GAMMAGARD LIQUID
-
human immunoglobulin g injection, solution
Baxter Healthcare Corporation
----------
GAMMAGARD LIQUID[Immune Globulin Intravenous (Human)] 10%
DESCRIPTION
GAMMAGARD LIQUID Immune Globulin Intravenous (Human), 10% is a ready-for-use sterile, liquid preparation of highly purified and concentrated immunoglobulin G (IgG) antibodies. The distribution of the IgG subclasses is similar to that of normal plasma.1,2 The Fc and Fab functions are maintained in GAMMAGARD LIQUID. Pre-kallikrein activator activity is not detectable. GAMMAGARD LIQUID contains 100 mg/mL protein. At least 98% of the protein is gammaglobulin, the average immunoglobulin A (IgA) concentration is 37µg/mL, and immunoglobulin M is present in trace amounts. GAMMAGARD LIQUID contains a broad spectrum of IgG antibodies against bacterial and viral agents. Glycine (0.25M) serves as a stabilizing and buffering agent, and there are no added sugars, sodium or preservatives. The pH is 4.6 to 5.1. The osmolality is 240 to 300 mOsmol/kg, which is similar to physiological osmolality (285 to 295 mOsmol/kg).3
GAMMAGARD LIQUID is manufactured from large pools of human plasma. Screening against potentially infectious agents begins with the donor selection process and continues throughout plasma collection and plasma preparation. Each individual plasma donation used in the manufacture of GAMMAGARD LIQUID is collected only at FDA approved blood establishments and is tested by FDA licensed serological tests for Hepatitis B Surface Antigen (HBsAg), and for antibodies to Human Immunodeficiency Virus (HIV-1/HIV-2) and Hepatitis C Virus (HCV) in accordance with U.S. regulatory requirements. As an additional safety measure, mini-pools of the plasma are tested for the presence of HIV-1 and HCV by FDA licensed Nucleic Acid Testing (NAT) and found negative. IgGs are purified from plasma pools using a modified Cohn-Oncley cold ethanol fractionation process, as well as cation and anion exchange chromatography.
To further improve the margin of safety, three dedicated, independent and effective virus inactivation/removal steps have been integrated into the manufacturing and formulation processes, namely solvent/detergent (S/D) treatment,4,5 35 nm nanofiltration,6,7 and a low pH incubation at elevated temperature.8,9 The S/D process includes treatment with an organic mixture of tri-n-butyl phosphate, octoxynol 9 and polysorbate 80 at 18°C to 25°C for a minimum of 60 minutes.
In vitro virus spiking studies have been used to validate the capability of the manufacturing process to inactivate and remove viruses. To establish the minimum applicable virus clearance capacity of the manufacturing process, these virus clearance studies were performed under extreme conditions (e.g., at minimum S/D concentrations, incubation time and temperature for the S/D treatment). Virus clearance studies for GAMMAGARD LIQUID performed in accordance with good laboratory practices (Table 1) have demonstrated that:
- S/D treatment inactivates the lipid-enveloped viruses investigated to below detection limits within minutes.
- 35 nm nanofiltration removes lipid-enveloped viruses to below detection limits and reduces the non-lipid enveloped viruses HAV and B19V. As determined by a polymerase chain reaction assay, nanofiltration reduced B19V by a mean log10 reduction factor of 4.8 genome equivalents.
- Treatment with low pH at elevated temperature of 30°C to 32°C inactivates lipid-enveloped viruses and encephalomyocarditis virus (EMCV, model for HAV) to below detection limits, and reduces mice minute virus (MMV, model for B19V).
|
Abbreviations: HIV-1, Human Immunodeficiency Virus Type 1; BVDV, Bovine Viral Diarrhea Virus (model for Hepatitis C Virus and other lipid enveloped RNA viruses); WNV, West Nile Virus; PRV, Pseudorabies Virus (model for lipid enveloped DNA viruses, including Hepatitis B Virus); EMCV, Encephalomyocarditis Virus (model for non-lipid enveloped RNA viruses, including Hepatitis A virus [HAV]); MMV, Mice Minute Virus (model for non-lipid enveloped DNA viruses, including B19 virus [B19V]); n.d. (not done), n.a. (not applicable). |
|||||||
|
|||||||
| Virus type | Enveloped RNA | Enveloped DNA | Non-enveloped RNA | Non-enveloped DNA | |||
| Family | Retroviridae | Flaviviridae | Herpesviridae | Picornaviridae | Parvoviridae | ||
| Virus | HIV-1 | BVDV | WNV | PRV | HAV | EMCV | MMV |
| SD treatment 35 nm nanofiltrationLow pH treatment | > 4.5 | > 6.2 | n.a. | > 4.8 | n.d. | n.d. | n.d |
| > 4.5 | > 5.1 | > 6.2 | > 5.6 | 5.7 | 1.4 | 2.0 | |
| > 5.8 | > 5.5 | > 6.0 | > 6.5 | n.d† | > 6.3 | 3.1 | |
| Overall log reduction factor (ORF) | > 14.8 | > 16.8 | > 12.2 | > 16.9 | 5.7† | > 7.7 | 5.1 |
CLINICAL PHARMACOLOGY
Clinical Efficacy
Use of GAMMAGARD LIQUID in patients with Primary Immunodeficiency is supported by the Phase 3 clinical study of subjects who were treated with 300 to 600 mg/kg every 21 to 28 days for 12 months. The 61 subjects in this study were between 6 to 72 years of age, 54% female and 46% male, and 93% Caucasian, 5% African-American, and 2% Asian. Three subjects were excluded from the per-protocol analysis due to non-study product related reasons. The primary efficacy endpoint was the annualized rate of specified acute serious bacterial infections, i.e., the mean number of specified acute serious bacterial infections per subject per year (see Table 2).
