COMIRNATY- covid-19 vaccine, mrna injection, suspension
Pfizer Laboratories Div Pfizer Inc
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use COMIRNATY safely and effectively. See full prescribing information for COMIRNATY.
COMIRNATY® (COVID-19 Vaccine, mRNA) suspension for injection, for intramuscular use Initial U.S. Approval: 2021 INDICATIONS AND USAGECOMIRNATY is a vaccine indicated for active immunization to prevent coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in individuals 16 years of age and older. (1) DOSAGE AND ADMINISTRATIONDOSAGE FORMS AND STRENGTHSSuspension for injection. After preparation, a single dose is 0.3 mL. (3) CONTRAINDICATIONSKnown history of a severe allergic reaction (e.g., anaphylaxis) to any component of COMIRNATY. (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or VAERS at 1-800-822-7967 or http://vaers.hhs.gov. See 17 for PATIENT COUNSELING INFORMATION. Revised: 8/2021 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
COMIRNATY is a vaccine indicated for active immunization to prevent coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in individuals 16 years of age and older.
2 DOSAGE AND ADMINISTRATION
For intramuscular injection only.
2.1 Preparation for Administration
Prior to Dilution
- COMIRNATY Multiple Dose Vial contains a volume of 0.45 mL, supplied as a frozen suspension that does not contain preservative. Each vial must be thawed and diluted prior to administration.
- Vials may be thawed in the refrigerator [2°C to 8°C (35°F to 46°F)] or at room temperature [up to 25°C (77°F)] [see How Supplied/Storage and Handling (16)].
- Refer to thawing instructions in the panels below.
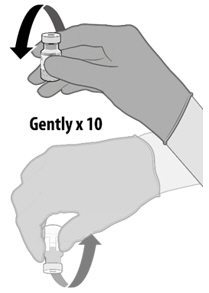
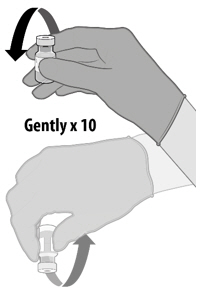
Dilution
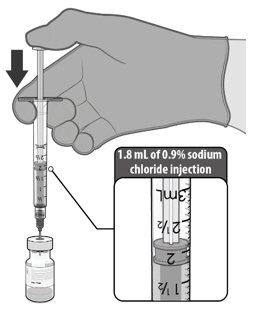
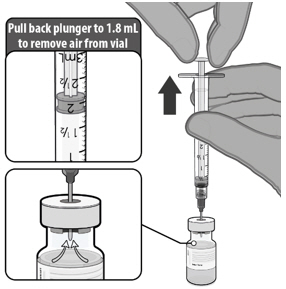
- Dilute the vial contents using 1.8 mL of sterile 0.9% Sodium Chloride Injection, USP to form COMIRNATY. Do not add more than 1.8 mL of diluent.
- ONLY use sterile 0.9% Sodium Chloride Injection, USP as the diluent. Do not use bacteriostatic 0.9% Sodium Chloride Injection or any other diluent.
- Vials of sterile 0.9% Sodium Chloride Injection, USP are provided but shipped separately. Use the provided diluent or another sterile 0.9% Sodium Chloride Injection, USP as the diluent.
- Provided diluent vials are single-use only; discard after 1.8 mL is withdrawn.
- If another sterile 0.9% Sodium Chloride Injection, USP is used as the diluent, discard after 1.8 mL is withdrawn.
- Do not dilute more than 1 vial of COMIRNATY using the same diluent vial.
- After dilution, 1 vial of COMIRNATY contains 6 doses of 0.3 mL each.
- Refer to dilution and dose preparation instructions in the panels below.
| THAWING PRIOR TO DILUTION | |
 |
|
 |
|
| DILUTION | |
 |
|
 |
|
 |
|
 |
|
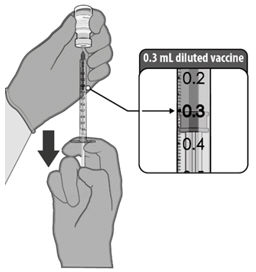
| PREPARATION OF INDIVIDUAL 0.3 mL DOSES OF COMIRNATY | |
 |
|
After dilution, vials of COMIRNATY contain 6 doses of 0.3 mL of vaccine. Low dead-volume syringes and/or needles can be used to extract 6 doses from a single vial. If standard syringes and needles are used, there may not be sufficient volume to extract a sixth dose from a single vial. Irrespective of the type of syringe and needle,
- each dose must contain 0.3 mL of vaccine.
- if the amount of vaccine remaining in the vial cannot provide a full dose of 0.3 mL, discard the vial and any excess volume.
- do not pool excess vaccine from multiple vials.
2.2 Administration Information
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The vaccine will be an off-white suspension. Do not administer if vaccine is discolored or contains particulate matter.
Administer a single 0.3 mL dose of COMIRNATY intramuscularly.
2.3 Vaccination Schedule
COMIRNATY is administered intramuscularly as a series of 2 doses (0.3 mL each) 3 weeks apart.
There are no data available on the interchangeability of COMIRNATY with other COVID-19 vaccines to complete the vaccination series. Individuals who have received 1 dose of COMIRNATY should receive a second dose of COMIRNATY to complete the vaccination series.
3 DOSAGE FORMS AND STRENGTHS
COMIRNATY is a suspension for injection. After preparation, a single dose is 0.3 mL.
4 CONTRAINDICATIONS
Do not administer COMIRNATY to individuals with known history of a severe allergic reaction (e.g., anaphylaxis) to any component of the COMIRNATY [see Description (11)].
5 WARNINGS AND PRECAUTIONS
5.1 Management of Acute Allergic Reactions
Appropriate medical treatment used to manage immediate allergic reactions must be immediately available in the event an acute anaphylactic reaction occurs following administration of COMIRNATY.
