PROLIA
-
denosumab injection
Amgen, Inc
----------
|
|||||||||||||||||||
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Treatment of Postmenopausal Women with Osteoporosis at High Risk for Fracture
Prolia is indicated for the treatment of postmenopausal women with osteoporosis at high risk for fracture, as defined by factors such as a history of osteoporotic fracture, or multiple risk factors for fracture; or patients who have failed or are intolerant to other available osteoporosis therapy. In postmenopausal women with osteoporosis, Prolia reduces the incidence of vertebral, nonvertebral and hip fractures [see Clinical Studies (14.1)].
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Prolia should be administered by a healthcare professional.
The recommended dose of Prolia is 60 mg administered as a single subcutaneous injection once every 6 months. Administer Prolia via subcutaneous injection in the upper arm, the upper thigh, or the abdomen. All patients should receive calcium 1000 mg daily and at least 400 IU vitamin D daily [see Warnings and Precautions (5.1)].
If a dose of Prolia is missed, administer the injection as soon as the patient is available. Thereafter, schedule injections every 6 months from the date of the last injection.
2.2 Preparation and Administration
Visually inspect Prolia for particulate matter and discoloration prior to administration whenever solution and container permit. Prolia is a clear, colorless to pale yellow solution that may contain trace amounts of translucent to white proteinaceous particles. Do not use if the solution is discolored or cloudy or if the solution contains many particles or foreign particulate matter.
Latex Allergy: People sensitive to latex should not handle the grey needle cap on the single-use prefilled syringe, which contains dry natural rubber (a derivative of latex).
Prior to administration, Prolia may be removed from the refrigerator and brought to room temperature (up to 25°C/77°F) by standing in the original container. This generally takes 15 to 30 minutes. Do not warm Prolia in any other way[see How Supplied/Storage and Handling (16)].
Instructions for Prefilled Syringe with Needle Safety Guard
IMPORTANT: In order to minimize accidental needlesticks, the Prolia single-use prefilled syringe will have a green safety guard; manually activate the safety guard after the injection is given.
DO NOT slide the green safety guard forward over the needle before administering the injection – it will lock in place and prevent injection.
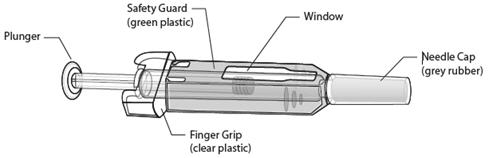
Activate the green safety guard (slide over the needle) after the injection.
The grey needle cap on the single use prefilled syringe contains dry natural rubber (a derivative of latex); people sensitive to latex should not handle the cap.
Step 1: Remove Grey Needle Cap
| Remove needle cap. | 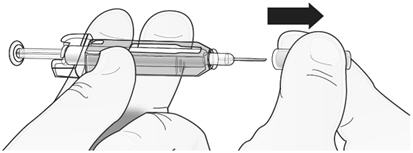 |
Step 2: Administer Injection
| Insert needle and inject all the liquid. | 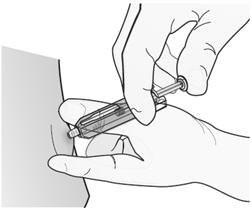 |
DO NOT put grey needle cap back on needle.
Step 3: Immediately Slide Green Safety Guard Over Needle
With the needle pointing away from you…
Hold the prefilled syringe by the clear plastic finger grip with one hand. Then, with the other hand, grasp the green safety guard by its base and gently slide it towards the needle until the green safety guard locks securely in place and/or you hear a “click.” DO NOT grip the green safety guard too firmly – it will move easily if you hold and slide it gently.
| Hold clear finger grip. | 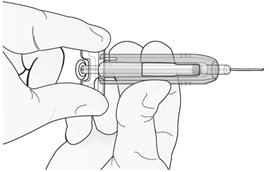 |
| Gently slide green safety guard over needle and lock securely in place. Do not grip green safety guard too firmly when sliding over needle. | 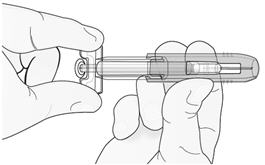 |
Immediately dispose of the syringe and needle cap in the nearest sharps container. DO NOT put the needle cap back on the used syringe.
Instructions for Single-use Vial
For administration of Prolia from the single-use vial, use a 27-gauge needle to withdraw and inject the 1 mL dose. Do not re-enter the vial. Discard vial and any liquid remaining in the vial.
3 DOSAGE FORMS AND STRENGTHS
- 1 mL of a 60 mg/mL solution in a single-use prefilled syringe
- 1 mL of a 60 mg/mL solution in a single-use vial
4 CONTRAINDICATIONS
4.1 Hypocalcemia
Pre-existing hypocalcemia must be corrected prior to initiating therapy with Prolia [see Warnings and Precautions (5.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Hypocalcemia and Mineral Metabolism
Hypocalcemia may be exacerbated by the use of Prolia. Pre-existing hypocalcemia must be corrected prior to initiating therapy with Prolia. In patients predisposed to hypocalcemia and disturbances of mineral metabolism (e.g. history of hypoparathyroidism, thyroid surgery, parathyroid surgery, malabsorption syndromes, excision of small intestine, severe renal impairment [creatinine clearance < 30 mL/min] or receiving dialysis), clinical monitoring of calcium and mineral levels (phosphorus and magnesium) is highly recommended.
