ranexa (Ranolazine) tablet, film coated, extended release
[CV Therapeutics, Inc.]
DESCRIPTION
Ranexa® (ranolazine) is available as an extended-release tablet for oral administration.
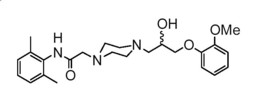
Ranolazine is a racemic mixture and chemically described as 1-piperazineacetamide, N-(2,6-dimethylphenyl)-4-[2-hydroxy-3-(2-methoxyphenoxy)propyl]-, (±)-. It has an empirical formula of C24H33N3O4, a molecular weight of 427.54 g/mole, and the following structural formula:
Ranolazine is a white to off-white solid. Ranolazine is soluble in dichloromethane and methanol; sparingly soluble in tetrahydrofuran, ethanol, acetonitrile, and acetone; slightly soluble in ethyl acetate, isopropanol, toluene, and ethyl ether; and very slightly soluble in water.
Ranexa is available for oral administration as film-coated, extended-release tablets containing 500 mg or 1000 mg of ranolazine. Inactive ingredients of the 500 mg tablet include carnauba wax, hypromellose, magnesium stearate, methacrylic acid copolymer (Type C), microcrystalline cellulose, polyethylene glycol, polysorbate 80, sodium hydroxide, titanium dioxide, and FD&C Yellow #6 Lake.
Inactive ingredients of the 1000 mg tablet include carnauba wax, hypromellose, lactose monohydrate, magnesium stearate, methacrylic acid copolymer (Type C), microcrystalline cellulose, polyethylene glycol, sodium hydroxide, titanium dioxide, triacetin, and Iron Oxide Yellow.
CLINICAL PHARMACOLOGY
Mechanism of Action
Ranexa has antianginal and anti-ischemic effects that do not depend upon reductions in heart rate or blood pressure. The mechanism of action of ranolazine is unknown. It does not increase the rate-pressure product, a measure of myocardial work, at maximal exercise.
Pharmacokinetics
Ranolazine is extensively metabolized in the gut and liver and its absorption is highly variable. For example, at a dose of 1000 mg b.i.d., the mean steady-state Cmax was 2569 ng/mL; 95% of Cmax values were between 420 and 6080 ng/mL. The pharmacokinetics of the (+) R- and (-) S-enantiomers of ranolazine are similar in healthy volunteers. The apparent terminal half-life of ranolazine is 7 hours. Steady state is generally achieved within 3 days of b.i.d. dosing with Ranexa. At steady state over the dose range 500 to 1000 mg b.i.d., Cmax and AUC0-τ increase slightly more than proportionally to dose, 2.2- and 2.4-fold, respectively. With twice daily dosing, the peak/trough ratio of the ranolazine plasma concentration is 1.6 to 3.0.
Absorption and Distribution
After oral administration of Ranexa peak plasma concentrations of ranolazine are reached between 2 and 5 hours. After oral administration of 14C-ranolazine as a solution, 73% of the dose is systemically available as ranolazine or metabolites. The bioavailability of ranolazine from Ranexa relative to that from a solution of ranolazine is 76%. Because ranolazine is a substrate of P-glycoprotein (P-gp), inhibitors of P-gp may increase the absorption of ranolazine.
Food (high-fat breakfast) has no important effect on the Cmax and AUC of ranolazine. Therefore, Ranexa may be taken without regard to meals. Over the concentration range of 0.25 to 10 µg/mL, ranolazine is approximately 62% bound to human plasma proteins.
Metabolism and Excretion
Following a single oral dose of ranolazine solution, approximately 75% of the dose is excreted in urine and 25% in feces. Ranolazine is metabolized rapidly and extensively in the liver and intestine; less than 5% is excreted unchanged in urine and feces. The pharmacologic activity of the metabolites has not been well characterized. After dosing to steady state with 500 mg to 1500 mg b.i.d., the four most abundant metabolites in plasma have AUC values ranging from about 5 to 33% that of ranolazine, and display apparent half-lives ranging from 6 to 22 hours. Ranolazine is metabolized mainly by CYP3A and to a lesser extent by CYP2D6.