5.2 Myocarditis and Pericarditis
Postmarketing data demonstrate increased risks of myocarditis and pericarditis, particularly within 7 days following the second dose. The observed risk is higher among males under 40 years of age than among females and older males. The observed risk is highest in males 12 through 17 years of age. Although some cases required intensive care support, available data from short-term follow-up suggest that most individuals have had resolution of symptoms with conservative management. Information is not yet available about potential long-term sequelae. The CDC has published considerations related to myocarditis and pericarditis after vaccination, including for vaccination of individuals with a history of myocarditis or pericarditis (https://www.cdc.gov/vaccines/covid-19/clinical-considerations/myocarditis.html).
5.3 Syncope
Syncope (fainting) may occur in association with administration of injectable vaccines, including COMIRNATY. Procedures should be in place to avoid injury from fainting.
6 ADVERSE REACTIONS
In clinical studies, the most commonly reported (≥10%) adverse reactions in participants 16 through 55 years of age following any dose were pain at the injection site (88.6%), fatigue (70.1%), headache (64.9%), muscle pain (45.5%), chills (41.5%), joint pain (27.5%), fever (17.8%), and injection site swelling (10.6%).
In clinical studies, the most commonly reported (≥10%) adverse reactions in participants 56 years of age and older following any dose were pain at the injection site (78.2%), fatigue (56.9%), headache, (45.9%), muscle pain (32.5%), chills (24.8%), joint pain (21.5%), injection site swelling (11.8%), fever (11.5%), and injection site redness (10.4%).
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice.
The safety of COMIRNATY was evaluated in participants 16 years of age and older in 2 clinical studies conducted in Germany (Study 1), United States, Argentina, Brazil, Turkey, South Africa, and Germany (Study 2). Study BNT162-01 (Study 1) was a Phase 2-part, dose-escalation trial that enrolled 60 participants, 18 through 55 years of age and 36 participants, 56 through 85 years of age. Study C4591001 (Study 2) is a Phase 1/2/3 multicenter, multinational, randomized, saline placebo-controlled, double-blinded (Phase 2/3), dose-finding, vaccine candidate-selection and efficacy study that has enrolled approximately 44,047 participants (22,026 COMIRNATY; 22,021 placebo) 16 years of age or older (including 378 and 376 participants 16 through 17 years of age in the vaccine and placebo groups, respectively). Upon issuance of the Emergency Use Authorization (December 11, 2020) for COMIRNATY, participants were unblinded to offer placebo participants COMIRNATY. Participants were unblinded in a phased manner over a period of months to offer placebo participants COMIRNATY. Study 2 also included 200 participants with confirmed stable human immunodeficiency virus (HIV) infection; HIV-positive participants are included in safety population disposition but are summarized separately in safety analyses. Confirmed stable HIV infection was defined as documented viral load <50 copies/mL and CD4 count >200 cells/mm3 within 6 months before enrollment, and on stable antiretroviral therapy for at least 6 months.
At the time of the analysis of the ongoing Study 2 with a data cut-off of March 13, 2021, there were 25,651 (58.2%) participants (13,031 COMIRNATY and 12,620 placebo) 16 years of age and older followed for ≥4 months after the second dose.
Participants 16 years and older in the reactogenicity subset were monitored for solicited local and systemic reactions and use of antipyretic medication after each vaccination in an electronic diary. Participants are being monitored for unsolicited adverse events, including serious adverse events, throughout the study [from Dose 1 through 1 month (all unsolicited adverse events) or 6 months (serious adverse events) after the last vaccination].
Demographic characteristics in Study 2 were generally similar with regard to age, gender, race, and ethnicity among participants who received COMIRNATY and those who received placebo. Overall, among the total participants who received either COMIRNATY or placebo, 50.9% were male, 49.1% were female, 79.3% were 16 through 64 years of age, 20.7% were 65 years of age and older, 82.0% were White, 9.6% were Black or African American, 25.9% were Hispanic/Latino, 4.3% were Asian, and 1.0% were American Indian or Alaska Native.
Local and Systemic Adverse Reactions Solicited in the Study 2
Table 1 and Table 2 present the frequency and severity of reported solicited local and systemic reactions, respectively, within 7 days following each dose of COMIRNATY and placebo in the subset of participants 16 through 55 years of age included in the safety population who were monitored for reactogenicity with an electronic diary.
Table 3 and Table 4 present the frequency and severity of reported solicited local and systemic reactions, respectively, within 7 days of each dose of COMIRNATY and placebo for participants 56 years of age and older.
In participants 16 through 55 years of age after receiving Dose 2, the mean duration of pain at the injection site was 2.5 days (range 1 to 70 days), for redness 2.2 days (range 1 to 9 days), and for swelling 2.1 days (range 1 to 8 days) for participants in the COMIRNATY group. In participants 56 years of age and older after receiving Dose 2, the mean duration of pain at the injection site was 2.4 days (range 1 to 36 days), for redness 3.0 days (range 1 to 34 days), and for swelling 2.6 days (range 1 to 34 days) for participants in the COMIRNATY group.