Hypocalcemia following Prolia administration is a significant risk in patients with severe renal impairment [creatinine clearance < 30 mL/min], or receiving dialysis. Instruct all patients with severe renal impairment, including those receiving dialysis, about the symptoms of hypocalcemia and the importance of maintaining calcium levels with adequate calcium and vitamin D supplementation.
Adequately supplement all patients with calcium and vitamin D [see Dosage and Administration (2.1), Contraindications (4.1), Adverse Reactions (6.1), and Patient Counseling Information (17.1)].
5.2 Serious Infections
In a clinical trial of over 7800 women with postmenopausal osteoporosis, serious infections leading to hospitalization were reported more frequently in the Prolia group than in the placebo group [see Adverse Reactions (6.1)]. Serious skin infections, as well as infections of the abdomen, urinary tract, and ear, were more frequent in patients treated with Prolia. Endocarditis was also reported more frequently in Prolia-treated subjects. The incidence of opportunistic infections was balanced between placebo and Prolia groups, and the overall incidence of infections was similar between the treatment groups. Advise patients to seek prompt medical attention if they develop signs or symptoms of severe infection, including cellulitis.
Patients on concomitant immunosuppressant agents or with impaired immune systems may be at increased risk for serious infections. Consider the benefit-risk profile in such patients before treating with Prolia. In patients who develop serious infections while on Prolia, prescribers should assess the need for continued Prolia therapy .
5.3 Dermatologic Adverse Reactions
In a large clinical trial of over 7800 women with postmenopausal osteoporosis, epidermal and dermal adverse events such as dermatitis, eczema, and rashes occurred at a significantly higher rate in the Prolia group compared to the placebo group. Most of these events were not specific to the injection site [see Adverse Reactions (6.1)]. Consider discontinuing Prolia if severe symptoms develop.
5.4 Osteonecrosis of the Jaw
Osteonecrosis of the jaw (ONJ), which can occur spontaneously, is generally associated with tooth extraction and/or local infection with delayed healing. ONJ has been reported in patients receiving denosumab [see Adverse Reactions (6.1)]. A routine oral exam should be performed by the prescriber prior to initiation of Prolia treatment. A dental examination with appropriate preventive dentistry should be considered prior to treatment with Prolia in patients with risk factors for ONJ such as invasive dental procedures (e.g., tooth extraction, dental implants, oral surgery), diagnosis of cancer, concomitant therapies (e.g., chemotherapy, corticosteroids), poor oral hygiene, and co-morbid disorders (e.g., periodontal and/or other pre-existing dental disease, anemia, coagulopathy, infection, ill-fitting dentures). Good oral hygiene practices should be maintained during treatment with Prolia.
For patients requiring invasive dental procedures, clinical judgment of the treating physician and/or oral surgeon should guide the management plan of each patient based on individual benefit-risk assessment.
Patients who are suspected of having or who develop ONJ while on Prolia should receive care by a dentist or an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition. Discontinuation of Prolia therapy should be considered based on individual benefit-risk assessment.
5.5 Suppression of Bone Turnover
In clinical trials in women with postmenopausal osteoporosis, treatment with Prolia resulted in significant suppression of bone remodeling as evidenced by markers of bone turnover and bone histomorphometry [see Clinical Pharmacology (12.2), Clinical Studies (14.1)]. The significance of these findings and the effect of long-term treatment with Prolia are unknown. The long-term consequences of the degree of suppression of bone remodeling observed with Prolia may contribute to adverse outcomes such as osteonecrosis of the jaw, atypical fractures, and delayed fracture healing. Monitor patients for these consequences.
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed below and also elsewhere in the labeling:
- Hypocalcemia [see Warnings and Precautions (5.1)]
- Serious Infections [see Warnings and Precautions (5.2)]
- Dermatologic Adverse Reactions [see Warnings and Precautions (5.3)]
- Osteonecrosis of the Jaw [see Warnings and Precautions (5.4)]
The most common adverse reactions reported with Prolia are back pain, pain in extremity, musculoskeletal pain, hypercholesterolemia, and cystitis.
The most common adverse reactions leading to discontinuation of Prolia are breast cancer, back pain, and constipation.
The Prolia Postmarketing Active Safety Surveillance Program is available to collect information from prescribers on specific adverse events. Please see www.proliasafety.com or call 1-800-772-6436 for more information about this program.
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice.
Treatment of postmenopausal women with osteoporosis
The safety of Prolia in the treatment of postmenopausal osteoporosis was assessed in a 3-year, randomized, double-blind, placebo-controlled, multinational study of 7808 postmenopausal women aged 60 to 91 years. A total of 3876 women were exposed to placebo and 3886 women were exposed to Prolia administered subcutaneously once every 6 months as a single 60 mg dose. All women were instructed to take at least 1000 mg of calcium and 400 IU of vitamin D supplementation per day.
The incidence of all-cause mortality was 2.3% (n = 90) in the placebo group and 1.8% (n = 70) in the Prolia group. The incidence of nonfatal serious adverse events was 24.2% in the placebo group and 25.0% in the Prolia group. The percentage of patients who withdrew from the study due to adverse events was 2.1% and 2.4% for the placebo and Prolia groups, respectively.
