SPORANOX
-
itraconazole capsule
Ortho-McNeil-Janssen Pharmaceuticals Inc.
----------
SPORANOX®
(itraconazole)
Capsules
SPORANOX ® (itraconazole) Capsules should not be administered for the treatment of onychomycosis in patients with evidence of ventricular dysfunction such as congestive heart failure (CHF) or a history of CHF. If signs or symptoms of congestive heart failure occur during administration of SPORANOX® Capsules, discontinue administration. When itraconazole was administered intravenously to dogs and healthy human volunteers, negative inotropic effects were seen. (See CLINICAL PHARMACOLOGY: Special Populations, CONTRAINDICATIONS, WARNINGS, PRECAUTIONS: Drug Interactions and ADVERSE REACTIONS: Post-marketing Experience for more information.)
Drug Interactions: Coadministration of cisapride, pimozide, quinidine, dofetilide, or levacetylmethadol (levomethadyl) with SPORANOX® (itraconazole) Capsules, Injection or Oral Solution is contraindicated SPORANOX®, a potent cytochrome P450 3A4 isoenzyme system (CYP3A4) inhibitor, may increase plasma concentrations of drugs metabolized by this pathway. Serious cardiovascular events, including QT prolongation, torsades de pointes, ventricular tachycardia, cardiac arrest, and/or sudden death have occurred in patients using cisapride, pimozide, levacetylmethadol (levomethadyl), or quinidine, concomitantly with SPORANOX® and/or other CYP3A4 inhibitors. See CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS: Drug Interactions for more information.
DESCRIPTION
SPORANOX® is the brand name for itraconazole, a synthetic triazole antifungal agent. Itraconazole is a 1:1:1:1 racemic mixture of four diastereomers (two enantiomeric pairs), each possessing three chiral centers. It may be represented by the following structural formula and nomenclature: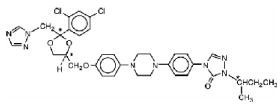
(±)-1-[(R*)-sec-butyl]-4-[p-[4-[p-[[(2R*,4S*)-2-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]-1-piperazinyl]phenyl]-Δ2-1,2,4-triazolin-5-one mixture with (±)-1-[(R*)-sec-butyl]-4-[p-[4-[p-[[(2S*,4R*)-2-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]-1-piperazinyl]phenyl]-Δ2-1,2,4-triazolin-5-one
(±)-1-[(RS)-sec-butyl]-4-[p-[4-[p-[[(2R,4S)-2-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]-1-piperazinyl]phenyl]-Δ2-1,2,4-triazolin-5-one
Itraconazole has a molecular formula of C35H38Cl2N8O4 and a molecular weight of 705.64. It is a white to slightly yellowish powder. It is insoluble in water, very slightly soluble in alcohols, and freely soluble in dichloromethane. It has a pKa of 3.70 (based on extrapolation of values obtained from methanolic solutions) and a log (n-octanol/water) partition coefficient of 5.66 at pH 8.1.
SPORANOX® Capsules contain 100 mg of itraconazole coated on sugar spheres. Inactive ingredients are hard gelatin capsule, hypromellose, polyethylene glycol (PEG) 20,000, starch, sucrose, titanium dioxide, FD&C Blue No. 1, FD&C Blue No. 2, D&C Red No. 22 and D&C Red No. 28.
CLINICAL PHARMACOLOGY
Pharmacokinetics and Metabolism:
NOTE: The plasma concentrations reported below were measured by high-performance liquid chromatography (HPLC) specific for itraconazole. When itraconazole in plasma is measured by a bioassay, values reported are approximately 3.3 times higher than those obtained by HPLC due to the presence of the bioactive metabolite, hydroxyitraconazole. (See MICROBIOLOGY.)
The pharmacokinetics of itraconazole after intravenous administration and its absolute oral bioavailability from an oral solution were studied in a randomized crossover study in 6 healthy male volunteers. The observed absolute oral bioavailability of itraconazole was 55%.