Special Populations
Age, Gender, and Race
A population pharmacokinetic evaluation of data from patients and healthy volunteers showed no clinically significant age- or gender-related effects on the pharmacokinetics of ranolazine. Dosage modifications for age or gender are, therefore, not required (see DOSAGE AND ADMINISTRATION). Requirements for dosage modification based on race have not been adequately assessed.
Pediatric
The pharmacokinetics of ranolazine have not been investigated in patients < 18 years of age.
Renal Insufficiency
In a pharmacokinetic study in patients with varying degrees of renal impairment, ranolazine plasma levels appeared to increase about 50%. The pharmacokinetics of ranolazine in patients on dialysis have not been assessed. In six subjects with severe renal impairment on Ranexa 500 mg b.i.d., mean diastolic blood pressure increased approximately 10 to 15 mm Hg.
Hepatic Insufficiency
The disposition of ranolazine administered in a dose of Ranexa 500 mg b.i.d. was studied in 16 subjects with mild or moderate hepatic impairment and 16 healthy volunteers. The plasma concentrations of ranolazine were increased in the subjects with mild (Child-Pugh Class A) and moderate (Child-Pugh Class B) liver impairment by factors of 1.3 and 1.6, respectively, relative to the healthy volunteers. In the same study, patients with mild and moderate hepatic impairment had increases in QTc that were larger than those of normal subjects at the same plasma ranolazine level (see Pharmacodynamic Effects).
Congestive Heart Failure
A population pharmacokinetic evaluation showed that congestive heart failure (CHF) (NYHA Class I to IV) had no significant effect on ranolazine pharmacokinetics.
Ranexa had minimal effects on heart rate and blood pressure in patients with angina and CHF NYHA Class I or II, and also in a study of 85 patients with CHF NYHA Class III or IV.
Diabetes Mellitus
A population pharmacokinetic evaluation of data from angina patients and healthy subjects showed no effect of diabetes on ranolazine pharmacokinetics.
Drug-Drug Interactions
Effects of Other Drugs on Ranolazine
In vivo studies in healthy volunteers confirm that ranolazine is primarily metabolized by CYP3A. Plasma levels of ranolazine with Ranexa 1000 mg b.i.d. are increased 3.2-fold by the potent CYP3A inhibitor ketoconazole co-administered at a dose of 200 mg b.i.d. Plasma levels of ranolazine with Ranexa 1000 mg b.i.d. are increased about 1.8- to 2.3-fold by the moderately potent CYP3A inhibitor diltiazem given in daily doses from 180 to 360 mg, respectively. Plasma levels of ranolazine with Ranexa 750 mg b.i.d. are increased about 2-fold by the CYP3A and P-gp inhibitor verapamil given at a dose of 120 mg t.i.d.
Ketoconazole, diltiazem, verapamil (also a P-gp inhibitor) and other potent or moderately potent CYP3A inhibitors should not be co-administered with Ranexa (see WARNINGS).
Less potent CYP3A inhibitors such as simvastatin (20 mg q.d.) and cimetidine (400 mg t.i.d.) do not increase the exposure to ranolazine in healthy volunteers receiving Ranexa.
No specific studies of ranolazine with CYP3A inducers have been conducted.
In vitro studies indicate that ranolazine is a P-gp substrate. Caution should be exercised when co-administering ranolazine and P-gp inhibitors such as ritonavir and cyclosporine (see PRECAUTIONS). The potent CYP2D6 inhibitor paroxetine, at a dose level of 20 mg q.d., increased ranolazine concentrations 1.2-fold in healthy volunteers receiving Ranexa 1000 mg b.i.d. No dose adjustment of Ranexa is necessary when it is co-administered with drugs inhibiting CYP2D6.
Plasma concentrations of ranolazine are not significantly altered by concomitant digoxin at 0.125 mg q.d.
Effects of Ranolazine on Other Drugs
In vitro studies indicate that ranolazine and its O-demethylated metabolite are inhibitors of CYP3A and CYP2D6. Ranolazine and its most abundant metabolites are not known to inhibit the metabolism of substrates for CYP1A2, 2C9, 2C19 or 2E1 in human liver microsomes, suggesting that ranolazine is unlikely to alter the pharmacokinetics of drugs metabolized by these enzymes.