Adverse reactions reported in ≥ 2% of postmenopausal women with osteoporosis and more frequently in the Prolia-treated women than in the placebo-treated women are shown in the table below.
| SYSTEM ORGAN CLASS Preferred Term | Prolia (N = 3886) n (%) | Placebo (N = 3876) n (%) |
| BLOOD AND LYMPHATIC SYSTEM DISORDERS | ||
| Anemia | 129 (3.3) | 107 (2.8) |
| CARDIAC DISORDERS | ||
| Angina pectoris | 101 (2.6) | 87 (2.2) |
| Atrial fibrillation | 79 (2.0) | 77 (2.0) |
| EAR AND LABYRINTH DISORDERS | ||
| Vertigo | 195 (5.0) | 187 (4.8) |
| GASTROINTESTINAL DISORDERS | ||
| Abdominal pain upper | 129 (3.3) | 111 (2.9) |
| Flatulence | 84 (2.2) | 53 (1.4) |
| Gastroesophageal reflux disease | 80 (2.1) | 66 (1.7) |
| GENERAL DISORDERS AND ADMINISTRATION SITE CONDITIONS | ||
| Edema peripheral | 189 (4.9) | 155 (4.0) |
| Asthenia | 90 (2.3) | 73 (1.9) |
| INFECTIONS AND INFESTATIONS | ||
| Cystitis | 228 (5.9) | 225 (5.8) |
| Upper respiratory tract infection | 190 (4.9) | 167 (4.3) |
| Pneumonia | 152 (3.9) | 150 (3.9) |
| Pharyngitis | 91 (2.3) | 78 (2.0) |
| Herpes zoster | 79 (2.0) | 72 (1.9) |
| METABOLISM AND NUTRITION DISORDERS | ||
| Hypercholesterolemia | 280 (7.2) | 236 (6.1) |
| MUSCULOSKELETAL AND CONNECTIVE TISSUE DISORDERS | ||
| Back pain | 1347 (34.7) | 1340 (34.6) |
| Pain in extremity | 453 (11.7) | 430 (11.1) |
| Musculoskeletal pain | 297 (7.6) | 291 (7.5) |
| Bone pain | 142 (3.7) | 117 (3.0) |
| Myalgia | 114 (2.9) | 94 (2.4) |
| Spinal osteoarthritis | 82 (2.1) | 64 (1.7) |
| NERVOUS SYSTEM DISORDERS | ||
| Sciatica | 178 (4.6) | 149 (3.8) |
| PSYCHIATRIC DISORDERS | ||
| Insomnia | 126 (3.2) | 122 (3.1) |
| SKIN AND SUBCUTANEOUS TISSUE DISORDERS | ||
| Rash | 96 (2.5) | 79 (2.0) |
| Pruritus | 87 (2.2) | 82 (2.1) |
Hypocalcemia
Decreases in serum calcium levels to less than 8.5 mg/dL were reported in 0.4% women in the placebo group and 1.7% women in the Prolia group at the month 1 visit. The nadir in serum calcium level occurs at approximately day 10 after Prolia dosing in subjects with normal renal function.
In clinical studies, subjects with impaired renal function were more likely to have greater reductions in serum calcium levels compared to subjects with normal renal function. In a study of 55 patients with varying degrees of renal function, serum calcium levels < 7.5 mg/dL or symptomatic hypocalcemia were observed in 5 subjects. These included no subjects in the normal renal function group, 10% of subjects in the CrCL 50 to 80 mL/min group, 29% of subjects in the CrCL <30 mL/min group, and 29% of subjects in the hemodialysis group. These subjects did not receive calcium and vitamin D supplementation. In a study of 4,550 postmenopausal women with osteoporosis, the mean change from baseline in serum calcium level 10 days after Prolia dosing was -5.5% in subjects with creatinine clearance < 30 mL/min vs. -3.1% in subjects with CrCL ≥ 30 mL/min.
Serious Infections
Receptor activator of nuclear factor kappa-B ligand (RANKL) is expressed on activated T and B lymphocytes and in lymph nodes. Therefore, a RANKL inhibitor such as Prolia may increase the risk of infection.
In the clinical study of 7808 postmenopausal women with osteoporosis, the incidence of infections resulting in death was 0.2% in both placebo and Prolia treatment groups. However, the incidence of nonfatal serious infections was 3.3% in the placebo group and 4.0% in the Prolia group. Hospitalizations due to serious infections in the abdomen (0.7% placebo vs. 0.9% Prolia), urinary tract (0.5% placebo vs. 0.7% Prolia), and ear (0.0% placebo vs. 0.1% Prolia) were reported. Endocarditis was reported in no placebo patients and 3 patients receiving Prolia.
Skin infections, including erysipelas and cellulitis, leading to hospitalization were reported more frequently in patients treated with Prolia (< 0.1% placebo vs. 0.4% Prolia).
There was no imbalance in the reporting of opportunistic infections.
Dermatologic Reactions
A significantly higher number of patients treated with Prolia developed epidermal and dermal adverse events (such as dermatitis, eczema and rashes), with these events reported in 8.2% of placebo and 10.8% of Prolia group (p < 0.0001). Most of these events were not specific to the injection site [see Warnings and Precautions (5.3)].
Osteonecrosis of the Jaw
ONJ has been reported in the osteoporosis clinical trial program in patients treated with Prolia[see Warnings and Precautions (5.4)].
Pancreatitis
Pancreatitis was reported in 4 patients (0.1%) in the placebo and 8 patients (0.2%) in the Prolia groups. Of these reports, one subject in the placebo group and all 8 subjects in the Prolia group had serious events including one death in the Prolia group. Several patients had a prior history of pancreatitis. The time from product administration to event occurrence was variable.
New Malignancies
The overall incidence of new malignancies was 4.3% in the placebo and 4.8% in the Prolia groups. New malignancies related to breast (0.7% placebo vs. 0.9% Prolia), reproductive (0.2% placebo vs. 0.5% Prolia) and gastrointestinal systems (0.6% placebo vs. 0.9% Prolia) were reported. A causal relationship to drug exposure has not been established.
