luveris (lutropin alfa)
[Serono, Inc.]
DESCRIPTION
Luveris® (lutropin alfa for injection) is a sterile lyophilized powder composed of recombinant human luteinizing hormone, r-hLH. r-hLH is a heterodimeric glycoprotein consisting of two non-covalently linked subunits (designated α and β) of 92 and 121 amino acids, respectively. The carbohydrate chain attachment to the r-hLH protein core occurs via N- but not O-linkage. The N glycosylation sites are Asn-52 and Asn-78 for the α–subunit and Asn-30 for the β–subunit. The β - chain has an N-glycosylation site and its structure and glycosylation pattern are very similar to that of pituitary-derived hLH.
The production process involves expansion of genetically modified Chinese Hamster Ovary (CHO) cells from an extensively characterized cell bank into large scale cell culture processing. Lutropin alfa is secreted by the CHO cells directly into the cell culture medium that is then purified using a series of chromatographic steps. The biological activity of lutropin alfa is determined using the Van Hell Bioassay described in the British Pharmacopoeia. The in vivo biological activity is determined using a house standard properly calibrated against the relevant international standard.
Luveris® is a sterile, lyophilized powder, which after reconstitution with Sterile Water for Injection, USP, is intended for subcutaneous (sc) administration. Each vial of Luveris® contains 82.5 IU lutropin alfa, 48 mg sucrose, 0.83 mg dibasic sodium phosphate dihydrate, 0.052 mg monobasic sodium phosphate monohydrate, 0.05 mg polysorbate 20, and 0.1 mg L-methionine. Phosphoric acid and/or sodium hydroxide are used to adjust the pH. After reconstitution with 1 mL of enclosed diluent, the product will deliver 75 IU of recombinant human lutropin alfa. The pH of the reconstituted solution is 7.5 to 8.5.
Therapeutic Class: Infertility
CLINICAL PHARMACOLOGY
The physicochemical, immunological, and biological activities of Luveris® are comparable to those of human pituitary LH. In the ovaries, during the follicular phase, LH stimulates theca cells to secrete androgens, which will be used as the substrate by granulosa cell aromatase enzyme to produce estradiol, supporting Follicle-Stimulating Hormone (FSH)-induced follicular development. Luveris® is administered concomitantly with Gonal-f® (follitropin alfa for injection) to stimulate development of a potentially competent follicle and to indirectly prepare the reproductive tract for implantation and pregnancy.
Pharmacokinetics
When given by intravenous administration, Luveris® demonstrates linear pharmacokinetics over the 300 to 40,000 IU dose range. Following a 75 IU dose, the concentration range is too small to allow proper quantification of the pharmacokinetic parameters. The disposition of r-hLH is adequately described by a biexponential model.
Following subcutaneous administration, the terminal half-life is slightly longer than after intravenous administration. Upon repeated daily administration, a modest accumulation takes place (accumulation ratio of 1.6 ± 0.8). Following administration of Luveris® 150 IU, r-hLH pharmacokinetics are described in Table 1.
| Parameter† | Luveris® 150 IU SC |
|---|---|
| †Cmax: peak concentration, tmax: time of Cmax, AUC: total area under the curve, t½: elimination half-life, ‡ median (range) | |
| Cmax (IU/L) | 1.1 ± 0.3 |
| tmax (h)‡ | 6 (3-9) |
| AUC (h·IU/L) | 44 ± 44 |
| t½ (h) | 14 ± 8 |
Absorption
Following subcutaneous administration of Luveris®, maximum serum concentration is reached after approximately 4 to 16 hours.
The mean absolute bioavailability of Luveris® following a single subcutaneous injection (at a much higher dose to allow proper quantification, i.e. 10,000 IU) to healthy female volunteers is 56 ± 23%, supported by an immunoassay method. There were no statistical differences between the intramuscular and subcutaneous routes of administration for Cmax, tmax, or bioavailability.
Distribution
Following an intravenous dose of 300 IU of Luveris®, a rapid distribution phase (t ½λ1 of approximately 1 hour) and a terminal half-life (t½) of approximately 11 hours were observed for r-hLH. The steady state volume of distribution (Vss) was approximately 10 L. Mean residence time (MRT) was approximately 6 hours.