| COMIRNATY Dose 1 N†=2899 n‡ (%) | Placebo Dose 1 N†=2908 n‡ (%) | COMIRNATY Dose 2 N†=2682 n‡ (%) | Placebo Dose 2 N†=2684 n‡ (%) |
|
|---|---|---|---|---|
| Notes: Reactions were collected in the electronic diary (e-diary) from Day 1 to Day 7 after vaccination. | ||||
| No Grade 4 solicited local reactions were reported in participants 16 through 55 years of age. | ||||
|
||||
| Redness§ | ||||
| Any (>2.0 cm) | 156 (5.4) | 28 (1.0) | 151 (5.6) | 18 (0.7) |
| Mild | 113 (3.9) | 19 (0.7) | 90 (3.4) | 12 (0.4) |
| Moderate | 36 (1.2) | 6 (0.2) | 50 (1.9) | 6 (0.2) |
| Severe | 7 (0.2) | 3 (0.1) | 11 (0.4) | 0 |
| Swelling§ | ||||
| Any (>2.0 cm) | 184 (6.3) | 16 (0.6) | 183 (6.8) | 5 (0.2) |
| Mild | 124 (4.3) | 6 (0.2) | 110 (4.1) | 3 (0.1) |
| Moderate | 54 (1.9) | 8 (0.3) | 66 (2.5) | 2 (0.1) |
| Severe | 6 (0.2) | 2 (0.1) | 7 (0.3) | 0 |
| Pain at the injection site¶ | ||||
| Any | 2426 (83.7) | 414 (14.2) | 2101 (78.3) | 312 (11.6) |
| Mild | 1464 (50.5) | 391 (13.4) | 1274 (47.5) | 284 (10.6) |
| Moderate | 923 (31.8) | 20 (0.7) | 788 (29.4) | 28 (1.0) |
| Severe | 39 (1.3) | 3 (0.1) | 39 (1.5) | 0 |
| COMIRNATY Dose 1 N†=2899 n‡ (%) | Placebo Dose 1 N†=2908 n‡ (%) | COMIRNATY Dose 2 N†=2682 n‡ (%) | Placebo Dose 2 N†=2684 n‡ (%) |
|
|---|---|---|---|---|
| Notes: Reactions and use of antipyretic or pain medication were collected in the electronic diary (e-diary) from Day 1 to Day 7 after each dose. | ||||
| No Grade 4 solicited systemic reactions were reported in participants 16 through 55 years of age. | ||||
|
||||
| Fever | ||||
| ≥38.0°C | 119 (4.1) | 25 (0.9) | 440 (16.4) | 11 (0.4) |
| ≥38.0°C to 38.4°C | 86 (3.0) | 16 (0.6) | 254 (9.5) | 5 (0.2) |
| >38.4°C to 38.9°C | 25 (0.9) | 5 (0.2) | 146 (5.4) | 4 (0.1) |
| >38.9°C to 40.0°C | 8 (0.3) | 4 (0.1) | 39 (1.5) | 2 (0.1) |
| >40.0°C | 0 | 0 | 1 (0.0) | 0 |
| Fatigue§ | ||||
| Any | 1431 (49.4) | 960 (33.0) | 1649 (61.5) | 614 (22.9) |
| Mild | 760 (26.2) | 570 (19.6) | 558 (20.8) | 317 (11.8) |
| Moderate | 630 (21.7) | 372 (12.8) | 949 (35.4) | 283 (10.5) |
| Severe | 41 (1.4) | 18 (0.6) | 142 (5.3) | 14 (0.5) |
| Headache§ | ||||
| Any | 1262 (43.5) | 975 (33.5) | 1448 (54.0) | 652 (24.3) |
| Mild | 785 (27.1) | 633 (21.8) | 699 (26.1) | 404 (15.1) |
| Moderate | 444 (15.3) | 318 (10.9) | 658 (24.5) | 230 (8.6) |
| Severe | 33 (1.1) | 24 (0.8) | 91 (3.4) | 18 (0.7) |
| Chills§ | ||||
| Any | 479 (16.5) | 199 (6.8) | 1015 (37.8) | 114 (4.2) |
| Mild | 338 (11.7) | 148 (5.1) | 477 (17.8) | 89 (3.3) |
| Moderate | 126 (4.3) | 49 (1.7) | 469 (17.5) | 23 (0.9) |
| Severe | 15 (0.5) | 2 (0.1) | 69 (2.6) | 2 (0.1) |
| Vomiting¶ | ||||
| Any | 34 (1.2) | 36 (1.2) | 58 (2.2) | 30 (1.1) |
| Mild | 29 (1.0) | 30 (1.0) | 42 (1.6) | 20 (0.7) |
| Moderate | 5 (0.2) | 5 (0.2) | 12 (0.4) | 10 (0.4) |
| Severe | 0 | 1 (0.0) | 4 (0.1) | 0 |
| Diarrhea# | ||||
| Any | 309 (10.7) | 323 (11.1) | 269 (10.0) | 205 (7.6) |
| Mild | 251 (8.7) | 264 (9.1) | 219 (8.2) | 169 (6.3) |
| Moderate | 55 (1.9) | 58 (2.0) | 44 (1.6) | 35 (1.3) |
| Severe | 3 (0.1) | 1 (0.0) | 6 (0.2) | 1 (0.0) |
| New or worsened muscle pain§ | ||||
| Any | 664 (22.9) | 329 (11.3) | 1055 (39.3) | 237 (8.8) |
| Mild | 353 (12.2) | 231 (7.9) | 441 (16.4) | 150 (5.6) |
| Moderate | 296 (10.2) | 96 (3.3) | 552 (20.6) | 84 (3.1) |
| Severe | 15 (0.5) | 2 (0.1) | 62 (2.3) | 3 (0.1) |
| New or worsened joint pain§ | ||||
| Any | 342 (11.8) | 168 (5.8) | 638 (23.8) | 147 (5.5) |
| Mild | 200 (6.9) | 112 (3.9) | 291 (10.9) | 82 (3.1) |
| Moderate | 137 (4.7) | 55 (1.9) | 320 (11.9) | 61 (2.3) |
| Severe | 5 (0.2) | 1 (0.0) | 27 (1.0) | 4 (0.1) |
| Use of antipyretic or pain medicationÞ | 805 (27.8) | 398 (13.7) | 1213 (45.2) | 320 (11.9) |
| COMIRNATY Dose 1 N†=2008 n‡ (%) | Placebo Dose 1 N†=1989 n‡ (%) | COMIRNATY Dose 2 N†=1860 n‡ (%) | Placebo Dose 2 N†=1833 n‡ (%) |