Immunogenicity
Denosumab is a human monoclonal antibody. As with all therapeutic proteins, there is potential for immunogenicity. Using an electrochemiluminescent bridging immunoassay, less than 1% (55 out of 8113) of patients treated with Prolia for up to 5 years tested positive for binding antibodies (including pre-existing, transient, and developing antibodies). None of the patients tested positive for neutralizing antibodies, as was assessed using a chemiluminescent cell-based in vitro biological assay. No evidence of altered pharmacokinetic profile, toxicity profile, or clinical response was associated with binding antibody development.
The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of a positive antibody (including neutralizing antibody) test result may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of antibodies to denosumab with the incidence of antibodies to other products may be misleading.
7 DRUG INTERACTIONS
No drug-drug interaction studies have been conducted with Prolia.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C
There are no adequate and well-controlled studies of Prolia in pregnant women. In genetically engineered mice in which RANK ligand (RANKL) was turned off by gene removal (a “knockout mouse”), absence of RANKL (the target of denosumab) caused fetal lymph node agenesis and led to postnatal impairment of dentition and bone growth. Pregnant RANKL knockout mice also showed altered maturation of the maternal mammary gland, leading to impaired lactation postpartum [see Use in Specific Populations (8.3)].
Prolia is approved only for use in postmenopausal women. Prolia should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Women who become pregnant during Prolia treatment are encouraged to enroll in Amgen's Pregnancy Surveillance Program. Patients or their physicians should call 1-800-77-AMGEN (1-800-772-6436) to enroll.
In an embryofetal developmental study, cynomolgus monkeys received subcutaneous denosumab weekly during organogenesis at doses up to 13-fold higher than the recommended human dose of 60 mg administered once every 6 months based on body weight (mg/kg). No evidence of maternal toxicity or fetal harm was observed. However, this study only assessed fetal toxicity during a period equivalent to the first trimester and fetal lymph nodes were not examined. Monoclonal antibodies are transported across the placenta in a linear fashion as pregnancy progresses, with the largest amount transferred during the third trimester. Potential adverse developmental effects resulting from exposures during the second and third trimesters have not been assessed in animals [see Nonclinical Toxicology (13.2)].
8.3 Nursing Mothers
It is not known whether Prolia is excreted into human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Prolia, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Maternal exposure to Prolia during pregnancy may impair mammary gland development and lactation based on animal studies in pregnant mice lacking the RANK/RANKL signaling pathway that have shown altered maturation of the maternal mammary gland, leading to impaired lactation postpartum [see Nonclinical Toxicology (13.2)].
8.4 Pediatric Use
Prolia is not recommended in pediatric patients. The safety and effectiveness of Prolia in pediatric patients have not been established.
Treatment with Prolia may impair bone growth in children with open growth plates and may inhibit eruption of dentition. In neonatal rats, inhibition of RANKL (the target of Prolia therapy) with a construct of osteoprotegerin bound to Fc (OPG-Fc) at doses ≤10 mg/kg was associated with inhibition of bone growth and tooth eruption. Adolescent primates dosed with denosumab at 10 and 50 times (10 and 50 mg/kg dose) higher than the recommended human dose of 60 mg administered once every 6 months, based on body weight (mg/kg), had abnormal growth plates [see Nonclinical Toxicology (13.2)].
8.5 Geriatric Use
Of the total number of patients in clinical studies of Prolia, 9943 patients (76%) were ≥ 65 years old, while 3576 (27%) were ≥ 75 years old. No overall differences in safety or efficacy were observed between these patients and younger patients and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
No dose adjustment is necessary in patients with renal impairment.
In clinical studies, patients with severe renal impairment (creatinine clearance < 30 mL/min) or receiving dialysis were at greater risk of developing hypocalcemia. Consider the benefit-risk profile when administering Prolia to patients with severe renal impairment or receiving dialysis. Clinical monitoring of calcium and mineral levels (phosphorus and magnesium) is highly recommended. Adequate intake of calcium and vitamin D is important in patients with severe renal impairment or receiving dialysis [see Warnings and Precautions (5.1), Adverse Reactions (6.1), and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No clinical studies have been conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of Prolia.
10 OVERDOSAGE
There is no experience with overdosage with Prolia.
11 DESCRIPTION
Prolia (denosumab) is a human IgG2 monoclonal antibody with affinity and specificity for human RANKL (receptor activator of nuclear factor kappa-B ligand). Denosumab has an approximate molecular weight of 147 kDa and is produced in genetically engineered mammalian (Chinese hamster ovary) cells.
Prolia is a sterile, preservative-free, clear, colorless to pale yellow solution.
Each 1 mL single-use prefilled syringe of Prolia contains 60 mg denosumab (60 mg/mL solution), 4.7% sorbitol, 17 mM acetate, 0.01% polysorbate 20, Water for Injection (USP), and sodium hydroxide to a pH of 5.2.
Each 1 mL single-use vial of Prolia contains 60 mg denosumab (60 mg/mL solution), 4.7% sorbitol, 17 mM acetate, Water for Injection, (USP), and sodium hydroxide to a pH of 5.2.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Prolia binds to RANKL, a transmembrane or soluble protein essential for the formation, function, and survival of osteoclasts, the cells responsible for bone resorption. Prolia prevents RANKL from activating its receptor, RANK, on the surface of osteoclasts and their precursors. Prevention of the RANKL/RANK interaction inhibits osteoclast formation, function, and survival, thereby decreasing bone resorption and increasing bone mass and strength in both cortical and trabecular bone.