| Number of Events | |
| Validated Infections* | |
| Bacteremia / Sepsis | 0 |
| Bacterial Meningitis | 0 |
| Osteomyelitis / Septic Arthritis | 0 |
| Bacterial Pneumonia | 0 |
| Visceral Abscess | 0 |
| Total | 0 |
| Hospitalizations Secondary to Infection | 0 |
| Mean Number of Validated Infections per Subject per Year | 0 |
| p-value† | p < 0.0001 |
| 95% Confidence Interval† | (0.000, 0.064) |
The secondary efficacy endpoints in this study were the annualized rate of other specified validated bacterial infections (see Table 3), and the number of hospitalizations secondary to all validated infectious complications (see Table 2 and Table 3).
| Number of Events | |
|
|
| Validated Infections * | |
| Urinary Tract Infection | 1 |
| Gastroenteritis | 1 |
| Lower Respiratory Tract Infection: Tracheobronchitis, Bronchiolitis Without Evidence of Pneumonia | 0 |
| Lower Respiratory Tract Infection: Other Infections (e.g., Lung Abscess, Empyema) | 0 |
| Otitis Media | 2 |
| Total | 4 |
| Hospitalizations Secondary to Infection | 0 |
| Mean Number of Validated Infections per Subject per Year | 0.07 |
| 95% Confidence Interval | (0.018, 0.168) |
In this study, there were no validated acute serious bacterial infections in any of the treated subjects. The annualized rate of acute serious bacterial infections was significantly less than (p < 0.0001) the rate of one infection per year, in accordance with recommendations by the FDA Blood Products Advisory Committee.10 Four of the 61 subjects reported a total of 4 other specified validated bacterial infections. None were serious or severe, none resulted in hospitalization, and all resolved completely.
The rate of all clinically-defined but non-validated infections was 3.4 infections per patient per year. These consisted primarily of recurrent episodes of commonly observed infections in this patient population - sinusitis, bronchitis, nasopharyngitis, urinary tract infections, and upper respiratory infections.
Pharmacokinetics
The overall pharmacokinetic characteristics of Immune Globulin Intravenous (Human) [IGIV] products are well-described in the literature.11,12 Following infusion, IGIV products show a biphasic decay curve. The initial (α) phase is characterized by an immediate post-infusion peak in serum IgG and is followed by rapid decay due to equilibration between the plasma and extravascular fluid compartments. The second (β) phase is characterized by a slower and constant rate of decay.
The commonly cited “normal” half-life of 18 to 25 days is based on studies in which tiny quantities of radiolabeled IgG are injected into healthy individuals.13,14 When radiolabeled IgG was injected into patients with hypogammaglobulinemia or agammaglobulinemia, highly variable half-lives ranging from 12 to 40 days were observed.13,14 In other radiolabeled studies, high serum concentrations of IgG, and hypermetabolism associated with fever and infection, have been seen to coincide with a shortened half-life of IgG.14-17
In contrast, however, pharmacokinetic studies in immunodeficient patients are based on the decline of IgG concentrations following infusions of large quantities of gammaglobulin. In such trials, investigators have reported uniformly prolonged half-lives of 26 to 35 days. 16,18,19-22
Pharmacokinetic parameters for GAMMAGARD LIQUID were determined from total IgG levels following the fourth infusion. A total of 61 subjects were enrolled and treated. Of these, 57 had sufficient pharmacokinetic data to be included in the dataset. Pharmacokinetic parameters are presented in Table 4.
| Parameter | Median | 95% Confidence Interval |
|
Abbreviations: AUC= area under the curve; Cmax=maximum concentration; Cmin= minimum concentration |
||
| Elimination Half-Life ( T ½ days) | 35 | (31, 42) |
| AUC0-21d (mg·days/dL) | 29139 | (27494, 30490) |
| Cmax (Peak, mg/dL) | 2050 | (1980, 2200) |
| Cmin (Trough, mg/dL) | 1030 | (939, 1110) |
| Incremental recovery (mg/dL)/(mg/kg) | 2.3 | (2.2, 2.6) |
Median IgG trough levels were maintained between 960 to 1120 mg/dL. These dosing regimens maintained serum trough IgG levels considerably above 450 mg/dL, which is consistent with levels considered to be effective in the treatment of patients with Primary Immunodeficiency.23,24 The elimination half-life of GAMMAGARD LIQUID of 35 days was similar to the half-lives reported for other IGIV products.13-15,17,25,26
INDICATIONS AND USAGE
Primary Immunodeficiency
GAMMAGARD LIQUID is indicated for the treatment of primary immunodeficiency disorders associated with defects in humoral immunity. These include but are not limited to congenital X-linked agammaglobulinemia, common variable immunodeficiency, Wiskott-Aldrich syndrome, and severe combined immunodeficiencies.15,22
CONTRAINDICATIONS
GAMMAGARD LIQUID is contraindicated in patients with known anaphylactic or severe hypersensitivity responses to Immune Globulin (Human).