The oral bioavailability of itraconazole is maximal when SPORANOX® (itraconazole) Capsules are taken with a full meal. The pharmacokinetics of itraconazole were studied in 6 healthy male volunteers who received, in a crossover design, single 100-mg doses of itraconazole as a polyethylene glycol capsule, with or without a full meal. The same 6 volunteers also received 50 mg or 200 mg with a full meal in a crossover design. In this study, only itraconazole plasma concentrations were measured. The respective pharmacokinetic parameters for itraconazole are presented in the table below:
| 50 mg (fed) | 100 mg (fed) | 100 mg (fasted) | 200 mg (fed) |
|
|---|---|---|---|---|
| * mean ± standard deviation | ||||
| Cmax
(ng/mL) | 45 ± 16* | 132 ± 67 | 38 ± 20 | 289 ± 100 |
| Tmax
(hours) | 3.2 ± 1.3 | 4.0 ± 1.1 | 3.3 ± 1.0 | 4.7 ± 1.4 |
| AUC0-∞
(ng·h/mL) | 567 ± 264 | 1899 ± 838 | 722 ± 289 | 5211 ± 2116 |
Doubling the SPORANOX® dose results in approximately a three-fold increase in the itraconazole plasma concentrations.
Values given in the table below represent data from a crossover pharmacokinetics study in which 27 healthy male volunteers each took a single 200-mg dose of SPORANOX® Capsules with or without a full meal:
| Itraconazole | Hydroxyitraconazole | |||
|---|---|---|---|---|
| Fed | Fasted | Fed | Fasted | |
| * mean ± standard deviation | ||||
| Cmax
(ng/mL) | 239 ± 85* | 140 ± 65 | 397 ± 103 | 286 ± 101 |
| Tmax
(hours) | 4.5 ± 1.1 | 3.9 ± 1.0 | 5.1 ± 1.6 | 4.5 ± 1.1 |
| AUC0-∞ (ng·h/mL) | 3423 ± 1154 | 2094 ± 905 | 7978 ± 2648 | 5191 ± 2489 |
| t1/2 (hours) | 21 ± 5 | 21 ± 7 | 12 ± 3 | 12 ± 3 |
Absorption of itraconazole under fasted conditions in individuals with relative or absolute achlorhydria, such as patients with AIDS or volunteers taking gastric acid secretion suppressors (e.g., H2 receptor antagonists), was increased when SPORANOX® Capsules were administered with a cola beverage. Eighteen men with AIDS received single 200-mg doses of SPORANOX® Capsules under fasted conditions with 8 ounces of water or 8 ounces of a cola beverage in a crossover design. The absorption of itraconazole was increased when SPORANOX® Capsules were coadministered with a cola beverage, with AUC0-24 and Cmax increasing 75% ± 121% and 95% ± 128%, respectively.
Thirty healthy men received single 200-mg doses of SPORANOX® Capsules under fasted conditions either 1) with water; 2) with water, after ranitidine 150 mg b.i.d. for 3 days; or 3) with cola, after ranitidine 150 mg b.i.d. for 3 days. When SPORANOX® Capsules were administered after ranitidine pretreatment, itraconazole was absorbed to a lesser extent than when SPORANOX® Capsules were administered alone, with decreases in AUC0-24 and Cmax of 39% ± 37% and 42% ± 39%, respectively. When SPORANOX® Capsules were administered with cola after ranitidine pretreatment, itraconazole absorption was comparable to that observed when SPORANOX® Capsules were administered alone. (See PRECAUTIONS: Drug Interactions.)
Steady-state concentrations were reached within 15 days following oral doses of 50 mg to 400 mg daily. Values given in the table below are data at steady-state from a pharmacokinetics study in which 27 healthy male volunteers took 200-mg SPORANOX® Capsules b.i.d. (with a full meal) for 15 days:
| Itraconazole | Hydroxyitraconazole | |
|---|---|---|
| * mean ± standard deviation | ||
| Cmax (ng/mL) | 2282 ± 514* | 3488 ± 742 |
| Cmin (ng/mL) | 1855 ± 535 | 3349 ± 761 |
| Tmax (hours) | 4.6 ± 1.8 | 3.4 ± 3.4 |
| AUC0-12 h (ng·h/mL) | 22569 ± 5375 | 38572 ± 8450 |
| t1/2 (hours) | 64 ± 32 | 56 ± 24 |
The plasma protein binding of itraconazole is 99.8% and that of hydroxyitraconazole is 99.5%. Following intravenous administration, the volume of distribution of itraconazole averaged 796 ± 185 liters.