12.2 Pharmacodynamics
In clinical studies, treatment with 60 mg of Prolia resulted in reduction in the bone resorption marker serum type 1 C-telopeptide (CTX) by approximately 85% by 3 days, with maximal reductions occurring by 1 month. CTX levels were below the limit of assay quantitation (0.049 ng/mL) in 39-68% of subjects 1-3 months after dosing of Prolia. At the end of each dosing interval, CTX reductions were partially attenuated from a maximal reduction of ≥ 87% to ≥ 45% (range: 45% to 80%), as serum denosumab levels diminished, reflecting the reversibility of the effects of Prolia on bone remodeling. These effects were sustained with continued treatment. Upon reinitiation, the degree of inhibition of CTX by Prolia was similar to that observed in patients initiating Prolia treatment.
Consistent with the physiological coupling of bone formation and resorption in skeletal remodeling, subsequent reductions in bone formation markers (i.e., osteocalcin and procollagen type 1 N-terminal peptide [P1NP]), were observed starting 1 month after the first dose of Prolia. After discontinuation of Prolia therapy, markers of bone resorption increased to levels 40-60% above pretreatment values but returned to baseline levels within 12 months.
12.3 Pharmacokinetics
In a study conducted in healthy male and female volunteers (n = 73, age range: 18 to 64 years) following a single subcutaneously administered Prolia dose of 60 mg after fasting (at least for 12 hours), the mean maximum denosumab concentration (Cmax) was 6.75 mcg/mL (standard deviation [SD] = 1.89 mcg/mL). The median time to maximum denosumab concentration (Tmax) was 10 days (range: 3 to 21 days). After Cmax, serum denosumab concentrations declined over a period of 4 to 5 months with a mean half-life of 25.4 days (SD = 8.5 days; n = 46). The mean area-under-the-concentration-time curve up to 16 weeks (AUC0-16 weeks) of denosumab was 316 mcg·day/mL (SD = 101 mcg·day/mL).
No accumulation or change in denosumab pharmacokinetics with time was observed upon multiple dosing of 60 mg subcutaneously administered once every 6 months.
Prolia pharmacokinetics were not affected by the formation of binding antibodies.
A population pharmacokinetic analysis was performed to evaluate the effects of demographic characteristics. This analysis showed no notable differences in pharmacokinetics with age (in postmenopausal women), race, or body weight (36 to 140 kg).
Drug Interactions
No drug-drug interaction studies have been conducted with Prolia.
Specific Populations
Gender:Mean serum denosumab concentration-time profiles observed in a study conducted in healthy men ≥ 50 years were similar to those observed in a study conducted in postmenopausal women using the same dose regimen.
Age:The pharmacokinetics of denosumab was not affected by age across all populations studied whose ages ranged from 28-87 years.
Race:The pharmacokinetics of denosumab was not affected by race.
Renal Impairment:In a study of 55 patients with varying degrees of renal function, including patients on dialysis, the degree of renal impairment had no effect on the pharmacokinetics of denosumab; thus, dose adjustment for renal impairment is not necessary.
Hepatic Impairment:No clinical studies have been conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of denosumab.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
The carcinogenic potential of denosumab has not been evaluated in long-term animal studies.
Mutagenicity
The genotoxic potential of denosumab has not been evaluated.
Impairment of Fertility
Denosumab had no effect on female fertility or male reproductive organs in monkeys at doses that were 13- to 50-fold higher than the recommended human dose of 60 mg administered once every 6 months, based on body weight (mg/kg).
13.2 Animal Toxicology and/or Pharmacology
Prolia is an inhibitor of osteoclastic bone resorption via inhibition of RANKL.
In ovariectomized monkeys, once-monthly treatment with denosumab suppressed bone turnover and increased bone mineral density (BMD) and strength of cancellous and cortical bone at doses 50-fold higher than the recommended human dose of 60 mg administered once every 6 months, based on body weight (mg/kg). Bone tissue was normal with no evidence of mineralization defects, accumulation of osteoid, or woven bone.
Adolescent primates treated with denosumab at doses > 10 times (10 and 50 mg/kg dose) higher than the recommended human dose of 60 mg administered once every 6 months, based on mg/kg, had abnormal growth plates, considered to be consistent with the pharmacological activity of denosumab [see Use in Specific Populations (8.4)].
Because the biological activity of denosumab in animals is specific to nonhuman primates, evaluation of genetically engineered (“knockout”) mice or use of other biological inhibitors of the RANK/RANKL pathway, namely OPG-Fc, provided additional information on the pharmacodynamic properties of denosumab. RANK/RANKL knockout mice exhibited absence of lymph node formation, as well as an absence of lactation due to inhibition of mammary gland maturation (lobulo-alveolar gland development during pregnancy). Neonatal RANK/RANKL knockout mice exhibited reduced bone growth and lack of tooth eruption. A corroborative study in 2-week-old rats given the RANKL inhibitor OPG-Fc also showed reduced bone growth, altered growth plates, and impaired tooth eruption. These changes were partially reversible in this model when dosing with the RANKL inhibitors was discontinued [see Use in Specific Populations (8.1, 8.4)].