The plasma levels of simvastatin, a CYP3A substrate, and its active metabolite are each increased about 2-fold in healthy subjects receiving simvastatin 80 mg q.d. and Ranexa 1000 mg b.i.d. (see DOSAGE AND ADMINISTRATION).
The pharmacokinetics of diltiazem are not affected by ranolazine in healthy volunteers receiving diltiazem 60 mg t.i.d. and Ranexa 1000 mg b.i.d.
The inhibitory effects of ranolazine on CYP2D6 have been evaluated in extensive metabolizers of dextromethorphan. The study showed that ranolazine and/or metabolites partially inhibit CYP2D6. Concomitant use of Ranexa with other drugs metabolized by CYP2D6, such as tricyclic antidepressants and antipsychotics, has not been formally studied, but lower doses of the other drug than usually prescribed may be required in the presence of ranolazine (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
In vitro studies suggest that ranolazine is a P-gp inhibitor. In vivo ranolazine increases digoxin concentrations 1.5-fold in healthy volunteers receiving Ranexa 1000 mg b.i.d. and digoxin 0.125 mg q.d. The dose of digoxin may have to be adjusted when ranolazine is co-administered with digoxin (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Pharmacodynamic Effects
Hemodynamic Effects
Minimal changes in mean heart rate (< 2 bpm) and systolic blood pressure (< 3 mm Hg) were observed in patients with chronic angina treated with Ranexa in controlled studies. Similar results were observed in subgroups of patients with CHF NYHA Class I or II, diabetes, or reactive airway disease, and in elderly patients. In six subjects with severe renal impairment on Ranexa 500 mg b.i.d., mean diastolic blood pressure increased approximately 10 to 15 mm Hg.
Electrocardiographic Effects
Dose and plasma concentration-related increases in the QTc interval (see WARNINGS), reductions in T wave amplitude and, in some cases, notched T waves, have been observed in patients treated with ranolazine. These effects are believed to be caused by ranolazine and not by its metabolites. The relationship between the change in QTc and ranolazine plasma concentrations is linear with a slope of about 2.6 msec/1000 ng/mL ranolazine. The variable blood levels attained after a given dose of ranolazine give a wide range of effects on QTc. At Tmax following repeat dosing at 1000 mg b.i.d., the mean change in QTc is about 6 msec, but in the 5% of the population with the highest plasma concentrations, the prolongation of QTc is at least 15 msec. In subjects with mild or moderate hepatic impairment, the relationship between plasma level of ranolazine and QTc is much steeper (see WARNINGS and CONTRAINDICATIONS).
Congestive heart failure, diabetes, renal impairment, and gender did not alter the slope of the QTc-concentration relationship of ranolazine.
CLINICAL STUDIES
Ranexa has been evaluated in patients with chronic angina who remained symptomatic despite treatment with the maximum dose of an antianginal agent. In the ERICA (Efficacy of Ranolazine In Chronic Angina) trial, 565 patients were randomized to receive an initial dose of Ranexa 500 mg b.i.d. or placebo for 1 week, followed by 6 weeks of treatment with Ranexa 1000 mg b.i.d. or placebo, in addition to concomitant treatment with amlodipine 10 mg q.d. In addition, 45% of the study population also received long-acting nitrates. Sublingual nitrates were used as needed to treat angina episodes. Results are shown in Table 1. Statistically significant decreases in angina attack frequency (p = 0.028) and nitroglycerin use (p = 0.014) were observed with Ranexa compared to placebo. These treatment effects appeared consistent across age and use of long-acting nitrates. The mean magnitude of effect was smaller in women.
| Placebo | Ranexa 1000 mg b.i.d. |
||
| Angina Frequency (attacks/week) | N | 281 | 277 |
| Mean | 4.3 | 3.3 | |
| Median | 2.4 | 2.2 | |
| Nitroglycerin Use (doses/week) | N | 281 | 277 |
| Mean | 3.6 | 2.7 | |
| Median | 1.7 | 1.3 | |
CARISA was a study in 823 chronic angina patients randomized to receive 12 weeks of treatment with twice-daily Ranexa 750 mg, 1000 mg, or placebo who also continued on daily doses of atenolol 50 mg, amlodipine 5 mg, or diltiazem CD 180 mg. Sublingual nitrates were used in this study as needed.