Patients with severe selective IgA deficiency (IgA < 0.05 g/L) may develop anti-IgA antibodies that can result in a severe anaphylactic reaction. Anaphylaxis can occur using GAMMAGARD LIQUID even though it contains low amounts of IgA (average concentration of 37μg/mL). These patients should be treated only if their IgA deficiency is associated with an immune deficiency for which therapy with intravenous immune globulin is clearly indicated. Such patients should only receive intravenous immune globulin with utmost caution and in a setting where supportive care is available for treating life-threatening reactions.
WARNINGS
Immune Globulin Intravenous (Human) products have been reported to be associated with renal dysfunction, acute renal failure, osmotic nephrosis, and death.27 Patients predisposed to acute renal failure include patients with any degree of pre-existing renal insufficiency, diabetes mellitus, age greater than 65, volume depletion, sepsis, paraproteinemia, or patients receiving known nephrotoxic drugs. Especially in such patients, IGIV products should be administered at the minimum concentration available and the minimum rate of infusion practicable. While these reports of renal dysfunction and acute renal failure have been associated with the use of many of the licensed IGIV products, those containing sucrose as a stabilizer accounted for a disproportionate share of the total number.
Glycine, an amino acid, is used as a stabilizer. GAMMAGARD LIQUID does not contain sucrose.
See PRECAUTIONS and DOSAGE AND ADMINISTRATION sections for important information intended to reduce the risk of acute renal failure.
Immune Globulin Intravenous (Human), 10% is made from human plasma. Products made from human plasma may contain infectious agents, such as viruses, that can cause disease. The risk that such products will transmit an infectious agent has been reduced by screening plasma donors for prior exposure to certain viruses, by testing for the presence of certain current virus infections, and by inactivating and/or removing certain viruses (see DESCRIPTION). Despite these measures, such products can still potentially transmit disease. Because this product is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses and theoretically, the Creutzfeldt-Jakob disease (CJD) agent. ALL infections thought by a physician possibly to have been transmitted by this product should be reported by the physician or other healthcare provider to Baxter Healthcare Corporation, at 1-800-423-2862 (in the U.S.). The physician should discuss the risks and benefits of this product with the patient.
GAMMAGARD LIQUID should only be administered intravenously. Other routes of administration have not been evaluated.
Immediate anaphylactic and hypersensitivity reactions are a remote possibility. Epinephrine and antihistamines should be available for treatment of any acute anaphylactoid reactions.
PRECAUTIONS
General
Some viruses, such as B19V (formerly known as Parvovirus B19) or Hepatitis A, are particularly difficult to remove or inactivate. B19V most seriously affects pregnant women, or immune-compromised individuals. Symptoms of B19V infection include fever, drowsiness, chills and runny nose followed about two weeks later by a rash and joint pain. Evidence of Hepatitis A may include several days to weeks of poor appetite, tiredness, and low-grade fever followed by nausea, vomiting and abdominal pain. Dark urine and a yellowed complexion are also common symptoms. Patients should be encouraged to consult their physician if such symptoms appear.
Components used in the packaging of this product are latex-free.
Renal Function
Periodic monitoring of renal function tests and urine output is particularly important in patients judged to have a potential increased risk for developing acute renal failure. Assure that patients are not volume depleted prior to the initiation of infusion of GAMMAGARD LIQUID. Renal function, including measurement of blood urea nitrogen (BUN)/serum creatinine, should be assessed prior to the initial infusion of IGIV products and again at appropriate intervals thereafter. If renal function deteriorates, discontinuation of the product should be considered.
For patients judged to be at risk of developing renal dysfunction, it may be prudent to reduce the rate of infusion to less than 3.3 mg IgG/kg/min (< 2 mL/kg/hr).
Hemolysis
IGIV products can contain blood group antibodies which may act as hemolysins and induce in vivo coating of red blood cells with immunoglobulin, causing a positive direct antiglobulin reaction and, rarely, hemolysis.28-30 Hemolytic anemia can develop subsequent to IGIV therapy due to enhanced red blood cells (RBC) sequestration (see ADVERSE REACTIONS).31 IGIV recipients should be monitored for clinical signs and symptoms of hemolysis (see PRECAUTIONS: Laboratory Tests).
Transfusion-Related Acute Lung Injury (TRALI)
There have been reports of noncardiogenic pulmonary edema (Transfusion Related Acute Lung Injury [TRALI]) in patients administered IGIV.32 TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever, and typically occurs within 1to 6 hours after transfusion. Patients with TRALI may be managed using oxygen therapy with adequate ventilatory support.
IGIV recipients should be monitored for pulmonary adverse reactions. If TRALI is suspected, appropriate tests should be performed for the presence of anti-neutrophil antibodies in both the product and patient serum (see PRECAUTIONS: Laboratory Tests).