Metabolism/Excretion
Following subcutaneous administration of Luveris®, r-hLH is eliminated from the body with a mean terminal half-life of about 18 hours. Total body clearance is approximately 2 to 3 L/h with less than 5 percent of the dose being excreted unchanged renally.
Pharmacodynamics
In the stimulation of follicular development, the primary effect resulting from administration of Luveris® is an increase in estradiol secretion by the follicles, the growth of which is stimulated by FSH.
Special populations
Pharmacokinetics of Luveris® in the geriatric or pediatric populations or in patients with renal or hepatic insufficiency have not been established.
Drug-Drug Interactions
There are no pharmacokinetic interactions with Gonal-f® (follitropin alfa for injection) when administered simultaneously. No drug-drug interaction studies have been conducted.
Clinical Studies:
The efficacy of Luveris® in infertile hypogonadotropic hypogonadal (HH) women with profound LH deficiency (LH < 1.2 IU/L) has been examined in two well-controlled, comparative studies and one non-comparative, extension study of stimulation of follicular development. In these studies, Luveris® was concomitantly administered with Gonal-f®.
Study 6253
Study 6253 was a randomized, open-label, dose-finding study conducted in Europe and Israel. Thirty-eight women were enrolled, randomized, and treated with 0, 25, 75 or 225 IU Luveris® concomitant with 150 IU Gonal-f® for up to 3 treatment cycles. Both women desiring pregnancy and those not desiring pregnancy were enrolled and randomized. Those wishing not to conceive used mechanical contraception.
The primary efficacy endpoint, follicular development, was a composite endpoint (the criteria for each component was to be met) defined by i) at least one follicle with a mean diameter ≥ 17 mm; ii) pre-ovulatory serum estradiol (E2) level ≥ 400 pmol/L (≥ 109 pg/mL); and iii) mid-luteal phase serum progesterone (P4)≥ 25 nmol/L (≥ 7.9 ng/mL). The results for follicular development are presented in Table 2. The data from subjects whose cycles were cancelled for the risk of OHSS before receiving hCG and before having a serum P4 drawn were analyzed both as a success and a failure.
| Follicular Development Rate Cycle Cancellation Due to Risk of OHSS(a) considered as: | No Luveris®& Gonal-f® (n=9) n (%) | 25 IU Luveris®& Gonal-f® (n=8) n (%) | 75 IU Luveris®& Gonal-f® (n=11) n (%) | 225 IU Luveris®& Gonal-f® (n=10) n (%) |
|---|---|---|---|---|
| (a) Cycles were cancelled due to the risk of OHSS when the E2 level exceeded 1,100 pg/mL and/or ≥ 3 follicles were ≥ 15 mm in diameter | ||||
| (b) Fisher’s Exact Test | ||||
| Success p-value vs. Gonal-f® aloneb | 1 (11%) | 2 (25%) | 7 (64%) | 7 (70%) |
| 0.028 | ||||
| Failure p-value vs. Gonal-f® aloneb | 1 (11%) | 2 (25%) | 5 (45%) | 4 (40%) |
| 0.157 | ||||
Table 3 presents secondary efficacy parameter results including the proportion of patients ovulating as determined from the mid-luteal phase P4 of ≥ 7.9 ng/mL (25 nmol/L), pre-ovulatory serum estradiol levels, and endometrial thickness.
| No Luveris®& Gonal-f® (n=9) | 25 IU Luveris®& Gonal-f® (n=8) | 75 IU Luveris®& Gonal-f® (n=11) | 225 IU Luveris®& Gonal-f® (n=10) | |
|---|---|---|---|---|
| (a) Study 6253 was not statistically powered to demonstrate differences in these parameters. | ||||
| (b) Ovulation [rate as measured by the proportion of patients with a serum P4≥25 nmol/L (7.9 ng/mL)] was a component of follicular development | ||||
| (c) day of hCG administration or last available assessment | ||||
| Ovulation(b), Number (n) and percentage (%) of subjects with mid-luteal P4≥ 7.9 ng/mL (25 nmol/L) | 1 (11%) | 2 (25%) | 5 (45%) | 5 (50%) |
| Pre-ovulatory serum estradiol levels(c), pg/mL, mean (SD) | 28 (46) | 59 (71) | 357 (473) | 701 (781) |
| Endometrial thickness(c) mm, mean (SD) | 3.8 (2.4) | 4.1 (2.9) | 6.7 (3.2) | 7.1 (2.5) |
Study 21008
Study 21008 was a double-blind, placebo-controlled, randomized, multinational, single cycle study conducted in the U.S., Canada, Israel, and Australia to assess the safety and efficacy of 75 IU Luveris® administered subcutaneously. Thirty-nine patients were randomized and treated in a 2:1 ratio to concomitant treatment with 75 IU Luveris® and 150 IU Gonal-f® (26 patients) or placebo and 150 IU Gonal-f® (13 patients). Only women desiring pregnancy were enrolled and randomized.