In this trial, statistically significant (p < 0.05) increases in modified Bruce treadmill exercise duration and time to angina were observed for each Ranexa dose versus placebo, at both trough (12 hours after dosing) and peak (4 hours after dosing) plasma levels, with minimal effects on blood pressure and heart rate. The changes vs placebo in exercise parameters are given in Table 2. Exercise treadmill results showed no increase in effect on exercise at the 1000 mg dose compared to the 750 mg dose. The effects of Ranexa on angina frequency and nitroglycerin use are shown in Table 3.
| Mean Difference from Placebo (sec) | ||
| Study | CARISA (N = 791) | |
| Ranexa Dose | 750 mg b.i.d. | 1000 mg b.i.d. |
|
*p-value ≤ 0.05 **p-value ≤ 0.005 |
||
| Exercise Duration Trough Peak |
24* 34** |
24* 26* |
| Time to Angina Trough Peak |
30* 38** |
26* 38** |
| Time to 1 mm ST Depression Trough Peak |
20 41** |
21 35** |
| Placebo | Ranexa 750 mg b.i.d. |
Ranexa 1000 mg b.i.d. |
||
| Angina Frequency (attacks/week) | N | 258 | 272 | 261 |
| Mean | 3.3 | 2.5 | 2.1 | |
| p-value vs placebo | — | 0.006 | < 0.001 | |
| Nitroglycerin Use (doses/week) | N | 252 | 262 | 244 |
| Mean | 3.1 | 2.1 | 1.8 | |
| p-value vs placebo | — | 0.016 | < 0.001 | |
Tolerance to ranolazine did not develop after 12 weeks of therapy. Rebound increases in angina, as measured by exercise duration, have not been observed following abrupt discontinuation of ranolazine.
Effects in Demographic Subsets
Gender
Effects on angina frequency and exercise tolerance were considerably smaller in women than in men. In CARISA, the improvement in Exercise Tolerance Test (ETT) in females was about 33% of that in males at the 1000 mg b.i.d. dose level. In ERICA, where the primary endpoint was angina attack frequency, the mean reduction in weekly angina attacks was 0.3 for females and 1.3 for males.
Age
No differences in efficacy were observed between younger and older patients. However, a higher incidence of adverse events were observed in elderly (≥ 75 years) patients on ranolazine (see PRECAUTIONS, Geriatric Use).
Race
There were insufficient numbers of non-Caucasian patients to allow for analyses of efficacy or safety by racial subgroup.
INDICATIONS AND USAGE
Ranexa is indicated for the treatment of chronic angina. Because Ranexa prolongs the QT interval, it should be reserved for patients who have not achieved an adequate response with other antianginal drugs. Ranexa should be used in combination with amlodipine, beta-blockers or nitrates. The effect on angina rate or exercise tolerance appeared to be smaller in women than men.
CONTRAINDICATIONS
Ranexa is contraindicated in patients:
- With pre-existing QT prolongation
- With hepatic impairment (Child-Pugh Classes A [mild], B [moderate] or C [severe]) (see CLINICAL PHARMACOLOGY, Hepatic Insufficiency, and Electrocardiographic Effects)
- On QT-prolonging drugs
- On potent and moderately potent CYP3A inhibitors, including diltiazem
WARNINGS
QT Prolongation
Ranolazine has been shown to prolong the QTc interval in a dose-related manner. While the clinical significance of the QTc prolongation in the case of ranolazine is unknown, other drugs with this potential have been associated with torsades de pointes-type arrhythmias and sudden death.
With repeat dosing, the mean effect on QTc of ranolazine 1000 mg b.i.d., at Tmax, is about 6 msec. However, in 5% of the population the prolongation of QTc is 15 msec. Age, weight, gender, race, heart rate, CHF NYHA Class I to IV, and diabetes have no significant effect on the relationship between ranolazine plasma level and increase in QTc. The relationship between ranolazine levels and QTc remains linear over a concentration range up to 4-fold greater than the concentrations produced by 1000 mg b.i.d., and is not affected by changes in heart rate. Doses > 1000 mg b.i.d. should not be used.