Thrombotic Events
Thrombotic events have been reported in association with IGIV (see ADVERSE REACTIONS).33-41 Patients at risk may include those with a history of atherosclerosis, multiple cardiovascular risk factors, advanced age, impaired cardiac output, and/or known or suspected hyperviscosity, hypercoagulable disorders and prolonged periods of immobilization. The potential risks and benefits of IGIV should be weighed against those of alternative therapies for all patients for whom IGIV administration is being considered. Baseline assessment of blood viscosity should be considered in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies (see PRECAUTIONS: Laboratory Tests).
Aseptic Meningitis Syndrome
An aseptic meningitis syndrome (AMS) has been reported to occur infrequently in association with IGIV treatment. Discontinuation of IGIV treatment has resulted in remission of AMS within several days without sequelae. The syndrome usually begins within several hours to two days following IGIV treatment. It is characterized by symptoms and signs including severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, and nausea and vomiting. Cerebrospinal fluid (CSF) studies are frequently positive with pleocytosis up to several thousand cells per cubic mm, predominantly from the granulocytic series, and elevated protein levels up to several hundred mg/dL. Patients exhibiting such symptoms and signs should receive a thorough neurological examination, including CSF studies, to rule out other causes of meningitis. AMS may occur more frequently in association with high dose (2 g/kg) IGIV treatment.
Laboratory Tests
If signs and/or symptoms of hemolysis are present after IGIV infusion, appropriate confirmatory laboratory testing should be done [see PRECAUTIONS].
If TRALI is suspected, appropriate tests should be performed for the presence of anti-neutrophil antibodies in both the product and patient serum [see PRECAUTIONS].
Because of the potentially increased risk of thrombosis, baseline assessment of blood viscosity should be considered in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies [see PRECAUTIONS].
Information For Patients
Patients should be instructed to immediately report symptoms of decreased urine output, sudden weight gain, fluid retention/edema, and/or shortness of breath (which may suggest kidney damage) to their physicians.
Drug Interactions
SeeDOSAGE AND ADMINISTRATION section.
Pregnancy Category C
Animal reproduction studies have not been conducted with GAMMAGARD LIQUID. It is also not known whether GAMMAGARD LIQUID can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. GAMMAGARD LIQUID should be given to a pregnant woman only if clearly indicated. Maternally administered IGIV products have been shown to cross the placenta, increasingly after 30 weeks gestation.42-44
Use in Pediatrics
The safety and efficacy of GAMMAGARD LIQUID has not been evaluated in neonates or infants.
ADVERSE REACTIONS
General
Various mild and moderate reactions, such as headache, fever, fatigue, chills, flushing, dizziness, urticaria, wheezing or chest tightness, nausea, vomiting, rigors, back pain, chest pain, muscle cramps, and changes in blood pressure may occur with infusions of Immune Globulin Intravenous (Human). In general, reported adverse reactions to GAMMAGARD LIQUID in patients with Primary Immunodeficiency are similar in kind and frequency to those observed with other IGIV products. Slowing or stopping the infusion usually allows the symptoms to disappear promptly. Although hypersensitivity reactions have not been reported in the clinical studies with GAMMAGARD LIQUID immediate anaphylactic and hypersensitivity reactions are a remote possibility. Epinephrine and antihistamines should be available for treatment of any acute anaphylactic reactions (see WARNINGS).
Clinical Study
Adverse experiences were examined among a total of 61 enrolled subjects with Primary Immunodeficiency who received at least one infusion of GAMMAGARD LIQUID during the Phase 3 multicenter clinical study. For this study, temporally associated adverse events are defined by the FDA as those occurring during or within 72 hours of completion of an infusion. Adverse drug reactions (ADR’s) are those adverse events that were deemed by the investigators as causally related to the infusion of GAMMAGARD LIQUID.
Of all adverse experiences, 15 events in 8 subjects were serious. Two serious events, two episodes of aseptic meningitis in one patient, were deemed to be possibly related to the infusion of GAMMAGARD LIQUID.
Among the 896 non-serious adverse experiences, 258 were judged by the investigator to be possibly or probably related to the infusion of GAMMAGARD LIQUID. Of these, 136 were mild, 106 were moderate, and 16 were severe. All of the severe non-serious adverse experiences were transient, did not lead to hospitalization, and resolved without complication. One subject withdrew from the study due to a non-serious adverse experience (papular rash).