|
|---|---|---|---|---|
| Notes: Reactions were collected in the electronic diary (e-diary) from Day 1 to Day 7 after vaccination. | ||||
| No Grade 4 solicited local reactions were reported in participants 56 years of age and older. | ||||
|
||||
| Redness§ | ||||
| Any (>2.0 cm) | 106 (5.3) | 20 (1.0) | 133 (7.2) | 14 (0.8) |
| Mild | 71 (3.5) | 13 (0.7) | 65 (3.5) | 10 (0.5) |
| Moderate | 30 (1.5) | 5 (0.3) | 58 (3.1) | 3 (0.2) |
| Severe | 5 (0.2) | 2 (0.1) | 10 (0.5) | 1 (0.1) |
| Swelling§ | ||||
| Any (>2.0 cm) | 141 (7.0) | 23 (1.2) | 145 (7.8) | 13 (0.7) |
| Mild | 87 (4.3) | 11 (0.6) | 80 (4.3) | 5 (0.3) |
| Moderate | 52 (2.6) | 12 (0.6) | 61 (3.3) | 7 (0.4) |
| Severe | 2 (0.1) | 0 | 4 (0.2) | 1 (0.1) |
| Pain at the injection site¶ | ||||
| Any (>2.0 cm) | 1408 (70.1) | 185 (9.3) | 1230 (66.1) | 143 (7.8) |
| Mild | 1108 (55.2) | 177 (8.9) | 873 (46.9) | 138 (7.5) |
| Moderate | 296 (14.7) | 8 (0.4) | 347 (18.7) | 5 (0.3) |
| Severe | 4 (0.2) | 0 | 10 (0.5) | 0 |
| COMIRNATY Dose 1 N†=2008 n‡ (%) | Placebo Dose 1 N†=1989 n‡ (%) | COMIRNATY Dose 2 N†=1860 n‡ (%) | Placebo Dose 2 N†=1833 n‡ (%) |
|
|---|---|---|---|---|
| Notes: Reactions and use of antipyretic or pain medication were collected in the electronic diary (e-diary) from Day 1 to Day 7 after each dose. | ||||
| The only Grade 4 solicited systemic reaction reported in participants 56 years of age and older was fatigue. | ||||
|
||||
| Fever | ||||
| ≥38.0°C | 26 (1.3) | 8 (0.4) | 219 (11.8) | 4 (0.2) |
| ≥38.0°C to 38.4°C | 23 (1.1) | 3 (0.2) | 158 (8.5) | 2 (0.1) |
| >38.4°C to 38.9°C | 2 (0.1) | 3 (0.2) | 54 (2.9) | 1 (0.1) |
| >38.9°C to 40.0°C | 1 (0.0) | 2 (0.1) | 7 (0.4) | 1 (0.1) |
| >40.0°C | 0 | 0 | 0 | 0 |
| Fatigue§ | ||||
| Any | 677 (33.7) | 447 (22.5) | 949 (51.0) | 306 (16.7) |
| Mild | 415 (20.7) | 281 (14.1) | 391 (21.0) | 183 (10.0) |
| Moderate | 259 (12.9) | 163 (8.2) | 497 (26.7) | 121 (6.6) |
| Severe | 3 (0.1) | 3 (0.2) | 60 (3.2) | 2 (0.1) |
| Grade 4 | 0 | 0 | 1 (0.1) | 0 |
| Headache§ | ||||
| Any | 503 (25.0) | 363 (18.3) | 733 (39.4) | 259 (14.1) |
| Mild | 381 (19.0) | 267 (13.4) | 464 (24.9) | 189 (10.3) |
| Moderate | 120 (6.0) | 93 (4.7) | 256 (13.8) | 65 (3.5) |
| Severe | 2 (0.1) | 3 (0.2) | 13 (0.7) | 5 (0.3) |
| Chills§ | ||||
| Any | 130 (6.5) | 69 (3.5) | 435 (23.4) | 57 (3.1) |
| Mild | 102 (5.1) | 49 (2.5) | 229 (12.3) | 45 (2.5) |
| Moderate | 28 (1.4) | 19 (1.0) | 185 (9.9) | 12 (0.7) |
| Severe | 0 | 1 (0.1) | 21 (1.1) | 0 |
| Vomiting¶ | ||||
| Any | 10 (0.5) | 9 (0.5) | 13 (0.7) | 5 (0.3) |
| Mild | 9 (0.4) | 9 (0.5) | 10 (0.5) | 5 (0.3) |
| Moderate | 1 (0.0) | 0 | 1 (0.1) | 0 |
| Severe | 0 | 0 | 2 (0.1) | 0 |
| Diarrhea# | ||||
| Any | 168 (8.4) | 130 (6.5) | 152 (8.2) | 102 (5.6) |
| Mild | 137 (6.8) | 109 (5.5) | 125 (6.7) | 76 (4.1) |
| Moderate | 27 (1.3) | 20 (1.0) | 25 (1.3) | 22 (1.2) |
| Severe | 4 (0.2) | 1 (0.1) | 2 (0.1) | 4 (0.2) |
| New or worsened muscle pain§ | ||||
| Any | 274 (13.6) | 165 (8.3) | 537 (28.9) | 99 (5.4) |
| Mild | 183 (9.1) | 111 (5.6) | 229 (12.3) | 65 (3.5) |
| Moderate | 90 (4.5) | 51 (2.6) | 288 (15.5) | 33 (1.8) |
| Severe | 1 (0.0) | 3 (0.2) | 20 (1.1) | 1 (0.1) |
| New or worsened joint pain§ | ||||
| Any | 175 (8.7) | 124 (6.2) | 353 (19.0) | 72 (3.9) |
| Mild | 119 (5.9) | 78 (3.9) | 183 (9.8) | 44 (2.4) |
| Moderate | 53 (2.6) | 45 (2.3) | 161 (8.7) | 27 (1.5) |
| Severe | 3 (0.1) | 1 (0.1) | 9 (0.5) | 1 (0.1) |
| Use of antipyretic or pain medicationÞ | 382 (19.0) | 224 (11.3) | 688 (37.0) | 170 (9.3) |
In participants with chronic, stable HIV infection the frequencies of solicited local and systemic adverse reactions were similar to or lower than those observed for all participants 16 years of age and older.