Itraconazole is metabolized predominately by the cytochrome P450 3A4 isoenzyme system (CYP3A4), resulting in the formation of several metabolites, including hydroxyitraconazole, the major metabolite. Results of a pharmacokinetics study suggest that itraconazole may undergo saturable metabolism with multiple dosing. Fecal excretion of the parent drug varies between 3-18% of the dose. Renal excretion of the parent drug is less than 0.03% of the dose. About 40% of the dose is excreted as inactive metabolites in the urine. No single excreted metabolite represents more than 5% of a dose. Itraconazole total plasma clearance averaged 381 ± 95 mL/minute following intravenous administration. (See CONTRAINDICATIONS and PRECAUTIONS: Drug Interactions for more information.)
Special Populations:
Renal Insufficiency:
Limited data are available on the use of oral itraconazole in patients with renal impairment. A pharmacokinetic study using a single 200-mg dose of itraconazole (four 50-mg capsules) was conducted in three groups of patients with renal impairment (uremia: n=7; hemodialysis: n=7; and continuous ambulatory peritoneal dialysis: n=5). In uremic subjects with a mean creatinine clearance of 13 mL/min. × 1.73 m2, the exposure, based on AUC, was slightly reduced compared with normal population parameters. This study did not demonstrate any significant effect of hemodialysis or continuous ambulatory peritoneal dialysis on the pharmacokinetics of itraconazole (Tmax, Cmax, and AUC0-8). Plasma concentration-versus-time profiles showed wide intersubject variation in all three groups. Caution should be exercised when the drug is administered in this patient population. (See PRECAUTIONS and DOSAGE AND ADMINISTRATION.)
Hepatic Insufficiency:
Itraconazole is predominantly metabolized in the liver. Patients with impaired hepatic function should be carefully monitored when taking itraconazole. A pharmacokinetic study using a single oral 100 mg capsule dose of itraconazole was conducted in 6 healthy and 12 cirrhotic subjects. A statistically significant reduction in mean Cmax (47%) and a twofold increase in the elimination half-life (37 ± 17 hours vs. 16 ± 5 hours) of itraconazole were noted in cirrhotic subjects compared with healthy subjects. However, overall exposure to itraconazole, based on AUC, was similar in cirrhotic patients and in healthy subjects. The prolonged elimination half-life of itraconazole observed in the single oral dose clinical trial with itraconazole capsules in cirrhotic patients should be considered when deciding to initiate therapy with other medications metabolized by CYP3A4. Data are not available in cirrhotic patients during long-term use of itraconazole. (See BOX WARNING, CONTRAINDICATIONS, PRECAUTIONS: Drug Interactions and DOSAGE AND ADMINISTRATION.)
Decreased Cardiac Contractility:
When itraconazole was administered intravenously to anesthetized dogs, a dose-related negative inotropic effect was documented. In a healthy volunteer study of SPORANOX® Injection (intravenous infusion), transient, asymptomatic decreases in left ventricular ejection fraction were observed using gated SPECT imaging; these resolved before the next infusion, 12 hours later. If signs or symptoms of congestive heart failure appear during administration of SPORANOX® Capsules, SPORANOX® should be discontinued. (See CONTRAINDICATIONS, WARNINGS, PRECAUTIONS: Drug Interactions and ADVERSE REACTIONS: Post-marketing Experience for more information.)
MICROBIOLOGY
Mechanism of Action:
In vitro studies have demonstrated that itraconazole inhibits the cytochrome P450-dependent synthesis of ergosterol, which is a vital component of fungal cell membranes.
Activity In Vitro and In Vivo:
Itraconazole exhibits in vitro activity against Blastomyces dermatitidis, Histoplasma capsulatum, Histoplasma duboisii, Aspergillus flavus, Aspergillus fumigatus, Candida albicans, and Cryptococcus neoformans. Itraconazole also exhibits varying in vitro activity against Sporothrix schenckii, Trichophyton species, Candida krusei, and other Candida species.