The primary efficacy endpoint, follicular development, was a composite endpoint (the criteria for each component was to be met), defined by: (i) at least one follicle with a mean diameter ≥17 mm, (ii) pre-ovulatory serum E2 level ≥ 109 pg/mL (≥ 400 pmol/L) and (iii) mid-luteal phase P4 level ≥ 7.9 ng/mL (≥25 nmol/L). The results for follicular development are presented in Table 4. Patients who did not meet one or more of the pre-specified component definitions but had a positive pregnancy test (serum hCG ≥ 10 mIU/mL) were considered as a success for follicular development. The data from subjects whose cycles were cancelled for the risk of OHSS before receiving hCG and before having a serum P4 drawn were analyzed both as a success and a failure.
| Follicular Development Rate | Placebo & Gonal-f® (n=13) n (%) | 75 IU Luveris®& Gonal-f® (n=26) n (%) |
|---|---|---|
| (a) Cycles were cancelled due to the risk of OHSS when the E2 level exceeded 1,100 pg/mL and/or ≥ 3 follicles were ≥ 15 mm in diameter | ||
| (b) Fisher’s Exact Test | ||
| Cycle Cancellation Due to Risk of OHSS(a) considered as Success | ||
| Successful Follicular Development | 2 (15%) | 17 (65%) |
| Failed Follicular Development | 11 (85%) | 9 (35%) |
| p-value vs. placebo(b) | 0.006 | |
| Cycle Cancellation Due to Risk of OHSS(a) considered as Failure | ||
| Successful Follicular Development | 1 (8%) | 11 (42%) |
| Failed Follicular Development | 12 (92%) | 15 (58%) |
| p-value vs. placebo(b) | 0.034 | |
Table 5 presents secondary efficacy parameter results including the proportion of patients ovulating as determined from the mid-luteal phase P4 of ≥ 7.9 ng/mL (25 nmol/L), pre-ovulatory serum estradiol levels, and endometrial thickness.
| Placebo & Gonal-f® (n=13) n (%) | 75 IU Luveris®& Gonal-f® (n=26) n (%) | |
|---|---|---|
| (a) Study 21008 was not statistically powered to demonstrate differences in these parameters. | ||
| (b) Ovulation [rate as measured by the proportion of patients with a serum P4≥25 nmol/L (7.9 ng/mL)] was a component of follicular development | ||
| (c) day of hCG administration or last available assessment | ||
| Ovulation(b) | 2 (15%) | 12 (46%) |
| Number (n) and percentage (%) of subjects with midluteal P4≥ 7.9 ng/mL (25 nmol/L) | ||
| Pre-ovulatory serum estradiol levels(c), pg/mL, mean (SD) | 78 (125) | 549 (673) |
| Endometrial thickness(c) | 5.0 (1.6) | 7.4 (3.1) |
Study 21415
Study 21415 was an open label, multi-center extension of Study 21008 to allow patients to continue treatment with 75 IU Luveris®. Thirty-one of the 39 patients from Study 21008 were treated for up to three cycles in Study 21415. Eleven had been treated with placebo and Gonal-f® and 20 had been treated with 75 IU Luveris® and Gonal-f® in Study 21008.
Treatment consisted of 75 IU Luveris® and a recommended starting dose of 75-150 IU Gonal-f®. Both Luveris® and Gonal-f® were administered subcutaneously once daily. After 7 days of treatment, if the patient’s response was considered sub-optimal based on follicle growth and serum estradiol levels, her physician could adjust the Gonal-f® dose in the range of 75-225 IU/day. As pregnancy was the treatment goal of the patients, multiple cycles of treatment and dose adjustment of Gonal-f® were allowed, consistent with established medical practice.