14 CLINICAL STUDIES
14.1 Postmenopausal Women with Osteoporosis
The efficacy and safety of Prolia in the treatment of postmenopausal osteoporosis was demonstrated in a 3-year, randomized, double-blind, placebo-controlled, trial. Enrolled women had a baseline BMD T-score between -2.5 and -4.0 at either the lumbar spine or total hip. Women with other diseases (such as rheumatoid arthritis, osteogenesis imperfecta, and Paget’s disease) or on therapies that affect bone were excluded from this study. The 7808 enrolled women were aged 60 to 91 years with a mean age of 72 years. Overall, the mean baseline lumbar spine BMD T-score was -2.8 and 23% of women had a vertebral fracture at baseline. Women were randomized to receive SC injections of either placebo (N = 3906) or Prolia 60 mg (N = 3902) once every 6 months. All women received at least 1000 mg calcium and 400 IU vitamin D supplementation daily.
The primary efficacy variable was the incidence of new morphometric (radiologically-diagnosed) vertebral fractures at 3 years. Vertebral fractures were diagnosed based on lateral spine radiographs (T4-L4) using a semiquantitative scoring method. Secondary efficacy variables included the incidence of hip fracture and nonvertebral fracture, assessed at 3 years.
Effect on Vertebral Fractures
Prolia significantly reduced the incidence of new morphometric vertebral fractures at 1, 2, and 3 years (p < 0.0001), as shown in Table 2. The incidence of new vertebral fractures at year 3 was 7.2% in the placebo-treated women compared to 2.3% for the Prolia-treated women. The absolute risk reduction was 4.8% and relative risk reduction was 68% for new morphometric vertebral fractures at year 3.
| Proportion of Women With Fracture (%)* |
Absolute Risk Reduction (%)† (95% CI) |
Relative Risk Reduction (%)† (95% CI) |
||
|
Placebo N = 3691 (%) |
Prolia N = 3702 (%) |
|||
| 0-1 Year | 2.2 | 0.9 | 1.4 (0.8, 1.9) | 61 (42, 74) |
| 0-2 Years | 5.0 | 1.4 | 3.5 (2.7, 4.3) | 71 (61, 79) |
| 0-3 Years | 7.2 | 2.3 | 4.8 (3.9, 5.8) | 68 (59, 74) |
Prolia was effective in reducing the risk for new morphometric vertebral fractures regardless of age, baseline rate of bone turnover, baseline BMD, baseline history of fracture, or prior use of a drug for osteoporosis.
Effect on Hip Fractures
The incidence of hip fracture was 1.2% for placebo-treated women compared to 0.7% for Prolia-treated women at year 3. The age-adjusted absolute risk reduction of hip fractures was 0.3% with a relative risk reduction of 40% at 3 years (p = 0.04) (Figure 1).
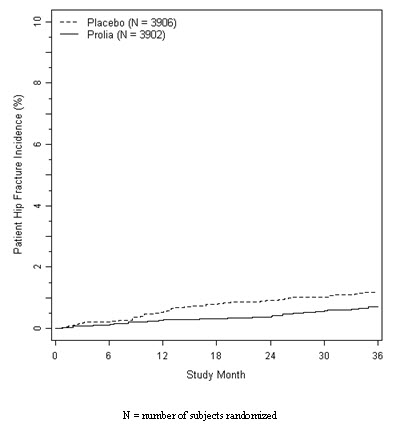
Figure 1. Cumulative Incidence of Hip Fractures Over 3 Years
Effect on Nonvertebral Fractures
Treatment with Prolia resulted in a significant reduction in the incidence of nonvertebral fractures (Table 3).
| Proportion of Women With Fracture (%)* |
Absolute Risk Reduction (%) (95% CI) |
Relative Risk Reduction (%) (95% CI) |
||
|
Placebo N = 3906 (%) |
Prolia N = 3902 (%) |
|||
| Nonvertebral fracture† | 8.0 | 6.5 | 1.5 (0.3, 2.7) | 20 (5, 33)‡ |
Effect on Bone Mineral Density (BMD)
Treatment with Prolia significantly increased BMD at all anatomic sites measured at 3 years. The treatment differences in BMD at 3 years were 8.8% at the lumbar spine, 6.4% at the total hip, and 5.2% at the femoral neck. Consistent effects on BMD were observed at the lumbar spine, regardless of baseline age, race, weight/body mass index (BMI), baseline BMD, and level of bone turnover.
After Prolia discontinuation, BMD returned to approximately baseline levels within 12 months.
Bone Histology and Histomorphometry
A total of 115 transiliac crest bone biopsy specimens were obtained from 92 postmenopausal women with osteoporosis at either month 24 and/or month 36 (53 specimens in Prolia group, 62 specimens in placebo group). Of the biopsies obtained, 115 (100%) were adequate for qualitative histology and 7 (6%) were adequate for full quantitative histomorphometry assessment.
Qualitative histology assessments showed normal architecture and quality with no evidence of mineralization defects, woven bone, or marrow fibrosis in patients treated with Prolia.
The presence of double tetracycline labeling in a biopsy specimen provides an indication of active bone remodeling, while the absence of tetracycline label suggests suppressed bone formation. In subjects treated with Prolia, 35% had no tetracycline label present at the month 24 biopsy and 38% had no tetracycline label present at the month 36 biopsy, while 100% of placebo-treated patients had double label present at both time points. When compared to placebo, treatment with Prolia resulted in virtually absent activation frequency and markedly reduced bone formation rates. However, the long-term consequences of this degree of suppression of bone remodeling are unknown.