Candida krusei, Candida glabrata and Candida tropicalis are generally the least susceptible Candida species, with some isolates showing unequivocal resistance to itraconazole in vitro. Itraconazole is not active against Zygomycetes (e.g., Rhizopus spp., Rhizomucor spp., Mucor spp. and Absidia spp.), Fusarium spp., Scedosporium spp. and Scopulariopsis spp.
The bioactive metabolite, hydroxyitraconazole, has not been evaluated against Histoplasma capsulatum, Blastomyces dermatitidis, Zygomycete, Fusarium spp., Scedosporium spp. and Scopulariopsis spp. Correlation between minimum inhibitory concentration (MIC) results in vitro and clinical outcome has yet to be established for azole antifungal agents.
Itraconazole administered orally was active in a variety of animal models of fungal infection using standard laboratory strains of fungi. Fungistatic activity has been demonstrated against disseminated fungal infections caused by Blastomyces dermatitidis, Histoplasma duboisii, Aspergillus fumigatus, Coccidioides immitis, Cryptococcus neoformans, Paracoccidioides brasiliensis, Sporothrix schenckii, Trichophyton rubrum, and Trichophyton mentagrophytes.
Itraconazole administered at 2.5 mg/kg and 5 mg/kg via the oral and parenteral routes increased survival rates and sterilized organ systems in normal and immunosuppressed guinea pigs with disseminated Aspergillus fumigatus infections. Oral itraconazole administered daily at 40 mg/kg and 80 mg/kg increased survival rates in normal rabbits with disseminated disease and in immunosuppressed rats with pulmonary Aspergillus fumigatus infection, respectively. Itraconazole has demonstrated antifungal activity in a variety of animal models infected with Candida albicans and other Candida species.
Resistance:
Isolates from several fungal species with decreased susceptibility to itraconazole have been isolated in vitro and from patients receiving prolonged therapy.
Several in vitro studies have reported that some fungal clinical isolates, including Candida species, with reduced susceptibility to one azole antifungal agent may also be less susceptible to other azole derivatives. The finding of cross-resistance is dependent on a number of factors, including the species evaluated, its clinical history, the particular azole compounds compared, and the type of susceptibility test that is performed. The relevance of these in vitro susceptibility data to clinical outcome remains to be elucidated.
Candida krusei, Candida glabrata and Candida tropicalis are generally the least susceptible Candida species, with some isolates showing unequivocal resistance to itraconazole in vitro.
Itraconazole is not active against Zygomycetes (e.g., Rhizopus spp., Rhizomucor spp., Mucor spp. and Absidia spp.), Fusarium spp., Scedosporium spp. and Scopulariopsis spp.
Studies (both in vitro and in vivo) suggest that the activity of amphotericin B may be suppressed by prior azole antifungal therapy. As with other azoles, itraconazole inhibits the 14C-demethylation step in the synthesis of ergosterol, a cell wall component of fungi. Ergosterol is the active site for amphotericin B. In one study the antifungal activity of amphotericin B against Aspergillus fumigatus infections in mice was inhibited by ketoconazole therapy. The clinical significance of test results obtained in this study is unknown.
INDICATIONS AND USAGE
SPORANOX® (itraconazole) Capsules are indicated for the treatment of the following fungal infections in immunocompromised and non-immunocompromised patients:
- Blastomycosis, pulmonary and extrapulmonary
- Histoplasmosis, including chronic cavitary pulmonary disease and disseminated, non-meningeal histoplasmosis, and
- Aspergillosis, pulmonary and extrapulmonary, in patients who are intolerant of or who are refractory to amphotericin B therapy.
Specimens for fungal cultures and other relevant laboratory studies (wet mount, histopathology, serology) should be obtained before therapy to isolate and identify causative organisms. Therapy may be instituted before the results of the cultures and other laboratory studies are known; however, once these results become available, antiinfective therapy should be adjusted accordingly.
SPORANOX® Capsules are also indicated for the treatment of the following fungal infections in non-immunocompromised patients:
- Onychomycosis of the toenail, with or without fingernail involvement, due to dermatophytes (tinea unguium), and
- Onychomycosis of the fingernail due to dermatophytes (tinea unguium).