Of the 31 patients, 27 (87.1%) achieved follicular development (Table 6), 20 (64.5%) achieved pregnancy (i.e., positive pregnancy test, serum hCG ≥ 10 mIU/mL), and 16 (51.6%) achieved clinical pregnancy. Of the 11 placebo patients from Study 21008, 7 (63.6%) achieved follicular development and 4 (36.4%) achieved clinical pregnancy when treated with 75 IU Luveris® and Gonal-f® in Cycle 1 of Study 21415.
| Follicular Development Rate | 75 IU Luveris®& Gonal-f® (n=31) | ||
|---|---|---|---|
| Cycle Cancellation Due to Risk of OHSS(a) considered as: | n (%) | ||
| Cycle 1 | Cycle 2 | Cycle 3 | |
| (a) Cycles were cancelled due to the risk of OHSS when the E2 level exceeded 1,100 pg/mL and/or ≥ 3 follicles were ≥ 15 mm in diameter | |||
| Success | 21 (67.7%) | 26 (83.9%) | 27 (87.1%) |
| Failure | 16 (51.6%) | 26 (83.9%) | 27 (87.1%) |
INDICATIONS AND USAGE
Luveris® (lutropin alfa for injection), concomitantly administered with Gonal-f® (follitropin alfa for injection), is indicated for stimulation of follicular development in infertile hypogonadotropic hypogonadal women with profound LH deficiency (LH < 1.2 IU/L). A definitive effect on pregnancy in this population has not been demonstrated. The safety and effectiveness of concomitant administration of Luveris® with any other preparation of recombinant human FSH or urinary human FSH is unknown.
Selection of Patients:
-
Patients should have baseline serum hormone levels of LH < 1.2 IU/L and FSH < 5 IU/L.
-
Before treatment with gonadotropins is instituted, a thorough gynecologic and endocrinologic evaluation must be performed. This should include an assessment of pelvic anatomy and exclusion of pregnancy.
-
Patients should have a negative progestin challenge test.
-
Patients in later reproductive life have a greater predisposition to endometrial carcinoma as well as a higher incidence of anovulatory disorders. A thorough diagnostic evaluation should always be performed in patients who demonstrate abnormal uterine bleeding or other signs of endometrial abnormalities before starting Luveris® and follitropin alfa therapy.
-
Evaluation of the partner’s fertility potential should be included in the initial evaluation.
CONTRAINDICATIONS
Luveris® (lutropin alfa for injection) is contraindicated in women who exhibit:
-
Prior hypersensitivity to hLH preparations or one of their excipients.
-
Primary ovarian failure.
-
Uncontrolled thyroid or adrenal dysfunction.
-
An uncontrolled organic intracranial lesion such as a pituitary tumor.
-
Abnormal uterine bleeding of undetermined origin (see " Selection of Patients").
-
Ovarian cyst or enlargement of undetermined origin (see " Selection of Patients").
-
Sex hormone dependent tumors of the reproductive tract and accessory organs.
-
Pregnancy.
WARNINGS
Gonadotropins, including Luveris® (lutropin alfa for injection), should only be used by physicians who are thoroughly familiar with infertility problems and their management. Like other gonadotropin products, Luveris® is a potent gonadotropic substance capable of contributing to the development of Ovarian Hyperstimulation Syndrome (OHSS) in women with or without pulmonary or vascular complications. Gonadotropin therapy requires a certain time commitment by physicians and supportive health professionals, and requires the availability of appropriate monitoring facilities (see " PRECAUTIONS/ Laboratory Tests"). Safe and effective use of Luveris® requires monitoring of ovarian response with serum estradiol and ovary ultrasound on a regular basis.
Overstimulation of the Ovary Following Gonadotropin Therapy:
Ovarian Enlargement:
Mild to moderate uncomplicated ovarian enlargement which may be accompanied by abdominal distension and/or abdominal pain may occur in patients treated with gonadotropins (such as Luveris®). These conditions generally regress without treatment within two or three weeks. Careful monitoring of ovarian response can further minimize the risk of overstimulation.
If the ovaries are abnormally enlarged on the last day of therapy with Luveris® and Gonal-f®, hCG should not be administered in this course of therapy. This will reduce the risk of development of Ovarian Hyperstimulation Syndrome.