16 HOW SUPPLIED/STORAGE AND HANDLING
Prolia is supplied in a single-use prefilled syringe with a safety guard or in a single-use vial. The grey needle cap on the single-use prefilled syringe contains dry natural rubber (a derivative of latex).
| 60 mg/1 mL in a single-use prefilled syringe | 1 per carton | NDC 55513-710-01 |
| 60 mg/1 mL in a single-use vial | 1 per carton | NDC 55513-720-01 |
Store Prolia in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton. Do not freeze. Prior to administration, Prolia may be allowed to reach room temperature (up to 25°C/77°F) in the original container. Once removed from the refrigerator, Prolia must not be exposed to temperatures above 25°C/77°F and must be used within 14 days. If not used within the 14 days, Prolia should be discarded. Do not use Prolia after the expiry date printed on the label.
Protect Prolia from direct light and heat.
Avoid vigorous shaking of Prolia.
17 PATIENT COUNSELING INFORMATION
17.1 Hypocalcemia
Adequately supplement patients with calcium and vitamin D and instruct them on the importance of maintaining serum calcium levels while receiving Prolia [see Warnings and Precautions (5.1) and Use in Specific Populations (8.6)]. Advise patients to seek prompt medical attention if they develop signs or symptoms of hypocalcemia.
17.2 Serious Infections
Advise patients to seek prompt medical attention if they develop signs or symptoms of infections, including cellulitis [see Warnings and Precautions (5.2].
17.3 Dermatologic Reactions
Advise patients to seek prompt medical attention if they develop signs or symptoms of dermatological reactions (dermatitis, rashes, and eczema) [see Warnings and Precautions (5.3)].
17.4 Osteonecrosis of the Jaw
Advise patients to maintain good oral hygiene during treatment with Prolia and to inform their dentist prior to dental procedures that they are receiving Prolia. Patients should inform their physician or dentist if they experience persistent pain and/or slow healing of the mouth or jaw after dental surgery [see Warnings and Precautions (5.4)].
17.5 Schedule of Administration
If a dose of Prolia is missed, administer the injection as soon as convenient. Thereafter, schedule injections every 6 months from the date of the last injection.
[Amgen Logo]
Manufactured by:
Amgen Manufacturing Limited, a subsidiary of Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-1799
This product, its production, and/or its use may be covered by one or more US Patents, including US Patent Nos. 6,740,522; 7,097,834; 7,364,736; and 7,411,050, as well as other patents or patents pending.
© 2010 Amgen Inc. All rights reserved.
1xxxxxx - v1
MEDICATION GUIDE
Prolia™ (PRÓ-lee-a)
(denosumab)
Injection
Read the Medication Guide that comes with Prolia before you start taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking with your doctor about your medical condition or treatment. Talk to your doctor if you have any questions about Prolia.
What is the most important information I should know about Prolia?
Prolia can cause serious side effects including:
-
Low calcium levels in your blood (hypocalcemia).
Prolia may lower the calcium levels in your blood. If you have low blood calcium before you start receiving Prolia, it may get worse during treatment. Your low blood calcium must be treated before you receive Prolia. Most people with low blood calcium levels do not have symptoms, but some people may have symptoms. Call your doctor right away if you have symptoms of low blood calcium such as:- Spasms, twitches, or cramps in your muscles
- Numbness or tingling in your fingers, toes, or around your mouth
Your doctor may prescribe calcium and vitamin D to help prevent low calcium levels in your blood while you take Prolia. Take calcium and vitamin D as your doctor tells you to.
-
Serious infections.
Serious infections in your skin, lower stomach area (abdomen), bladder, or ear may happen if you take Prolia. Inflammation of the inner lining of the heart (endocarditis) due to an infection also may happen more often in people who take Prolia. You may need to go to the hospital for treatment if you develop an infection.
Prolia is a medicine that may affect your immune system. People who have weakened immune system or take medicines that affect the immune system may have an increased risk for developing serious infections.
Call your doctor right away if you have any of the following symptoms of infection:- Fever or chills
- Skin that looks red or swollen and is hot or tender to touch
- Severe abdominal pain
- Frequent or urgent need to urinate or burning feeling when you urinate
-
Skin problems.
Skin problems such as inflammation of your skin (dermatitis), rash, and eczema may happen if you take Prolia. Call your doctor if you have any of the following symptoms of skin problems that do not go away or get worse:- Redness
- Itching
- Small bumps or patches (rash)
- Your skin is dry or feels like leather
- Blisters that ooze or become crusty
- Skin peeling
-
Severe jaw bone problems (osteonecrosis).
Severe jaw bone problems may happen when you take Prolia. Your doctor should examine your mouth before you start Prolia. Your doctor may tell you to see your dentist before you start Prolia. It is important for you to practice good mouth care during treatment with Prolia.
Call your doctor right away if you have any of these side effects.
What is Prolia?
Prolia is a prescription medicine used to treat osteoporosis (thinning and weakening of bone) in women after menopause (“change of life”) who
- Have an increased risk for fractures (broken bones).
- Cannot use another osteoporosis medicine or other osteoporosis medicines did not work well.
Who should not receive Prolia?
Do not take Prolia if you have been told by your doctor that your blood calcium level is too low.
What should I tell my doctor before receiving Prolia?
Before taking Prolia, tell your doctor if you:
- Have low blood calcium.
- Cannot take daily calcium and vitamin D.
- Had parathyroid or thyroid surgery (glands located in your neck).
- Have been told you have trouble absorbing minerals in your stomach or intestines (malabsorption syndrome).
- Have kidney problems or are on kidney dialysis.
- Plan to have dental surgery or teeth removed.