There are no studies examining the effects of ranolazine in patients with pre-existing QT prolongation or receiving other QT-prolonging drugs. Because of possible additive effects on the QT interval, ranolazine should be avoided in patients with known QT prolongation (including congenital long QT syndrome, uncorrected hypokalemia), known history of ventricular tachycardia and in patients receiving drugs that prolong the QTc interval, such as Class Ia (e.g., quinidine) and Class III (e.g., dofetilide, sotalol) antiarrhythmics, and antipsychotics (e.g., thioridazine, ziprasidone).
Because the QTc-prolonging effect is increased approximately 3-fold in patients with hepatic dysfunction, ranolazine is contraindicated in patients with mild, moderate or severe liver disease (see Special Populations and Hepatic Insufficiency).
Ranolazine is primarily metabolized by CYP3A. Use of ranolazine with potent or moderately potent inhibitors of CYP3A should be avoided because concomitant administration will increase ranolazine plasma levels and QTc prolongation. These inhibitors include ketoconazole and other azole antifungals, diltiazem, verapamil, macrolide antibiotics, HIV protease inhibitors and grapefruit juice or grapefruit-containing products.
Tumor Promotion
A published study reported that ranolazine promoted tumor formation and progression to malignancy when given to transgenic APC(min/+) mice at a dose of 30 mg/kg twice daily (see REFERENCES). The clinical significance of this finding is unclear (see PRECAUTIONS, Carcinogenesis, Mutagenesis, Impairment of Fertility).
PRECAUTIONS
Co-administration of ranolazine and digoxin increases the plasma concentrations of digoxin by approximately 1.5-fold and the dose of digoxin may have to be reduced accordingly. The dose of other P-gp substrates may have to be reduced as well when ranolazine is co-administered.
Ranolazine can inhibit the activity of CYP2D6 and thus the metabolism of drugs that are mainly metabolized by this enzyme, for example tricyclic antidepressants and some antipsychotics, may be impaired and exposure to these drugs increased. The dose of such drugs may have to be reduced when ranolazine is co-administered.
In vitro studies indicate that ranolazine is a P-gp substrate. Caution should be exercised when co-administering ranolazine and P-gp inhibitors such as ritonavir or cyclosporine.
Use in Patients with Congestive Heart Failure
No dosage adjustment for Ranexa is required in patients with CHF (NYHA Class I to IV) (see CLINICAL PHARMACOLOGY and Special Populations).
Use in Patients with Diabetes Mellitus
No dosage adjustment is required in patients with diabetes (see CLINICAL PHARMACOLOGY and Special Populations).
Use in Patients with Severe Renal Impairment
Ranexa increases blood pressure by about 15 mm Hg in patients with severe renal impairment. Blood pressure should be monitored regularly after initiation of Ranexa in such patients.
Laboratory Tests
Average elevations of serum creatinine by 0.1 mg/dL have been observed in angina patients treated with ranolazine. In patients with renal impairment, the percentage increase in creatinine from pretreatment values was of the same magnitude as in angina patients; BUN did not increase. These elevations have a rapid onset, show no signs of progression during long-term therapy, and are reversible after discontinuation of ranolazine. The results of a special renal function study in healthy volunteers receiving Ranexa 1000 mg b.i.d. showed that the glomerular filtration rate was not affected by ranolazine. The elevated creatinine levels are likely due to a blockage of creatinine’s tubular secretion by ranolazine or one of its metabolites. Urinalysis results are unaffected by ranolazine.
Transient eosinophilia was observed infrequently on ranolazine. Small mean decreases in hematocrit (1.2%) were also observed on ranolazine in controlled studies; however, there was no evidence of occult fecal blood loss.
Information for Patients
To ensure safe and effective use of Ranexa, the following information and instructions should be communicated to the patient when appropriate.