Ovarian Hyperstimulation Syndrome (OHSS):
OHSS is a medical event distinct from uncomplicated ovarian enlargement. Severe OHSS may progress rapidly (within 24 hours to several days) to become a serious medical event. It is characterized by an apparent dramatic increase in vascular permeability which can result in a rapid accumulation of fluid in the peritoneal cavity, thorax, and potentially, the pericardium. The early warning signs of development of OHSS are severe pelvic pain, nausea, vomiting, and weight gain. The following symptomatology has been seen with cases of OHSS: abdominal pain, abdominal distension, gastrointestinal symptoms including nausea, vomiting and diarrhea, severe ovarian enlargement, weight gain, dyspnea, and oliguria. Clinical evaluation may reveal hypovolemia, hemoconcentration, electrolyte imbalances, ascites, hemoperitoneum, pleural effusions, hydrothorax, acute pulmonary distress, and thromboembolic events (see " Pulmonary and Vascular Complications"). Transient liver function test abnormalities that are suggestive of hepatic dysfunction have been reported in association with Ovarian Hyperstimulation Syndrome (OHSS). These liver function test abnormalities may be accompanied by morphological changes on liver biopsy.
In hypogonadotropic hypogonadal women with profound LH and FSH deficiency from five clinical trials, four cases of OHSS were reported in 4 of 70 (5.7%) patients treated with 75 IU Luveris® and Gonal-f® and one case was reported in 1 of 31 (3.2%) patients treated with Gonal-f® alone. Among women treated with any dose of Luveris® in these studies, five of 96 (5.2%) patients reported 6 cases of OHSS after treatment with Luveris® and Gonal-f®.
OHSS may be more severe and more protracted if pregnancy occurs. OHSS develops rapidly; therefore, patients should be followed for at least two weeks after hCG administration. Most often, OHSS occurs after treatment has been discontinued and reaches its maximum severity at seven to ten days following treatment. Usually, OHSS resolves spontaneously with the onset of menses. If there is evidence that OHSS may be developing prior to hCG administration (see " PRECAUTIONS/ Laboratory Tests"), hCG must be withheld.
If severe OHSS occurs, treatment with gonadotropins must be stopped and the patient should be hospitalized.
A physician experienced in the management of this syndrome, or who is experienced in the management of fluid and electrolyte imbalances should be consulted.
Multiple Births:
Patients should be advised of the potential risk of multiple births before starting treatment.
Pulmonary and Vascular Complications:
As with other gonadotropin products, a potential for the occurrence of arterial thromboembolism exists.
PRECAUTIONS
GENERAL PRECAUTIONS
Careful attention should be given to the diagnosis of infertility in candidates for Luveris® therapy. (See " INDICATIONS AND USAGE / Selection of Patients").
INFORMATION FOR PATIENTS
Prior to therapy with Luveris®, patients should be informed of the duration of treatment and monitoring of their condition that will be required. The risks of ovarian hyperstimulation syndrome and multiple births (see “ WARNINGS”) and other possible adverse reactions (see “ Adverse Reactions”) should also be discussed.
LABORATORY TESTS:
In most instances, treatment of women with LH and FSH results only in follicular recruitment and development. In the absence of an endogenous LH surge, hCG is given when monitoring of the patient indicates that sufficient follicular development has occurred. This may be estimated by ultrasound alone or in combination with measurement of serum estradiol levels. The combination of both ultrasound and serum estradiol measurement are useful for monitoring the development of follicles, for timing of the ovulatory trigger, as well as for detecting ovarian enlargement and minimizing the risk of the Ovarian Hyperstimulation Syndrome and multiple gestation. It is recommended that the number of growing follicles be confirmed using ultrasonography because serum estrogens do not give an indication of the size or number of follicles.
With the exception of confirmation of pregnancy, the clinical confirmation of ovulation is obtained by direct and indirect indices of progesterone production. The indices most generally used are as follows:
-
A rise in basal body temperature
-
Increase in serum progesterone and
-
Menstruation following a shift in basal body temperature
When used in conjunction with the indices of progesterone production, sonographic visualization of the ovaries will assist in determining if ovulation has occurred. Sonographic evidence of ovulation may include the following:
-
Fluid in the cul-de-sac
-
Ovarian stigmata
-
Collapsed follicle
-
Secretory endometrium
Accurate interpretation of the indices of ovulation requires a physician who is experienced in the interpretation of these tests.