Unsolicited Adverse Events
Overall, 11,253 (51.1%) participants in the COMIRNATY group and 11,316 (51.4%) participants in the placebo group had follow-up time between ≥4 months to <6 months after Dose 2 in the blinded placebo-controlled follow-up period with an additional 1,778 (8.1%) and 1,304 (5.9%) with ≥6 months of blinded follow-up time in the COMIRNATY and placebo groups, respectively.
A total of 12,006 (54.5%) participants originally randomized to COMIRNATY had ≥6 months total (blinded and unblinded) follow-up after Dose 2.
In an analysis of all unsolicited adverse events reported following any dose, through 1 month after Dose 2, in participants 16 years of age and older (N=43,847; 21,926 COMIRNATY group vs. 21,921 placebo group), those assessed as adverse reactions not already captured by solicited local and systemic reactions were nausea (274 vs. 87), malaise (130 vs. 22), lymphadenopathy (83 vs. 7), asthenia (76 vs. 25), decreased appetite (39 vs. 9), hyperhidrosis (31 vs. 9), lethargy (25 vs. 6), and night sweats (17 vs. 3).
In analyses of all unsolicited adverse events in Study 2 from Dose 1 up to the participant unblinding date, 58.2% of study participants had at least 4 months of follow-up after Dose 2. Among participants 16 through 55 years of age who received at least one dose of study vaccine, 12,995 of whom received COMIRNATY and 13,026 of whom received placebo, unsolicited adverse events were reported by 4,396 (33.8%) participants in the COMIRNATY group and 2,136 (16.4%) participants in the placebo group. In a similar analysis in participants 56 years of age and older that included 8,931 COMIRNATY recipients and 8,895 placebo recipients, unsolicited adverse events were reported by 2,551 (28.6%) participants in the COMIRNATY group and 1,432 (16.1%) participants in the placebo group. Among participants with confirmed stable HIV infection that included 100 COMIRNATY recipients and 100 placebo recipients, unsolicited adverse events were reported by 29 (29%) participants in the COMIRNATY group and 15 (15%) participants in the placebo group. The higher frequency of reported unsolicited adverse events among COMIRNATY recipients compared to placebo recipients was primarily attributed to events that are consistent with adverse reactions solicited among participants in the reactogenicity subset (Table 3 and Table 4).
Throughout the placebo-controlled safety follow-up period, Bell's palsy (facial paralysis) was reported by 4 participants in the COMIRNATY group and 2 participants in the placebo group. Onset of facial paralysis was Day 37 after Dose 1 (participant did not receive Dose 2) and Days 3, 9, and 48 after Dose 2. In the placebo group the onset of facial paralysis was Day 32 and Day 102. Currently available information is insufficient to determine a causal relationship with the vaccine. In the analysis of blinded, placebo-controlled follow-up, there were no other notable patterns or numerical imbalances between treatment groups for specific categories of non-serious adverse events (including other neurologic or neuro-inflammatory, and thrombotic events) that would suggest a causal relationship to COMIRNATY. In the analysis of unblinded follow-up, there were no notable patterns of specific categories of non-serious adverse events that would suggest a causal relationship to COMIRNATY.
Serious Adverse Events
In Study 2, among participants 16 through 55 years of age who had received at least 1 dose of vaccine or placebo (COMIRNATY =12,995; placebo = 13,026), serious adverse events from Dose 1 up to the participant unblinding date in ongoing follow-up were reported by 103 (0.8%) COMIRNATY recipients and 117 (0.9%) placebo recipients. In a similar analysis, in participants 56 years of age and older (COMIRNATY = 8,931; placebo = 8,895), serious adverse events were reported by 165 (1.8%) COMIRNATY recipients and 151 (1.7%) placebo recipients who received at least 1 dose of COMIRNATY or placebo, respectively. In these analyses, 58.2% of study participants had at least 4 months of follow-up after Dose 2. Among participants with confirmed stable HIV infection serious adverse events from Dose 1 up to the participant unblinding date in ongoing follow-up were reported by 2 (2%) COMIRNATY recipients and 2 (2%) placebo recipients.
In the analysis of blinded, placebo-controlled follow-up, there were no notable patterns between treatment groups for specific categories of serious adverse events (including neurologic, neuro-inflammatory, and thrombotic events) that would suggest a causal relationship to COMIRNATY. In the analysis of unblinded follow-up, there were no notable patterns of specific categories of serious adverse events that would suggest a causal relationship to COMIRNATY.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postmarketing use of COMIRNATY, including under Emergency Use Authorization. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to vaccine exposure.
Cardiac Disorders: myocarditis, pericarditis
Gastrointestinal Disorders: diarrhea, vomiting
Immune System Disorders: severe allergic reactions, including anaphylaxis, and other hypersensitivity reactions (e.g., rash, pruritus, urticaria, angioedema)
Musculoskeletal and Connective Tissue Disorders: pain in extremity (arm)
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to COMIRNATY during pregnancy. Women who are vaccinated with COMIRNATY during pregnancy are encouraged to enroll in the registry by visiting https://mothertobaby.org/ongoing-study/covid19-vaccines/.
Risk Summary
All pregnancies have a risk of birth defect, loss, or other adverse outcomes. In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. Available data on COMIRNATY administered to pregnant women are insufficient to inform vaccine-associated risks in pregnancy.
A developmental toxicity study has been performed in female rats administered the equivalent of a single human dose of COMIRNATY on 4 occasions; twice prior to mating and twice during gestation. These studies revealed no evidence of harm to the fetus due to the vaccine (see Animal Data).
Data
Animal Data
In a developmental toxicity study, 0.06 mL of a vaccine formulation containing the same quantity of nucleoside-modified messenger ribonucleic acid (mRNA) (30 mcg) and other ingredients included in a single human dose of COMIRNATY was administered to female rats by the intramuscular route on 4 occasions: 21 and 14 days prior to mating, and on gestation days 9 and 20. No vaccine-related adverse effects on female fertility, fetal development, or postnatal development were reported in the study.