Prior to initiating treatment, appropriate nail specimens for laboratory testing (KOH preparation, fungal culture, or nail biopsy) should be obtained to confirm the diagnosis of onychomycosis.
(See CLINICAL PHARMACOLOGY: Special Populations, CONTRAINDICATIONS, WARNINGS, and ADVERSE REACTIONS: Post-marketing Experience for more information.)
Description of Clinical Studies:
Blastomycosis:
Analyses were conducted on data from two open-label, non-concurrently controlled studies (N=73 combined) in patients with normal or abnormal immune status. The median dose was 200 mg/day. A response for most signs and symptoms was observed within the first 2 weeks, and all signs and symptoms cleared between 3 and 6 months. Results of these two studies demonstrated substantial evidence of the effectiveness of itraconazole for the treatment of blastomycosis compared with the natural history of untreated cases.
Histoplasmosis:
Analyses were conducted on data from two open-label, non-concurrently controlled studies (N=34 combined) in patients with normal or abnormal immune status (not including HIV-infected patients). The median dose was 200 mg/day. A response for most signs and symptoms was observed within the first 2 weeks, and all signs and symptoms cleared between 3 and 12 months. Results of these two studies demonstrated substantial evidence of the effectiveness of itraconazole for the treatment of histoplasmosis, compared with the natural history of untreated cases.
Histoplasmosis in HIV-infected patients:
Data from a small number of HIV-infected patients suggested that the response rate of histoplasmosis in HIV-infected patients is similar to that of non-HIV-infected patients. The clinical course of histoplasmosis in HIV-infected patients is more severe and usually requires maintenance therapy to prevent relapse.
Aspergillosis:
Analyses were conducted on data from an open-label, “single-patient-use” protocol designed to make itraconazole available in the U.S. for patients who either failed or were intolerant of amphotericin B therapy (N=190). The findings were corroborated by two smaller open-label studies (N=31 combined) in the same patient population. Most adult patients were treated with a daily dose of 200 to 400 mg, with a median duration of 3 months. Results of these studies demonstrated substantial evidence of effectiveness of itraconazole as a second-line therapy for the treatment of aspergillosis compared with the natural history of the disease in patients who either failed or were intolerant of amphotericin B therapy.
Onychomycosis of the toenail:
Analyses were conducted on data from three double-blind, placebo-controlled studies (N=214 total; 110 given SPORANOX® Capsules) in which patients with onychomycosis of the toenails received 200 mg of SPORANOX® Capsules once daily for 12 consecutive weeks. Results of these studies demonstrated mycologic cure, defined as simultaneous occurrence of negative KOH plus negative culture, in 54% of patients. Thirty-five percent (35%) of patients were considered an overall success (mycologic cure plus clear or minimal nail involvement with significantly decreased signs) and 14% of patients demonstrated mycologic cure plus clinical cure (clearance of all signs, with or without residual nail deformity). The mean time to overall success was approximately 10 months. Twenty-one percent (21%) of the overall success group had a relapse (worsening of the global score or conversion of KOH or culture from negative to positive).
Onychomycosis of the fingernail:
Analyses were conducted on data from a double-blind, placebo-controlled study (N=73 total; 37 given SPORANOX® Capsules) in which patients with onychomycosis of the fingernails received a 1-week course (pulse) of 200 mg of SPORANOX® Capsules b.i.d., followed by a 3-week period without SPORANOX®, which was followed by a second 1-week pulse of 200 mg of SPORANOX® Capsules b.i.d. Results demonstrated mycologic cure in 61% of patients. Fifty-six percent (56%) of patients were considered an overall success and 47% of patients demonstrated mycologic cure plus clinical cure. The mean time to overall success was approximately 5 months. None of the patients who achieved overall success relapsed.
CONTRAINDICATIONS
Congestive Heart Failure:
SPORANOX® (itraconazole) Capsules should not be administered for the treatment of onychomycosis in patients with evidence of ventricular dysfunction such as congestive heart failure (CHF) or a history of CHF. (See CLINICAL PHARMACOLOGY: Special Populations, WARNINGS, PRECAUTIONS: Drug Interactions-Calcium Channel Blockers, and ADVERSE REACTIONS: Post-marketing Experience.)