CARCINOGENESIS, MUTAGENESIS, IMPAIRMENT OF FERTILITY
Long-term studies to evaluate the carcinogenic potential of Luveris® in animals have not been performed. In vitro mutagenicity testing of Luveris® in bacteria and mammalian cell lines, chromosome aberration assay in human lymphocytes and in vivo mouse micronucleus have shown no indication of genetic defects. Impaired fertility has been reported in animals exposed to high doses of lutropin alfa; increased pre- and post-implantation losses were observed in female rats and rabbits given lutropin alfa at doses of 10 IU/kg/day and higher.
PREGNANCY
Pregnancy: Category X. When administered to rats during the late period of pregnancy, doses of 10 IU/kg/day and higher were also shown to affect the postnatal survival and growth of the newborns. There was no evidence of teratogenic effect in either rats or rabbits. See “ CONTRAINDICATIONS” section. Luveris® is contraindicated in women who are pregnant and may cause fetal harm when administered to a pregnant woman. Reproductive toxicity studies performed in female rats and rabbits showed that lutropin alfa at doses of 10 IU/kg/day and greater caused an increase in pre- and post-implantation losses.
NURSING MOTHERS
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised if Luveris® is administered to a nursing woman.
PEDIATRIC PATIENTS
Luveris®is not indicated in pediatric patients. Safety and effectiveness in pediatric patients have not been established.
GERIATRIC PATIENTS
Luveris® is not indicated in geriatric patients. Safety and effectiveness in geriatric patients have not been established.
ADVERSE REACTIONS
The safety of Luveris® was examined in six clinical studies that treated 170 infertile women with hypogonadotropic hypogonadism of whom 152 received Luveris® and Gonal-f® in 283 treatment cycles. Adverse events reported by ≥2% of patients (regardless of causality) treated with any dose of Luveris® (25, 75, 150, 225 IU) are listed in Table 7.
| 0 IU Luveris®& Gonal-f® | 75 IU Luveris®& Gonal-f® | All doses of Luveris®& Gonal-f® | ||
|---|---|---|---|---|
| Patients (n=43) | Patients (n=118) | Patients (n=152) | ||
| n (%) | n (%) | n (%) | ||
| (a) Study 6905 was a randomized, open-label, dose-finding study to assess the efficacy and safety of Luveris® administered with 150 IU Gonal-f® for induction of follicular development in HH women. | ||||
| (b) Study 7798 was an uncontrolled, multicenter, dose-finding study to assess the efficacy and safety of Luveris® administered with 150 IU Gonal-f® for induction of follicular development in LH and FSH deficient anovulatory women in Germany. | ||||
| (c) Study 8297 was an uncontrolled, multicenter, dose-finding study to assess the efficacy and safety of Luveris® administered with 150 IU Gonal-f® for induction of follicular development in HH women in Spain. | ||||
| Patients With Events | 20 (46.5) | 50 (42.4) | 72 (47.4) | |
| Headache | 2 (4.7) | 12 (10.2) | 15 (9.9) | |
| Nausea | 0 | 8 (6.8) | 11 (7.2) | |
| Ovarian Hyperstimulation | 1 (2.3) | 7 (5.9) | 9 (5.9) | |
| Breast Pain Female | 4 (9.3) | 6 (5.1) | 9 (5.9) | |
| Abdominal Pain | 5 (11.6) | 6 (5.1) | 13 (8.6) | |
| Ovarian Cyst | 4 (9.3) | 6 (5.1) | 8 (5.3) | |
| Flatulence | 3 (7.0) | 5 (4.2) | 6 (3.9) | |
| Injection Site Reaction | 2 (4.7) | 4 (3.4) | 6 (3.9) | |
| Dysmenorrhoea | 1 (2.3) | 2 (1.7) | 4 (2.6) | |
| Ovarian Disorder | 0 | 2 (1.7) | 3 (2.0) | |
| Diarrhoea | 1 (2.3) | 3 (2.5) | 3 (2.0) | |
| Constipation | 0 | 3 (2.5) | 3 (2.0) | |
| Pain | 3 (7.0) | 3 (2.5) | 6 (3.9) | |
| Fatigue | 0 | 3 (2.5) | 5 (3.3) | |
| Upper Resp Tract Infection | 2 (4.7) | 1 (0.8) | 3 (2.0) | |
The following medical events have been reported subsequent to pregnancies resulting from administration of gonadotropins for ovulation induction in controlled clinical studies:
-
Spontaneous Abortion
-
Ectopic Pregnancy
-
Premature Labor
-
Postpartum Fever
-
Congenital abnormalities
There is no evidence that use of any gonadotropin drug product for treatment of infertility is associated with an increased risk of congenital malformations.