Of the 345 temporally related adverse experiences, those occurring in > 5% of subjects are shown in Table 5. Of these events, only headache occurred in association with more than 5% of infusions. All events were expected based on past experiences with intravenous gammaglobulin products.
| Event | By Infusion | By Subject | ||
| Number | Percentage | Number | Percentage | |
|
||||
| Headache | 57 | 6.90 | 22 | 36.1 |
| Fever | 19 | 2.30 | 13 | 21.3 |
| Fatigue | 18 | 2.18 | 10 | 16.4 |
| Vomiting | 10 | 1.21 | 9 | 14.8 |
| Chills | 14 | 1.69 | 8 | 13.1 |
| Infusion site events | 8 | 0.97 | 8 | 13.1 |
| Nausea | 9 | 1.09 | 6 | 9.8 |
| Dizziness | 7 | 0.85 | 6 | 9.8 |
| Pain in Extremity | 7 | 0.85 | 5 | 8.2 |
| Diarrhea | 7 | 0.85 | 5 | 8.2 |
| Cough | 5 | 0.61 | 5 | 8.2 |
| Pruritus | 5 | 0.61 | 4 | 6.5 |
| Pharyngeal Pain | 5 | 0.61 | 4 | 6.5 |
The majority (227/258) of the non-serious adverse experiences deemed related to study product were considered expected based on previous experience with IGIV products and 31 were considered unexpected. In virtually every case, these unexpected events were either consistent with the subject’s specific type of immunodeficiency or with the subject’s medical history prior to entering the study. A total of 14 hospitalizations occurred during the study but none were related to infection.
Hematology and clinical chemistry parameters were monitored in all subjects prior to each infusion throughout the 12-month period of study. Mean values for all laboratory parameters remained consistent throughout the study period. Three of the hematology values in one subject were outside of the normal range and reported as non-serious adverse experiences that resolved completely. These were a red cell count of 3.9 x106/μL, hematocrit of 31%, and white cell count of 3.88 x 103/μL. All spontaneously returned to baseline. One subject had an elevated BUN (45 mg/dL) and creatinine (1.4 mg/dL) on one occasion that were reported as non-serious adverse experiences and resolved completely. These values improved to 30 mg/dL and 0.8 mg/dL, respectively, by the next infusion. Six of the patients had a single, transient elevation in serum transaminases. Two additional patients had persistent elevations in transaminases, ALT and AST, which were present at the initiation of the study, prior to the infusion of GAMMAGARD LIQUID. There was no other evidence of liver abnormalities. None of the hematology or chemistry laboratory abnormalities that occurred during the course of the study required clinical intervention and none had clinical consequences.
During the Phase 3 clinical study, viral safety was assessed by serological screening for HBsAg and antibodies to HCV and HIV-1 and HIV-2 prior to, during, and at the end of the study and by Polymerase Chain Reaction (PCR) tests for HBV, HCV, and HIV-1 genomic sequences prior to and at the end of the study. None of the 61 treated subjects were positive prior to study entry and none converted from negative to positive during the 12-month period of study.
Postmarketing:
The following is a list of adverse reactions that have been identified and reported during the post-approval use of IGIV products:
Post-marketing ADRs Identified and Reported Post-Approval
Respiratory
Cyanosis, hypoxemia, pulmonary edema, dyspnea, bronchospasm
Cardiovascular
thromboembolism, hypotension
Neurological
seizures, tremor
Hematologic
hemolysis, positive direct antiglobulin (Coombs) test
General/Body as a Whole
pyrexia, rigors
Musculoskeletal
back pain
Gastrointestinal
hepatic dysfunction, abdominal pain
Rare and Uncommon Adverse Events:
Respiratory
apnea, Acute Respiratory Distress Syndrome (ARDS), Transfusion Related Acute Lung Injury (TRALI)
Integumentary
bullous dermatitis, epidermolysis, erythema multiforme, Stevens-Johnson syndrome
Cardiovascular
cardiac arrest, vascular collapse
Neurological
coma, loss of consciousness
Hematologic
pancytopenia, leukopenia
Because postmarketing reporting of these reactions is voluntary and the at-risk populations are of uncertain size, it is not always possible to reliably estimate the frequency of the reaction to establish a causal relationship to exposure to the product. Such is also the case with literature reports authored independently45 (see PRECAUTIONS).
DOSAGE AND ADMINISTRATION
GAMMAGARD LIQUID should be at room temperature during administration.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. Do not use if particulate matter and/or discoloration is observed. Only clear or slightly opalescent and colorless or pale yellow solutions are to be administered. GAMMAGARD LIQUID should only be administered intravenously. Other routes of administration have not been evaluated. The use of an in-line filter is optional.
For patients with Primary Immunodeficiency, monthly doses of approximately 300 to 600 mg/kg infused at 3 to 4 week intervals are commonly used.23,24 As there are significant differences in the half-life of IgG among patients with Primary Immunodeficiency, the frequency and amount of immunoglobulin therapy may vary from patient to patient. The proper amount can be determined by monitoring clinical response. The minimum serum concentration of IgG necessary for protection varies among patients and has not been established by controlled clinical studies.
Rate of Administration
During the first infusion of the Phase 3 clinical study, GAMMAGARD LIQUID was infused at an initial rate of 0.5 mL/kg/hr (0.8 mg/kg/min). The rate was gradually increased every 30 minutes to a rate of 5.0 mL/kg/hr (8.9 mg/kg/min) if it was well tolerated. However, some patients completed the infusion before the maximum rate could be obtained. During subsequent infusions the initial rate and the rate of escalation were based on their previous infusion history; however, the maximum rate attained during the first infusion was used throughout the remainder of the study. The mean rate attained by all patients was 4.3 mL/kg/hr. Fifty-eight subjects (95%) achieved a maximum rate of 4.0 mL/kg/hr or greater and of these, 16 subjects (26%) attained a rate of 5.0 mL/kg/hr.