8.2 Lactation
Risk Summary
It is not known whether COMIRNATY is excreted in human milk. Data are not available to assess the effects of COMIRNATY on the breastfed infant or on milk production/excretion. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for COMIRNATY and any potential adverse effects on the breastfed child from COMIRNATY or from the underlying maternal condition. For preventive vaccines, the underlying maternal condition is susceptibility to disease prevented by the vaccine.
8.4 Pediatric Use
Safety and effectiveness of COMIRNATY in individuals 16 through 17 years of age is based on safety and effectiveness data in this age group and in adults [see Adverse Reactions (6) and Clinical Studies (14.1)].
The safety and effectiveness of COMIRNATY in individuals younger than 16 years of age have not been established.
8.5 Geriatric Use
Of the total number of COMIRNATY recipients in Study 2 as of March 13, 2021 (N = 22,026), 20.7% (n = 4,552) were 65 years of age and older and 4.2% (n = 925) were 75 years of age and older [see Clinical Studies (14.1)]. No overall differences in safety or effectiveness were observed between these recipients and younger recipients.
11 DESCRIPTION
COMIRNATY (COVID-19 Vaccine, mRNA) is a sterile suspension for injection for intramuscular use. COMIRNATY is supplied as a frozen suspension in multiple dose vials; each vial must be diluted with 1.8 mL of sterile 0.9% Sodium Chloride Injection, USP prior to use to form the vaccine. Each dose of COMIRNATY contains 30 mcg of a nucleoside-modified messenger RNA (mRNA) encoding the viral spike (S) glycoprotein of SARS-CoV-2.
Each 0.3 mL dose of the COMIRNATY also includes the following ingredients: lipids (0.43 mg ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate), 0.05 mg 2-(polyethylene glycol 2000)-N,N-ditetradecylacetamide, 0.09 mg 1,2-distearoyl-sn-glycero-3-phosphocholine, and 0.2 mg cholesterol), 0.01 mg potassium chloride, 0.01 mg monobasic potassium phosphate, 0.36 mg sodium chloride, 0.07 mg dibasic sodium phosphate dihydrate, and 6 mg sucrose. The diluent (0.9% Sodium Chloride Injection, USP) contributes an additional 2.16 mg sodium chloride per dose.
COMIRNATY does not contain preservative.
The vial stoppers are not made with natural rubber latex.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
COMIRNATY has not been evaluated for the potential to cause carcinogenicity, genotoxicity, or impairment of male fertility. In a developmental toxicity study in rats with COMIRNATY there were no vaccine-related effects on female fertility [see Use in Specific Populations (8.1)].
14 CLINICAL STUDIES
Efficacy in Participants 16 Years of Age and Older
Study 2 is an ongoing, multicenter, multinational, randomized, placebo-controlled, observer-blind, dose-finding, vaccine candidate–selection, and efficacy study in participants 12 years of age and older. Randomization was stratified by age: 12 through 15 years of age, 16 through 55 years of age, or 56 years of age and older, with a minimum of 40% of participants in the ≥56-year stratum. The study excluded participants who were immunocompromised and those who had previous clinical or microbiological diagnosis of COVID-19. Participants with preexisting stable disease, defined as disease not requiring significant change in therapy or hospitalization for worsening disease during the 6 weeks before enrollment, were included as were participants with known stable infection with HIV, hepatitis C virus (HCV), or hepatitis B virus (HBV).
In Study 2, based on data accrued through March 13, 2021, approximately 44,000 participants 16 years of age and older were randomized equally and received 2 doses of COMIRNATY or placebo. Participants are planned to be followed for up to 24 months, for assessments of safety and efficacy against COVID-19.
Overall, among the total participants who received COMIRNATY or placebo, 51.4% or 50.3% were male and 48.6% or 49.7% were female, 79.1% or 79.2% were 16 through 64 years of age, 20.9% or 20.8% were 65 years of age and older, 81.9% or 82.1% were White, 9.5% or 9.6% were Black or African American, 1.0% or 0.9% were American Indian or Alaska Native, 4.4% or 4.3% were Asian, 0.3% or 0.2% Native Hawaiian or other Pacific Islander, 25.6% or 25.4% were Hispanic/Latino, 73.9% or 74.1% were non-Hispanic/Latino, 0.5% or 0.5% did not report ethnicity, 46.0% or 45.7% had comorbidities [participants who have 1 or more comorbidities that increase the risk of severe COVID-19 disease: defined as subjects who had at least one of the Charlson comorbidity index category or body mass index (BMI) ≥30 kg/m2], respectively. The mean age at vaccination was 49.8 or 49.7 years and median age was 51.0 or 51.0 in participants who received COMIRNATY or placebo, respectively.
Efficacy Against COVID-19
The population for the analysis of the protocol pre-specified primary efficacy endpoint included 36,621 participants 12 years of age and older (18,242 in the COMIRNATY group and 18,379 in the placebo group) who did not have evidence of prior infection with SARS-CoV-2 through 7 days after the second dose. The population in the protocol pre-specified primary efficacy analysis included all participants 12 years of age and older who had been enrolled from July 27, 2020, and followed for the development of COVID-19 through November 14, 2020. Participants 18 through 55 years of age and 56 years of age and older began enrollment from July 27, 2020, 16 through 17 years of age began enrollment from September 16, 2020, and 12 through 15 years of age began enrollment from October 15, 2020.
For participants without evidence of SARS-CoV-2 infection prior to 7 days after Dose 2, vaccine efficacy against confirmed COVID-19 occurring at least 7 days after Dose 2 was 95.0% (95% credible interval: 90.3, 97.6), which met the pre-specified success criterion. The case split was 8 COVID-19 cases in the COMIRNATY group compared to 162 COVID-19 cases in the placebo group.
The population for the updated vaccine efficacy analysis included participants 16 years of age and older who had been enrolled from July 27, 2020, and followed for the development of COVID-19 during blinded placebo-controlled follow-up through March 13, 2021, representing up to 6 months of follow-up after Dose 2. There were 12,796 (60.8%) participants in the COMIRNATY group and 12,449 (58.7%) in the placebo group followed for ≥4 months after Dose 2 in the blinded placebo-controlled follow-up period.