The following adverse reactions have been previously reported during menotropin therapy:
-
Pulmonary and vascular complications (see " WARNINGS")
-
Adnexal torsion (as a complication of ovarian enlargement)
-
Mild to moderate ovarian enlargement
-
Hemoperitoneum
There have been infrequent reports of ovarian neoplasms, both benign and malignant, in women who have undergone multiple drug regimens for ovulation induction; however, a causal relationship has not been established.
OVERDOSAGE
Aside from possible ovarian hyperstimulation and multiple gestations (see " WARNINGS"), there is no information on the consequences of overdosage with Luveris®.
DOSAGE AND ADMINISTRATION
For Subcutaneous Use Only
Dosage
It is recommended that 75 IU Luveris® be concomitantly administered subcutaneously with 75 IU to 150 IU Gonal-f® as two separate injections in the initial treatment cycle. Concomitant administration of Luveris® with Gonal-f® was studied in the clinical trials for Luveris®. The safety and effectiveness of concomitant administration of Luveris® with any other preparation of recombinant human FSH or urinary human FSH is unknown. Luveris® and Gonal-f® should be administered daily until adequate follicular development is indicated by ovary ultrasonography and serum estradiol. Treatment duration should not normally exceed 14 days unless signs of imminent follicular development are present.
To complete follicular development and effect ovulation in the absence of an endogenous LH surge, human chorionic gonadotropin (hCG) should be given one day after the last dose of Luveris® and Gonal-f®. Treatment with hCG should be withheld if the ovaries are abnormally enlarged or if excessive estradiol production has occurred. If the ovaries are abnormally enlarged or abdominal pain occurs, treatment with Luveris® and Gonal-f® should be discontinued and hCG should not be administered, and the patient should be advised not to have intercourse; this may reduce the chances of developing Ovarian Hyperstimulation Syndrome and, should spontaneous ovulation occur, reduce the chances of multiple gestation. A follow-up visit should be conducted in the luteal phase.
Doses administered in subsequent cycles should be individualized for each patient based on her response in the preceding cycle. Doses of Gonal-f® greater than 225 IU per day are not routinely recommended. As in the initial cycle, hCG must be given to complete follicular development and induce ovulation. The precautions described above should be followed to minimize the chance of developing Ovarian Hyperstimulation Syndrome.
The couple should be encouraged to have intercourse daily, beginning on the day prior to hCG administration until ovulation becomes apparent in the indices used for the determination of progestational activity.
In light of the indices and parameters mentioned, it should become obvious that, unless a physician is willing to devote considerable time to these patients and be familiar with and conduct the necessary laboratory studies, he/she should not prescribe Luveris®.
Administration:
Dissolve the contents of one vial of Luveris® in 1 mL Sterile Water for Injection, USP. Gonal-f® should be reconstituted and administered as directed in the prescriber labeling for this product. Administer entire contents of each vial SUBCUTANEOUSLY as separate injections. For single use. Use immediately after reconstitution. Any unused reconstituted material should be discarded. Mix gently. Do not shake.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration.
Directions for Administration of Luveris®:
Luveris® and Gonal-f® may be self-administered by the patient. Follow the directions below for reconstituting and injecting as separate injections of Luveris® and Gonal-f®. Gonal-f® should be reconstituted and administered as directed in the prescriber labeling for this product. Any unused reconstituted material should be discarded.
Step 1: Prepare the vials
Wash your hands thoroughly with soap and water.
Begin by opening the carton of Luveris®. Remove the plastic flip-tops from the vial of Luveris® powder and the vial of diluent provided with Luveris®. After removing the plastic flip-tops with your thumb, wipe the rubber stoppers with alcohol. The rubber stoppers should not be touched after they are wiped.