In general, it is recommended that patients beginning therapy with IGIV or switching from one IGIV product to another be started at the lower rates and then advanced to the maximal rate if they have tolerated several infusions at intermediate rates of infusion. It is important to individualize rates for each patient.
As noted in the WARNINGS section, patients who have underlying renal disease or who are judged to be at risk of developing thrombotic events should not be infused rapidly with any IGIV product. Although there are no prospective studies demonstrating that any concentration or rate of infusion is completely safe, it is believed that risk is decreased at lower rates of infusion.46 Therefore, as a guideline, it is recommended that these patients who are judged to be at risk of renal dysfunction or thrombotic complications be gradually titrated up to a more conservative maximal rate of less than 3.3 mgIgG/kg/min (< 2mL/kg/hr).
A rate of administration that is too rapid may cause flushing and changes in pulse rate and blood pressure. Slowing or stopping the infusion usually results in the prompt disappearance of signs. The infusion may then be resumed at a rate that is comfortable for the patient.
Drug Interactions
Antibodies in IGIV products may interfere with patient responses to live vaccines, such as those for measles, mumps and rubella.47,48,49 The immunizing physician should be informed of recent therapy with IGIV products so that appropriate precautions can be taken.
Admixtures of GAMMAGARD LIQUID with other drugs and intravenous solutions have not been evaluated. It is recommended that GAMMAGARD LIQUID be administered separately from other drugs or medications that the patient may be receiving. The product should not be mixed with IGIV products from other manufacturers.
Normal saline should not be used as a diluent. If dilution is preferred, GAMMAGARD LIQUID may be diluted with 5% dextrose in water (D5W).50 No other drug interactions or compatibilities have been evaluated.
HOW SUPPLIED
GAMMAGARD LIQUID is supplied in single use bottles as follows:
| NDC Number | Volume | Grams |
| 0944-2700-02 | 10 mL | 1.0 |
| 0944-2700-03 | 25 mL | 2.5 |
| 0944-2700-04 | 50 mL | 5.0 |
| 0944-2700-05 | 100 mL | 10.0 |
| 0944-2700-06 | 200 mL | 20.0 |
STORAGE
Refrigeration: 36 months storage at refrigerated temperature 2° to 8°C (36°-46°F). Do not freeze.
Room Temperature: 12 months storage at room temperature 25°C, (77°F) within the first 24 months of the date of manufacture. See below for detailed storage information.
The total storage time of GAMMAGARD LIQUID depends on the point of time the vial is transferred to room temperature. Examples for total storage times are illustrated in Figure 1. The new expiration date must be recorded on the package when the product is transferred to room temperature.
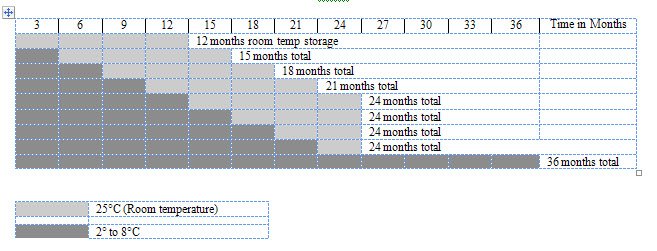
Figure 1: Storage Guidelines Months for Date of Manufacture
Storage Details:
- Example 1: If the product is taken out of the refrigerator after 3 months from date of manufacture, it can be stored for 12 months at room temperature. Total storage time is 15 months.
- Example 2: If the product is taken out of the refrigerator after 21 months from the date of manufacture, it can be stored for 3 additional months at room temperature. Total storage time is 24 months.
- After 24 months from date of manufacture, product cannot be stored at room temperature.
REFERENCES
- Skvaril F. Qualitative and quantitative aspects of IgG subclasses in i.v. immunoglobulin preparations. In: Nydegger UE, ed. Immunotherapy. London: Academic Press; 1981:118-122.
- French M. Serum IgG subclasses in normal adults. Monogr Allergy. 1986;19:100-107.
- Lacy CF, Armstrong LL, Goldman MP, Lance LL. Appendix: Abbreviations and Measurements. Drug Information Handbook. Lexi-Comp; 1999:1254.
- Horowitz B, Prince AM, Hamman J, Watklevicz C. Viral safety of solvent/detergent-treated blood products. Blood Coagul Fibrinolysis. 1994;5 Suppl 3:S21-S28.
- Kreil TR, Berting A, Kistner O, Kindermann J. West Nile virus and the safety of plasma derivatives: verification of high safety margins, and the validity of predictions based on model virus data. Transfusion. 2003;43:1023-1028.
- Hamamoto Y, Harada S, Kobayashi S, et al. A novel method for removal of human immunodeficiency virus: filtration with porous polymeric membranes. Vox Sang. 1989;56:230-236.
- Yuasa T, Ishikawa G, Manabe S, Sekiguchi S, Takeuchi K, Miyamura T. The particle size of hepatitis C virus estimated by filtration through microporous regenerated cellulose fibre. J Gen Virol. 1991;72 ( Pt 8):2021-2024.