SARS-CoV-2 variants of concern identified from COVID-19 cases in this study include B.1.1.7 (Alpha) and B.1.351 (Beta). Representation of identified variants among cases in vaccine versus placebo recipients did not suggest decreased vaccine effectiveness against these variants.
The updated vaccine efficacy information is presented in Table 5.
| Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhea; vomiting). | |||
|
|||
| First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence of prior SARS-CoV-2 infection* | |||
| Subgroup | COMIRNATY N†=19,993 Cases n1‡ Surveillance Time§ (n2¶) | Placebo N†=20,118 Cases n1‡ Surveillance Time§ (n2¶) | Vaccine Efficacy % (95% CI#) |
| All participantsf | 77 6.092 (19,711) | 833 5.857 (19,741) | 91.1 (88.8, 93.1) |
| 16 through 64 years | 70 4.859 (15,519) | 709 4.654 (15,515) | 90.5 (87.9, 92.7) |
| 65 years and older | 7 1.233 (4192) | 124 1.202 (4226) | 94.5 (88.3, 97.8) |
| First COVID-19 occurrence from 7 days after Dose 2 in participants with or without* evidence of prior SARS-CoV-2 infection | |||
| Subgroup | COMIRNATY N†=21,047 Cases n1‡ Surveillance Time§ (n2¶) | Placebo N†=21,210 Cases n1‡ Surveillance Time§ (n2¶) | Vaccine Efficacy % (95% CI#) |
| All participants | 81 6.340 (20,533) | 854 6.110 (20,595) | 90.9 (88.5, 92.8) |
| 16 through 64 years | 74 5.073 (16,218) | 726 4.879 (16,269) | 90.2 (87.5, 92.4) |
| 65 years and older | 7 1.267 (4315) | 128 1.232 (4326) | 94.7 (88.7, 97.9) |
Subgroup analyses of vaccine efficacy (although limited by small numbers of cases in some subgroups) did not suggest meaningful differences in efficacy across genders, ethnic groups, geographies, or for participants with obesity or medical comorbidities associated with high risk of severe COVID-19.
Efficacy Against Severe COVID-19
Efficacy analyses of secondary efficacy endpoints supported benefit of COMIRNATY in preventing severe COVID-19. Vaccine efficacy against severe COVID-19 is presented only for participants with or without prior SARS-CoV-2 infection (Table 6) as the COVID-19 case counts in participants without prior SARS-CoV-2 infection were the same as those in participants with or without prior SARS-CoV-2 infection in both the COMIRNATY and placebo groups.
| Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhea; vomiting). | |||
|
|||
| Vaccine Efficacy – First Severe COVID-19 Occurrence | |||
| COMIRNATY Cases n1§ Surveillance Time¶ (n2#) | Placebo Cases n1§ Surveillance Time¶ (n2#) | Vaccine Efficacy % (95% CIÞ) |
|
| 7 days after Dose 2Þ | 1 6.353 (20,540) | 21 6.237 (20,629) | 95.3 (70.9, 99.9) |
| Vaccine Efficacy – First Severe COVID-19 Occurrence Based on CDC Definition | |||
| COMIRNATY Cases n1§ Surveillance Time¶ (n2#) | Placebo Cases n1§ Surveillance Time¶ (n2#) | Vaccine Efficacy % (95% CIÞ) |
|
| 7 days after Dose 2Þ | 0 6.345 (20,513) | 31 6.225 (20,593) | 100 (87.6, 100.0) |
16 HOW SUPPLIED/STORAGE AND HANDLING
COMIRNATY Suspension for Intramuscular Injection, Multiple Dose Vials are supplied in a carton containing 25 multiple dose vials (NDC 0069-1000-03) or 195 multiple dose vials (NDC 0069-1000-02). A 0.9% Sodium Chloride Injection, USP diluent is provided but shipped separately, and should be stored at controlled room temperature 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature]. The provided 0.9% Sodium Chloride Injection, USP diluent will be supplied either as cartons of 10 mL single-use vials manufactured by Hospira, Inc (NDC 0409-4888-10), or 2 mL single-use vials manufactured by Fresenius Kabi USA, LLC (NDC 63323-186-02).
After dilution, 1 vial contains 6 doses of 0.3 mL.
During storage, minimize exposure to room light, and avoid exposure to direct sunlight and ultraviolet light.
Do not refreeze thawed vials.
Frozen Vials Prior to Use
Cartons of COMIRNATY Multiple Dose Vials arrive in thermal containers with dry ice. Once received, remove the vial cartons immediately from the thermal container and preferably store in an ultra-low temperature freezer between -90°C to -60°C (-130°F to -76°F) until the expiry date printed on the label. Alternatively, vials may be stored at -25°C to -15°C (-13°F to 5°F) for up to 2 weeks. Vials must be kept frozen and protected from light, in the original cartons, until ready to use. Vials stored at -25°C to -15°C (-13°F to 5°F) for up to 2 weeks may be returned 1 time to the recommended storage condition of -90°C to -60°C (-130°F to -76°F). Total cumulative time the vials are stored at -25°C to -15°C (-13°F to 5°F) should be tracked and should not exceed 2 weeks.
If an ultra-low temperature freezer is not available, the thermal container in which COMIRNATY arrives may be used as temporary storage when consistently re-filled to the top of the container with dry ice. Refer to the re-icing guidelines packed in the original thermal container for instructions regarding the use of the thermal container for temporary storage. The thermal container maintains a temperature range of -90°C to -60°C (-130°F to -76°F). Storage of the vials between -96°C to -60°C (-141°F to -76°F) is not considered an excursion from the recommended storage condition.