Patients should be advised:
- that Ranexa is only for patients not responding adequately to other antianginal drugs
- that Ranexa may produce changes in the electrocardiogram (QTc interval prolongation)
- to inform their physician of any personal or family history of QTc prolongation, congenital long QT syndrome, or proarrhythmic conditions such as hypokalemia
- that Ranexa should be avoided in patients receiving drugs that prolong the QTc interval such as Class Ia (e.g., quinidine) or Class III (e.g., dofetilide, sotalol) antiarrhythmic agents, erythromycin, and certain antipsychotics (e.g., thioridazine, ziprasidone)
- that Ranexa should be avoided in patients receiving drugs that are potent or moderately potent inhibitors of CYP3A, including, for example, ketoconazole, HIV protease inhibitors, macrolide antibiotics, diltiazem, and verapamil
- that grapefruit juice or grapefruit products should be avoided when taking Ranexa
- that doses of Ranexa higher than 1000 mg twice a day should not be used
- that if a dose of Ranexa is missed, the usual dose should be taken at the next scheduled time. The next dose should not be doubled
- that Ranexa will not abate an acute angina episode
- that Ranexa should generally be avoided in patients with mild, moderate or severe liver impairment
- that Ranexa should generally be avoided in patients with severe renal impairment
- to inform their physician of any other medications when taken concurrently with Ranexa, including over-the-counter medications
- to contact their physician if they experience palpitations or fainting spells while taking Ranexa
- that Ranexa may cause dizziness and lightheadedness; therefore, patients should know how they react to this drug before they operate an automobile, or machinery, or engage in activities requiring mental alertness or coordination
- that Ranexa may be taken with or without meals
- that Ranexa tablets should be swallowed whole and not crushed, broken, or chewed
Drug-Drug Interactions
(also see CLINICAL PHARMACOLOGY, Drug-Drug Interactions, and DOSAGE AND ADMINISTRATION)
Pharmacokinetic Interactions: Effects of Other Drugs on Ranolazine
Ketoconazole
As a potent inhibitor of CYP3A, ketoconazole (200 mg b.i.d.) increases average steady-state plasma concentrations of ranolazine 3.2-fold. Ranexa should not be used during treatment with ketoconazole (see CONTRAINDICATIONS).
Diltiazem
As a moderate inhibitor of CYP3A, diltiazem (180 to 360 mg daily) causes dose-dependent mean increases in average ranolazine steady-state concentrations of about 1.8- to 2.3-fold.
Verapamil
Verapamil 120 mg t.i.d. increases ranolazine steady-state plasma concentrations about 2-fold.
Cimetidine
Co-administration of cimetidine does not increase the plasma concentrations of ranolazine. No dose adjustment of Ranexa is required in patients treated with cimetidine.
Digoxin
Co-administration of digoxin does not increase the plasma concentration of ranolazine. No dose adjustment of Ranexa is required in patients treated with digoxin.
Paroxetine
Paroxetine, a potent inhibitor of CYP2D6, increased average steady-state plasma concentrations of ranolazine 1.2-fold. No dose adjustment of Ranexa is required in patients treated with paroxetine or other CYP2D6 inhibitors.
Pharmacokinetic Interactions: Effects of Ranolazine on Other Drugs
Digoxin
As a result of an interaction at the P-gp level, co-administration of ranolazine and digoxin results in a 1.5-fold elevation of digoxin plasma concentrations. The dose of digoxin may have to be adjusted when ranolazine is co-administered with digoxin.
Simvastatin
Co-administration of ranolazine and simvastatin results in about a 2-fold increase in plasma concentrations of simvastatin, and its active metabolite.
Warfarin
Ranolazine has no significant effect on the pharmacokinetics of (+) R- and (-) S- warfarin.
Drug/Laboratory Test Interactions
Ranolazine is not known to interfere with any laboratory test.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Ranolazine demonstrated no mutagenic potential in the following assays: Ames bacterial mutation assay, Saccharomyces assay for mitotic gene conversion, chromosomal aberrations assay in Chinese hamster ovary (CHO) cells, mammalian CHO/HGPRT gene mutation assay and mouse and rat bone marrow micronucleus assays.
There was no evidence of carcinogenic potential in 21 to 24 month studies in mice or rats. The highest oral doses used in the carcinogenicity studies were 150 mg/kg/day for 21 months in rats (900 mg/m2/day) and 50 mg/kg/day for 24 months in mice (150 mg/m2/day). These doses are equivalent to 0.8 and 0.1 times, respectively, the maximum recommended human dose (MRHD) of 2 grams on a mg/m2 basis and represent the maximum tolerated doses in these species (see WARNINGS, Tumor Promotion).
There are no adequate studies assessing the effect of ranolazine on fertility or reproductive capacity.
Pregnancy
Pregnancy Category C
There are no adequate studies assessing the effect of ranolazine on the developing fetus.