- Kempf C, Jentsch P, Poirier B, et al. Virus inactivation during production of intravenous immunoglobulin. Transfusion. 1991;31:423-427.
- Louie RE, Galloway CJ, Dumas ML, Wong MF, Mitra G. Inactivation of hepatitis C virus in low pH intravenous immunoglobulin. Biologicals. 1994;22:13-19.
- Golding B. IGIV Clinical Endpoints. Presented at: Blood Products Advisory Committee, 65th Meeting. 17 March 2000. Silver Spring, MD.
- Schiff RI. Intraveneous immunoglobulins for treatment of antibody deficiencies. In: Good RA, Lindenlaub E, eds. The Nature, Cellular, and Biochemical Basis and Management of Immunodeficiencies. Symposium Vernried, West Germany. 21-25 September. Stuttgart: F. K. Schattauer Verlag; 1986:523-541.
- Morell A. Pharmacokinetics of intravenous immunoglobulin preparations. In: Lee ML, Strand V, eds. Intravenous Immunoglobulins in Clinical Practice. New York: M. Dekker, Inc.; 1997:1-18.
- Morell A, Skvaril F. Structure and Biological Properties of Immunoglobulins and γ-Globulin Preparations. II. Properties of γ-Globulin Preparations, Schweizerishche Medizinische Wochenschrift 1980; 110:80-85.
- Waldmann TA, Strober W. Metabolism of immunoglobulins. Prog Allergy. 1969;13:1-110.
- Stiehm ER. Standard and special human immune serum globulins as therapeutic agents. Pediatrics. 1979;63:301-319.
- Lee ML, Mankarious S, Ochs H, Fischer S, Wedgwood RJ. The pharmacokinetics of total IgG, IgG subclasses, and type specific antibodies in immunodeficient patients. Immunol Invest. 1991;20:193-198.
- Buckley RH. Immunoglobulin replacement therapy: indications and contraindications for use and variable IgG levels achieved. In: Alving BM, Finlayson JS, eds. Immunoglobulins: characteristics and use of intravenous preparations. Washington, D.C.: US Department of Health and Human Services; 1979:3-8.
- Mankarious S, Lee M, Fischer S, et al. The half-lives of IgG subclasses and specific antibodies in patients with primary immunodeficiency who are receiving intravenously administered immunoglobulin. J Lab Clin Med. 1988;112:634-640.
- Pirofsky B. Safety and toxicity of a new serum immunoglobulin G intravenous preparation, IGIV pH 4.25. Rev Infect Dis. 1986;8 Suppl 4:S457-63.
- Pirofsky B. Clinical use of a new pH 4.25 intravenous immunoglobulin preparation (Gamimune-N). J Infect. 1987;15 Suppl 1:29-37.
- Schiff RI. Half-life and clearance of pH 6.8 and pH 4.25 immunoglobulin G intravenous preparations in patients with primary disorders of humoral immunity. Rev Infect Dis. 1986;8 Suppl 4:S449-56.
- Schiff RI, Rudd C. Alterations in the half-life and clearance of IgG during therapy with intravenous gamma-globulin in 16 patients with severe primary humoral immunodeficiency. J Clin Immunol. 1986;6:256-264.
- Eijkhout HW, Der Meer JW, Kallenberg CG, et al. The effect of two different dosages of intravenous immunoglobulin on the incidence of recurrent infections in patients with primary hypogammaglobulinemia. A randomized, double-blind, multicenter crossover trial. Ann Intern Med. 2001;135:165-174.
- Roifman CM, Gelfand EW. Replacement therapy with high dose intravenous gamma-globulin improves chronic sinopulmonary disease in patients with hypogammaglobulinemia. Pediatr Infect Dis J. 1988;7:S92-S96.
- Ballow M, Berger M, Bonilla FA, et al. Pharmacokinetics and tolerability of a new intravenous immunoglobulin preparation, IGIV-C, 10% (Gamunex, 10%). Vox Sang. 2003;84:202-210.
- Ochs HD, Pinciaro PJ, The Octagam Study Group. Octagam((R)) 5%, an intravenous IgG product, is efficacious and well tolerated in subjects with primary immunodeficiency diseases. J Clin Immunol. 2004;24:309-314.
- Cayco AV, Perazella MA, Hayslett JP. Renal insufficiency after intravenous immune globulin therapy: a report of two cases and an analysis of the literature. J Am Soc Nephrol. 1997;8:1788-1794.
- Copelan EA, Strohm PL, Kennedy MS, Tutschka PJ. Hemolysis following intravenous immune globulin therapy. Transfusion. 1986;26:410-412.
- Wilson JR, Bhoopalam H, Fisher M. Hemolytic anemia associated with intravenous immunoglobulin. Muscle Nerve. 1997;20:1142-1145.
- Thomas MJ, Misbah SA, Chapel HM, Jones M, Elrington G, Newsom-Davis J. Hemolysis after high-dose intravenous Ig. Blood. 1993;82:3789.
- Kessary-Shoham H, Levy Y, Shoenfeld Y, Lorber M, Gershon H. In vivo administration of intravenous immunoglobulin (IVIg) can lead to enhanced erythrocyte sequestration. J Autoimmun. 1999;13:129-135.
