaria, exfoliative dermatitis, vesiculobullous rash; Rare: psoriasis.
Special Senses –Frequent: abnormal vision, taste perversion; Infrequent: tinnitus, amblyopia, deafness; Rare: ophthalmitis, diplopia, photophobia.
Urogenital System –Frequent: impotence; Infrequent – metrorrhagia, urinary frequency, urinary tract infection, sexual function abnormal, urinary incontinence, vaginitis; Rare: kidney calculus, abnormal ejaculation, hematuria, menorrhagia, nocturia, polyuria, urinary urgency.
Serious Adverse Events Observed During the Non-US Postmarketing Evaluation of CAMPRAL® (acamprosate calcium)
Although no causal relationship to CAMPRAL® has been found, the serious adverse event of acute kidney failure has been reported to be temporally associated with CAMPRAL® treatment in at least 3 patients and is not described elsewhere in the labeling.
DRUG ABUSE AND DEPENDENCE
Controlled Substance Class
Acamprosate calcium is not a controlled substance.
Physical and Psychological Dependence
CAMPRAL® did not produce any evidence of withdrawal symptoms in patients in clinical trials at therapeutic doses. Post marketing data, collected retrospectively outside the U.S., have provided no evidence of CAMPRAL® abuse or dependence.
OVERDOSAGE
In all reported cases of acute overdosage with CAMPRAL® (total reported doses of up to 56 grams of acamprosate calcium), the only symptom that could be reasonably associated with CAMPRAL® was diarrhea. Hypercalcemia has not been reported in cases of acute overdose. A risk of hypercalcemia should be considered in chronic overdosage only. Treatment of overdose should be symptomatic and supportive.
DOSAGE AND ADMINISTRATION
The recommended dose of CAMPRAL® is two 333 mg tablets (each dose should total 666 mg) taken three times daily. Although dosing may be done without regard to meals, dosing with meals was employed during clinical trials and is suggested as an aid to compliance in those patients who regularly eat three meals daily. A lower dose may be effective in some patients.
Treatment with CAMPRAL® should be initiated as soon as possible after the period of alcohol withdrawal, when the patient has achieved abstinence, and should be maintained if the patient relapses. CAMPRAL® should be used as part of a comprehensive psychosocial treatment program.
Dosage in Renal Impairment: For patients with moderate renal impairment (creatinine clearance of 30-50 mL/min), a starting dose of one 333 mg tablet taken three times daily is recommended. Patients with severe renal impairment (creatinine clearance of ≤30 mL/min) should not be given CAMPRAL®.
HOW SUPPLIED
CAMPRAL® 333 mg tablets are enteric-coated, white, round, biconvex tablets, identified with “333” debossed on one side.
Opaque HDPE bottles of 180-NDC #0456-3330-01
Dose Pak of 180-NDC #0456-3330-60
10 X 10 Unit Dose-NDC #0456-3330-63
Rx only
Storage:
Store at 25ºC (77ºF); excursions permitted to 15º - 30ºC (59º - 86ºF).
Manufactured by:
Merck Santé s.a.s.
Subsidiary of Merck KGaA, Darmstadt, Germany
37, rue Saint-Romain
69008 LYON FRANCE
Manufactured for FOREST PHARMACEUTICALS, Inc. Subsidiary of Forest Laboratories, Inc.
St. Louis, MO 63045
08/05
| Campral (acamprosate calcium) | |||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||
Revised: 10/2007FOREST PHARMACEUTICALS, Inc.