Step 2: Withdraw the Water into the Syringe
Carefully remove the needle cover. Do not touch the needle or allow the needle to touch any surface. After removing the needle cover, draw air into the syringe by slowly pulling back the plunger to the 1 cc mark. Place the vial of diluent on a hard, flat surface. Carefully insert the needle through the rubber stopper into the vial with the sterile water (diluent). Gently inject the air into the vial (the injected air creates pressure, which makes withdrawing the solution easier). Without removing the needle, turn the vial upside down and withdraw all of the water into the syringe, making sure the tip of the needle remains in the water. Remove the needle from the vial.
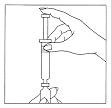
Step 3: Inject the Water into the Luveris® Vial
Place the vial containing the Luveris® powder on a hard, flat surface. Insert the needle through the rubber stopper into the vial. Keep the syringe in a straight, upright position as you insert it through the center of the rubber stopper, or it may be difficult to depress the plunger. After inserting the needle, slowly inject the sterile water (diluent) by depressing the plunger on the syringe into the vial of Luveris® powder.
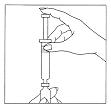
Step 4: Gently Dissolve the Luveris® Powder
Leaving the needle in the vial, gently rotate the vial between your fingers until all of the powder is dissolved. Do not shake. Check that the solution is clear and colorless. Do not use if the solution is cloudy, discolored, or contains particles.
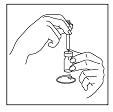
Step 5: Withdraw the Luveris® solution from the Vial
Without removing the needle, turn the vial upside down and withdraw all of the Luveris® solution into the syringe. Make sure the tip of the needle remains in the solution by slowly backing the needle out of the vial to withdraw as much of the solution as possible. Next, remove the needle from the vial.
Step 6: Replace needle and remove air bubbles in the syringe
Recap the syringe needle and twist the cap and needle off of syringe. Twist a new needle onto the end of the syringe and carefully remove the cap of the needle. To remove any air bubbles in the syringe, point the needle up and gently tap the syringe. When all the bubbles float to the top, slightly push the plunger until a small drop or two of solution begins to appear from the tip of the needle.
Step 7: Recap the syringe needle
Recap the syringe needle. Do not touch the needle or allow the needle to touch any surface. Carefully lay the syringe down on a flat, clean surface.
Step 8: Carefully clean the injection site
Suitable injection sites on the stomach (a few inches above or below the navel) will be advised by your fertility specialist. Occasionally your fertility specialist may suggest an alternative site. Make yourself comfortable by sitting or lying down. Carefully clean the injection site with an alcohol wipe and allow it to air-dry.

Step 9: Administer your injection
Remove the needle cap from the syringe needle. Hold the syringe like a pencil. With your other hand, pinch the skin together. Using a dart-like motion, insert the needle at a 45° to 90° angle (just under the skin) into the pad of tissue as shown or as directed by your doctor, nurse, or pharmacist. Do not inject into a vein.
Release the hand pinching the skin and depress the plunger in a slow, steady motion until all the medication is injected.
Step 10: Gently withdraw the needle
Withdraw the needle.
Step 11: Storage and clean up
Discard the used needle and syringe into your safety container. Place gauze over the injection site. If any bleeding occurs, apply gentle pressure. If bleeding does not stop within a few minutes, place a clean piece of gauze over the injection site and cover it with an adhesive bandage. Remember that your injection materials must be kept sterile and cannot be reused.
HOW SUPPLIED
Luveris® (lutropin alfa for injection) is supplied in a sterile, lyophilized single dose vial containing 82.5 IU Luveris® to deliver 75 IU Luveris®, after reconstitution with the diluent.
The following package combination is available:
- 1 vial Luveris® 75 IU and 1 vial 1 mL Sterile Water for Injection, USP, NDC 44087-1375-1
Storage: Vials may be stored refrigerated or at room temperature (2˚-25˚ C / 36˚-77˚ F). Protect from light.
Store in original package. Use immediately after reconstitution. Discard unused material.
Rx Only
Manufactured for: Serono, Inc. Rockland, MA 02370 USA
For more information, call toll-free 1-866-538-7879.
May 2005
| Luveris (lutropin alfa) | |||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
Revised: 12/2005Serono, Inc.