- Rizk A, Gorson KC, Kenney L, Weinstein R. Transfusion-related acute lung injury after the infusion of IVIG. Transfusion. 2001;41:264-268.
- Brannagan TH, III, Nagle KJ, Lange DJ, Rowland LP. Complications of intravenous immune globulin treatment in neurologic disease. Neurology. 1996;47:674-677.
- Dalakas MC. High-dose intravenous immunoglobulin and serum viscosity: risk of precipitating thromboembolic events. Neurology. 1994;44:223-226.
- ElKayam O, Paran D, Milo R, et al. Acute myocardial infarction associated with high dose intravenous immunoglobulin infusion for autoimmune disorders. A study of four cases. Ann Rheum Dis. 2000;59:77-80.
- Gomperts ED, Darr F. Rapid infusion of intravenous immunoglobulin in patients with neuromuscular diseases. Neurology. 2002;58:1444.
- Haplea SS, Farrar JT, Gibson GA, Laskin M, Pizzi LT, Ashbury AK. Thromboembolic events associated with intravenous immunoglobulin therapy [abstract]. Neurology. 1997;48:A54.
- Harkness K, Howell SJ, Davies-Jones GA. Encephalopathy associated with intravenous immunoglobulin treatment for Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 1996;60:586.
- Kwan T, Keith P. Stroke following intravenous immunoglobulin infusion in a 28-year-old male with common variable immune deficiency: a case report and literature review. Can J Allergy Clin Immunol. 1999;4:250-253.
- Wolberg AS, Kon RH, Monroe DM, Hoffman M. Coagulation factor XI is a contaminant in intravenous immunoglobulin preparations. Am J Hematol. 2000;65:30-34.
- Woodruff RK, Grigg AP, Firkin FC, Smith IL. Fatal thrombotic events during treatment of autoimmune thrombocytopenia with intravenous immunoglobulin in elderly patients. Lancet. 1986;2:217-218.
- Hammarstrom L, Smith CI. Placental transfer of intravenous immunoglobulin. Lancet. 1986;1:681.
- Morell A, Sidiropoulos D, Herrmann U, et al. IgG subclasses and antibodies to group B streptococci, pneumococci, and tetanus toxoid in preterm neonates after intravenous infusion of immunoglobulin to the mothers. Pediatr Res. 1986;20:933-936.
- Sidiropoulos D, Herrmann U, Jr., Morell A, von Muralt G, Barandun S. Transplacental passage of intravenous immunoglobulin in the last trimester of pregnancy. J Pediatr. 1986;109:505-508.
- Pierce LR, Jain N. Risks associated with the use of intravenous immunoglobulin. Transfusion Med Rev. 2003;17:241-251.
- Tan E, Hajinazarian M, Bay W, Neff J, Mendell JR. Acute renal failure resulting from intravenous immunoglobulin therapy. Arch Neurol. 1993;50:137-139.
- Morbidity and Mortality Weekly Report. Measles, Mumps, and Rubella; Vaccine use and strategies for elimination of measles, rubella, and congenital rubella syndrome and control of mumps. Recommendations for the Advisory Committee on Immunization Practices (ACIP). 98 A.D.;47.
- Peter G. Summary of major changes in the 1994 Red Book: American Academy of Pediatrics. Report of the Committee on Infectious Disease. Pediatrics. 1994;93:1000-1002.
- Siber GR, Werner BG, Halsey NA, et al. Interference of immune globulin with measles and rubella immunization. J Pediatr. 1993;122:204-211.
- Data on file, Baxter Healthcare Corporation.
To enroll in the confidential, industry-wide Patient Notification System, call 1-888-UPDATE U (1-888-873-2838)
Baxter and GAMMAGARD LIQUID are trademarks of Baxter International Inc.,
Baxter Healthcare Corporation
Westlake Village, CA 91362 USA
U.S. License No. 140
Revised Oct 2009
PACKAGE LABEL - PRINCIPAL DISPLAY PANEL
GAMMAGARD LIQUID 10g unit carton
NDC 0944-2700-06
Immune Globulin Intravenous
(Human) 10%
GAMMAGARD LIQUID
10g/100mL
Solution for Infusion
Baxter (logo)

GAMMAGARD LIQUID 10g vial label
NDC 0944-2700-05
single-dose container
Immune Globulin Intravenous
(Human) 10%
GAMMAGARD LIQUID
10g/100mL
Solution for Infusion
Refrigeration: 36 months storage at refrigerated temperature 2°-8°C (36°F-46°F). Do not freeze.
Room temperature: 12 months storage at room temperature 25°C (77°F) within the first 24 months for the date of manufacture.
See package insert for detailed storage information.
Latex free.
For intravenous use only
Rx Only
Baxter Healthcare Corporation,
Westlake Village, CA 91362 USA,
U.S. License No. 140
| GAMMAGARD LIQUID
human immunoglobulin g injection, solution |
|||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| BLA | BLA125105 | 07/30/2010 | |
| Labeler - Baxter Healthcare Corporation (039121363) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Baxter SA | 370191025 | MANUFACTURE | |