There are no adequate well-controlled studies in pregnant women. Ranexa should be used during pregnancy only when the potential benefit to the patient justifies the potential risk to the fetus.
Nursing Mothers
It is not known whether ranolazine is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions from ranolazine in nursing infants, a decision should be made whether to discontinue nursing or to discontinue Ranexa, taking into account the importance of the drug to the mother.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Geriatric Use
Of the chronic angina patients treated with ranolazine in controlled studies, 496 (48%) were ≥ 65 years of age, and 114 (11%) were ≥ 75 years of age. No overall differences in efficacy were observed between older and younger patients. There were no differences in safety for patients ≥ 65 years compared to younger patients, but patients ≥ 75 years of age on ranolazine, compared to placebo, appeared to have a higher incidence of adverse events, serious adverse events, and drug discontinuations due to adverse events. In controlled ranolazine studies, the placebo-subtracted incidence of any adverse event in patients ≥ 75 years old treated with ranolazine was 23%, and 11% discontinued ranolazine due to unacceptable adverse events. In CARISA and ERICA, the most commonly reported placebo-subtracted adverse events in patients ≥ 75 years old on ranolazine included constipation (19%), nausea (6%), and dizziness (6%). In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
ADVERSE REACTIONS
Of the patients treated with Ranexa, 1,026 were enrolled in three double-blind, placebo-controlled, randomized studies of up to 12 weeks duration. In addition, upon study completion, 746 patients received continued treatment with Ranexa in open-label, long-term studies; 639 patients were exposed to Ranexa for more than 1 year, 578 patients for more than 2 years, and 372 for more than 3 years. Subgroup evaluations in patients with reactive airway disease, CHF, and diabetes were also conducted. These conditions did not alter the general nature or frequency of treatment-emergent adverse events observed in the broader ranolazine-treated population.
In controlled clinical trials of angina patients, the most frequently reported treatment-emergent adverse events (> 4%), occurring more often with ranolazine than placebo, were dizziness (6.2%), headache (5.5%), constipation (4.5%), and nausea (4.4%). In open-label, long-term treatment studies, a similar adverse event profile was observed in patients treated with ranolazine.
About 6% of patients discontinued treatment with Ranexa due to an adverse event in controlled studies in angina patients compared to about 3% on placebo. The most common adverse events that led to discontinuation more frequently on ranolazine than placebo were dizziness (1.3% versus 0.1%), and nausea (1% versus 0%), asthenia, constipation and headache (each about 0.5% versus 0%).
Small, reversible elevations in serum creatinine and BUN levels have been observed in clinical studies with ranolazine. These elevations were observed without evidence of renal toxicity (see PRECAUTIONS and Laboratory Tests).
Adverse Events Occurring at an Incidence of ≥ 2% Among Ranexa-treated Angina Patients in the CARISA and ERICA Trials
The most commonly observed treatment-emergent adverse events for chronic angina patients from CARISA and ERICA that occurred more frequently with ranolazine than placebo are shown in Table 4.
| Number (%) of Angina Patients | ||
| Placebo (N = 552) |
Ranexa* (N = 835) |
|
| *Doses include 500 mg b.i.d., 750 mg b.i.d., and 1000 mg b.i.d. | ||
| Gastrointestinal Disorders | ||
| Constipation | 9 (2) | 63 (8) |
| Nausea | 5 (1) | 33 (4) |
| Nervous System Disorders | ||
| Dizziness | 12 (2) | 41 (5) |
| Headache | 11 (2) | 22 (3) |
The dose-related adverse events of dizziness and syncope are shown in Table 5.
| Number (%) of Angina Patients | |||
| Placebo (N = 552) |
Ranexa 750 mg b.i.d. (N = 279) |
Ranexa 1000 mg b.i.d. (N = 556) |
|
| Dizziness | 12 (2) | 10 (4) | 31 (6) |
| Syncope | 0 | 0 | 4 (0.7) |
Adverse Events Occurring Among All Ranolazine-treated Patients with Chronic Angina
A total of 2,018 patients with chronic angina were treated with ranolazine in controlled clinical trials.
The following additional adverse events occurred at an incidence of > 0.5 to < 2.0% in patients treated with ranolazine and were more frequent than the incidence observed in placebo-treated patients.