Transportation of Frozen Vials
If local redistribution is needed and full cartons containing vials cannot be transported at -90°C to -60°C (-130°F to -76°F), vials may be transported at -25°C to -15°C (-13°F to 5°F). Any hours used for transport at -25°C to -15°C (-13°F to 5°F) count against the 2-week limit for storage at -25°C to -15°C (-13°F to 5°F). Frozen vials transported at -25°C to -15°C (-13°F to 5°F) may be returned 1 time to the recommended storage condition of -90°C to -60°C (-130°F to -76°F).
Thawed Vials Before Dilution
Thawed Under Refrigeration
Thaw and then store undiluted vials in the refrigerator [2°C to 8°C (35°F to 46°F)] for up to 1 month. A carton of 25 vials or 195 vials may take up to 2 or 3 hours, respectively, to thaw in the refrigerator, whereas a fewer number of vials will thaw in less time.
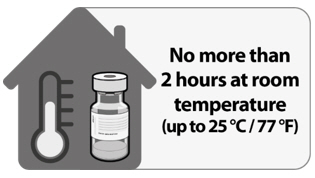
Thawed at Room Temperature
For immediate use, thaw undiluted vials at room temperature [up to 25°C (77°F)] for 30 minutes. Thawed vials can be handled in room light conditions.
Vials must reach room temperature before dilution.
Undiluted vials may be stored at room temperature for no more than 2 hours.
Transportation of Thawed Vials
Available data support transportation of 1 or more thawed vials at 2°C to 8°C (35°F to 46°F) for up to 12 hours.
Vials After Dilution
After dilution, store vials between 2°C to 25°C (35°F to 77°F) and use within 6 hours from the time of dilution. During storage, minimize exposure to room light, and avoid exposure to direct sunlight and ultraviolet light. Any vaccine remaining in vials must be discarded after 6 hours. Do not refreeze.
17 PATIENT COUNSELING INFORMATION
Inform vaccine recipient of the potential benefits and risks of vaccination with COMIRNATY.
Inform vaccine recipient of the importance of completing the two dose vaccination series.
There is a pregnancy exposure registry for COMIRNATY. Encourage individuals exposed to COMIRNATY around the time of conception or during pregnancy to register by visiting https://mothertobaby.org/ongoing-study/covid19-vaccines/.
Advise vaccine recipient to report any adverse events to their healthcare provider or to the Vaccine Adverse Event Reporting System at 1-800-822-7967 and www.vaers.hhs.gov.
This product's labeling may have been updated. For the most recent prescribing information, please visit https://dailymed.nlm.nih.gov/dailymed/.
Manufactured for
BioNTech Manufacturing GmbH
An der Goldgrube 12
55131 Mainz, Germany

Manufactured by
Pfizer Inc., New York, NY 10017
LAB-1448-1.0
US Govt. License No. 2229
PRINCIPAL DISPLAY PANEL - 6 Dose Vial Label
COVID-19 Vaccine, mRNA
COMIRNATY®
Rx only
After dilution, vial contains 6 doses of 0.3 mL
For intramuscular use. Contains no preservative.
DILUTE BEFORE USE. Discard 6 hours after
dilution when stored at 2 to 25°C (35 to 77°F).
Dilution date and time:
US License No. 2229
BioNTech Manufacturing GmbH & Pfizer Inc.

PRINCIPAL DISPLAY PANEL - 195 Vial Carton Label
NDC 0069-1000-02
COVID-19 Vaccine, mRNA
COMIRNATY®
Suspension for Injection, for Intramuscular Use
195 Multiple Dose Vials
(after dilution each vial contains 6 doses of 0.3 mL)
STORAGE: Prior to dilution, store at -90°C to -60°C (-130°F to -76°F). Store in this carton to protect from light.
DOSAGE AND ADMINISTRATION: Diluent (sterile 0.9% Sodium Chloride Injection, USP) supplied separately.
Use the provided diluent or an alternate brand of sterile 0.9% Sodium Chloride Injection, USP.
After dilution, each vial contains 6 doses of 0.3 mL.
Please see prescribing information for additional details including instructions for preparation, dosage and administration.
MUST BE DILUTED BEFORE USE with sterile 0.9% Sodium Chloride Injection, USP.
After dilution, store the vaccine at 2°C to 25°C (35°F to 77°F).
Discard after 6 hours. Contains no preservative.
See prescribing information or scan QR code.
BIONTECH
Pfizer
Manufactured for
BioNTech Manufacturing GmbH
An der Goldgrube 12
55131 Mainz, Germany
Manufactured by
Pfizer Inc.
New York, NY 10017
US License No. 2229
Rx only
LOT:
EXP:
PAA177818
PRINCIPAL DISPLAY PANEL - 25 Vial Carton
NDC 0069-1000-03
COVID-19 Vaccine, mRNA
COMIRNATY®
Suspension for Injection, for Intramuscular Use
25 Multiple Dose Vials
(after dilution each vial contains 6 doses of 0.3 mL)
MUST BE DILUTED BEFORE USE with sterile
0.9% Sodium Chloride Injection, USP.
After dilution, store the vaccine at 2°C to 25°C (35°F to 77°F).
Discard after 6 hours.

| COMIRNATY
covid-19 vaccine, mrna injection, suspension |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Pfizer Laboratories Div Pfizer Inc (134489525) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Manufacturing Belgium NV | 370156507 | ANALYSIS(0069-1000) , MANUFACTURE(0069-1000) , PACK(0069-1000) , LABEL(0069-1000) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pharmacia & Upjohn Company LLC | 618054084 | ANALYSIS(0069-1000) , MANUFACTURE(0069-1000) , PACK(0069-1000) , LABEL(0069-1000) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Wyeth BioPharma Division of Wyeth Pharmaceuticals LLC | 174350868 | ANALYSIS(0069-1000) , API MANUFACTURE(0069-1000) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Inc | 004954111 | ANALYSIS(0069-1000) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Ireland Pharmaceuticals | 985586408 | ANALYSIS(0069-1000) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Hospira Zagreb d.o.o. | 500625201 | ANALYSIS(0069-1000) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| SGS Lab Simon | 283063907 | ANALYSIS(0069-1000) | |