- Are pregnant or plan to become pregnant. Prolia may harm your unborn baby. Tell your doctor right away if you become pregnant while taking Prolia.
Pregnancy Surveillance Program: Prolia is not intended for use in pregnant women. If you become pregnant while taking Prolia, talk to your doctor about enrolling with Amgen’s Pregnancy Surveillance Program or call 1-800-772-6436 (l-800-77-AMGEN). The purpose of this program is to collect information about women who have become pregnant while taking Prolia.
- Are breast-feeding or plan to breast-feed. It is not known if Prolia passes into your breast milk. You and your doctor should decide if you will take Prolia or breast-feed. You should not do both.
Tell your doctor about all the medicines you take, including prescription and nonprescription drugs, vitamins, and herbal supplements.
Know the medicines you take. Keep a list of medicines with you to show to your doctor or pharmacist when you get a new medicine.
How will I receive Prolia?
- Prolia is an injection that will be given to you by a healthcare professional. Prolia is injected under your skin (subcutaneous).
- You will receive Prolia 1 time every 6 months.
- You should take calcium and vitamin D as your doctor tells you to while you receive Prolia.
- If you miss a dose of Prolia, you should receive your injection as soon as you can.
- Take good care of your teeth and gums while you receive Prolia. Brush and floss your teeth regularly.
- Tell your dentist that you are receiving Prolia before you have dental work.
What are the possible side effects of Prolia?
Prolia may cause serious side effects.
- See “What is the most important information I should know about Prolia?”
- Long-term effects on bone: It is not known if the use of Prolia over a long period of time may cause slow healing of broken bones or unusual fractures.
The most common side effects of Prolia are:
- Back pain
- Pain in your arms and legs
- High cholesterol
- Muscle pain
- Bladder infection
These are not all the possible side effects of Prolia. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I handle Prolia if I need to pick it up from a pharmacy?
- Keep Prolia in a refrigerator at 36°F to 46°F (2°C to 8°C) in the original carton.
- Do not freeze Prolia.
- When you remove Prolia from the refrigerator, Prolia must be kept at room temperature [up to 77°F (25°C)] in the original carton and must be used within 14 days.
- Do not keep Prolia at temperatures above 77°F (25°C). Warm temperatures will affect how Prolia works.
- Do not shake Prolia.
- Keep Prolia in the original carton to protect from light.
Keep Prolia and all medicines out of reach of children.
General information about Prolia
Do not give Prolia to other people even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about Prolia. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about Prolia that is written for health professionals.
For more information, go to www.Prolia.com or call Amgen at 1-800-772-6436.
What are the ingredients in Prolia?
Active ingredient: denosumab
Inactive ingredients: sorbitol, acetate, polysorbate 20 (prefilled syringe only), Water for Injection (USP), and sodium hydroxide
What is osteoporosis?
Osteoporosis is a disease in which the bones become thin and weak, increasing the chance of having a broken bone. Osteoporosis usually causes no symptoms until a fracture happens. The most common fractures are in the spine (backbone). They can shorten height, even without causing pain. Over time, the spine can become curved or deformed and the body bent over. Fractures from osteoporosis can also happen in almost any bone in the body, for example: the wrist, rib, or hip. Once you have had a fracture, the chance for more fractures greatly increases.
The following risk factors increase your chance of getting fractures from osteoporosis:
- Past broken bones from osteoporosis
- Very low bone mineral density (BMD)
- Frequent falls
- Limited movement, such as using a wheelchair
- Medical conditions likely to cause bone loss, such as some kinds of arthritis
- Taking steroid medicines called glucocorticoids, such as prednisone
- Other medicines that may cause bone loss, for example: seizure medicines (such as phenytoin), blood thinners (such as heparin), high doses of vitamin A
What can I do to treat osteoporosis?
There are many steps you can take to treat osteoporosis. Taking Prolia, along with calcium and vitamin D, may be one option for you.
Amgen Manufacturing Limited, a subsidiary of Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-1799
This Medication Guide has been approved by the US Food and Drug Administration.
1xxxxxx - v1
Issued: 06/2010
PACKAGE LABEL - PRINCIPAL DISPLAY PANEL - PREFILLED SYRINGE, 60 MG
1 x 60 mg Single Use Prefilled Syringe
NDC 55513-710-01
AMGEN®
proliaTM
(denosumab)
60 mg/mL
60 mg/mL Injection – For Subcutaneous Use Only.
Single Use Prefilled Syringe. Discard unused portion.
Sterile Solution – No Preservative.
Rx Only
Refrigerate at 2° to 8°C (36° to 46°F). Do not freeze. Avoid excessive shaking. Protect from direct light and heat.
This Product Contains Dry Natural Rubber.
Manufactured by: Amgen Manufacturing Ltd., a subsidiary of Amgen Inc. Thousand Oaks, CA 91320-1799

| PROLIA
denosumab injection |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| BLA | BLA125320 | 06/05/2010 | |
| Labeler - Amgen, Inc (039976196) |
| Registrant - Amgen, Inc (039976196) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Amgen, Inc | 071629633 | MANUFACTURE, ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Boehringer Ingelheim Pharma GmbH and Co. KG | 340700520 | MANUFACTURE, ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Amgen, Inc | 048053201 | MANUFACTURE, ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Amgen Manufacturing Ltd | 785800020 | MANUFACTURE, ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Amgen, Inc | 039976196 | MANUFACTURE, ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Amgen Fremont Inc | 960193530 | ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Charles River Laboratories Inc. | 177728660 | ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| WUXI APPTEC INC | 136584468 | ANALYSIS | |