Cardiac Disorders – palpitations
Ear and Labyrinth Disorders – tinnitus, vertigo
Gastrointestinal Disorders – abdominal pain, dry mouth, vomiting
General Disorders and Administrative Site Adverse Events – peripheral edema
Respiratory, Thoracic and Mediastinal Disorders – dyspnea
Other more rare (≤ 0.5%) but potentially medically important adverse events observed more frequently with ranolazine than placebo treatment in controlled studies included: bradycardia, hematuria, hypoesthesia, hypotension, orthostatic hypotension, paresthesia, tremor, and blurred vision.
DRUG ABUSE AND DEPENDENCE
Ranolazine does not have any potential for abuse or dependence.
OVERDOSAGE
No cases of intentional or accidental overdose with ranolazine have been reported. In the event of overdose, the expected symptoms would be dizziness, nausea/vomiting, diplopia, paresthesia, and confusion. Syncope with prolonged loss of consciousness may develop. Because the QTc interval increases with ranolazine plasma concentration, continuous ECG monitoring may be warranted in the event of overdose. If required, general supportive measures should be initiated.
Since ranolazine is about 62% bound to plasma proteins, complete clearance of ranolazine by hemodialysis is not likely.
DOSAGE AND ADMINISTRATION
Ranexa dosing should be initiated at 500 mg b.i.d. and increased to 1000 mg b.i.d., as needed, based on clinical symptoms. The maximum recommended daily dose of Ranexa is 1000 mg b.i.d. Baseline and follow-up ECGs should be obtained to evaluate effects on QT interval. Use of QT-prolonging drugs and drugs that increase plasma concentrations of ranolazine should be avoided (see CONTRAINDICATIONS, WARNINGS and PRECAUTIONS, Drug-Drug Interactions).
The dose of simvastatin and digoxin and other P-gp substrates may have to be reduced when ranolazine is co-administered. Dose adjustments of Ranexa are generally not required on the basis of age or gender, or in patients with CHF or diabetes mellitus.
The concomitant use of Ranexa with other commonly administered cardiovascular medications (amlodipine, beta-blockers, nitrates, anti-hypertensive agents) is well-tolerated.
If a dose of Ranexa is missed, the prescribed dose should be taken at the next scheduled time. The next dose should not be doubled.
Ranexa may be taken with or without meals. Ranexa tablets should be swallowed whole and not crushed, broken, or chewed.
HOW SUPPLIED
Ranexa (ranolazine) is supplied as film-coated, oblong-shaped extended-release tablets containing the following amount of ranolazine:
| Strength | Color | Marking | ||
| 500 mg | light orange | CVT500 | ||
| 1000 mg | pale yellow | CVT1000 |
Ranexa (ranolazine extended-release tablets) is available in:
| Strength | NDC Code | ||
| Unit-of-Use Bottle (60 Tablets) | 500 mg | 67159-112-03 | |
| Pharmacy Bottle (500 Tablets) | 500 mg | 67159-112-04 | |
| Unit-of-Use Bottle (60 Tablets) | 1000 mg | 67159-114-03 | |
| Pharmacy Bottle (500 Tablets) | 1000 mg | 67159-114-04 |
Store at 25°C (77°F) with excursion permitted to 15° to 30°C (59° to 86°F).
REFERENCES
1. M.A. Suckow et al. The anti-ischemia agent ranolazine promotes the development of intestinal tumors in APC(min/+) mice. Cancer Letters 209(2004):165–169.
Issued: June 2007
Manufactured for:
CV Therapeutics, Inc.
Palo Alto, CA 94304 USA
By:
DSM Pharmaceuticals, Inc.
Greenville, NC 27834 USA
Ranexa is a registered trademark of CV Therapeutics, Inc.
For additional information, contact CVT Therapeutics at 1-877-CVT-7171 or drug.info@cvt.com.
U.S. Patent Numbers 4,567,264; 6,303,607; 6,369,062; 6,479,496; 6,503,911; 6,525,057; 6,562,826; 6,617,328; 6,620,814; 6,852,724; 6,864,258
©2006, CV Therapeutics, Inc.
Rx only
L000008
0607
| Ranexa (Ranolazine) | ||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Ranexa (Ranolazine) | |||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||
Revised: 08/2007CV Therapeutics, Inc.