Drug Interactions:
Concomitant administration of SPORANOX® (itraconazole) Capsules, Injection, or Oral Solution and certain drugs metabolized by the cytochrome P450 3A4 isoenzyme system (CYP3A4) may result in increased plasma concentrations of those drugs, leading to potentially serious and/or life-threatening adverse events. Cisapride, oral midazolam, nisoldipine, pimozide, quinidine, dofetilide, triazolam and levacetylmethadol (levomethadyl) are contraindicated with SPORANOX®. HMG CoA-reductase inhibitors metabolized by CYP3A4, such as lovastatin and simvastatin, are also contraindicated with SPORANOX®. Ergot alkaloids metabolized by CYP3A4 such as dihydroergotamine, ergometrine (ergonovine), ergotamine and methylergometrine (methylergonovine) are contraindicated with SPORANOX®. (See BOX WARNING, and PRECAUTIONS: Drug Interactions.)
SPORANOX® should not be administered for the treatment of onychomycosis to pregnant patients or to women contemplating pregnancy.
SPORANOX® is contraindicated for patients who have shown hypersensitivity to itraconazole or its excipients. There is no information regarding cross-hypersensitivity between itraconazole and other azole antifungal agents. Caution should be used when prescribing SPORANOX® to patients with hypersensitivity to other azoles.
WARNINGS
SPORANOX® (itraconazole) Capsules and SPORANOX® Oral Solution should not be used interchangeably. This is because drug exposure is greater with the Oral Solution than with the Capsules when the same dose of drug is given. In addition, the topical effects of mucosal exposure may be different between the two formulations. Only the Oral Solution has been demonstrated effective for oral and/or esophageal candidiasis.
Hepatic Effects:
SPORANOX® has been associated with rare cases of serious hepatotoxicity, including liver failure and death. Some of these cases had neither pre-existing liver disease nor a serious underlying medical condition, and some of these cases developed within the first week of treatment. If clinical signs or symptoms develop that are consistent with liver disease, treatment should be discontinued and liver function testing performed. Continued SPORANOX® use or reinstitution of treatment with SPORANOX® is strongly discouraged unless there is a serious or life-threatening situation where the expected benefit exceeds the risk. (See PRECAUTIONS: Information for Patients and ADVERSE REACTIONS.)
Cardiac Dysrhythmias:
Life-threatening cardiac dysrhythmias and/or sudden death have occurred in patients using cisapride, pimozide, levacetylmethadol (levomethadyl), or quinidine concomitantly with SPORANOX® and/or other CYP3A4 inhibitors. Concomitant administration of these drugs with SPORANOX® is contraindicated. (See BOX WARNING, CONTRAINDICATIONS, and PRECAUTIONS: Drug Interactions.)
Cardiac Disease:
SPORANOX® Capsules should not be administered for the treatment of onychomycosis in patients with evidence of ventricular dysfunction such as congestive heart failure (CHF) or a history of CHF. SPORANOX® Capsules should not be used for other indications in patients with evidence of ventricular dysfunction unless the benefit clearly outweighs the risk.
For patients with risk factors for congestive heart failure, physicians should carefully review the risks and benefits of SPORANOX® therapy. These risk factors include cardiac disease such as ischemic and valvular disease; significant pulmonary disease such as chronic obstructive pulmonary disease; and renal failure and other edematous disorders. Such patients should be informed of the signs and symptoms of CHF, should be treated with caution, and should be monitored for signs and symptoms of CHF during treatment. If signs or symptoms of CHF appear during administration of SPORANOX® Capsules, discontinue administration.
Itraconazole has been shown to have a negative inotropic effect. When itraconazole was administered intravenously to anesthetized dogs, a dose-related negative inotropic effect was documented. In a healthy volunteer study of SPORANOX® Injection (intravenous infusion), transient, asymptomatic decreases in left ventricular ejection fraction were observed using gated SPECT imaging; these resolved before the next infusion, 12 hours later.
SPORANOX® has been associated with reports of congestive heart failure. In post-marketing experience, heart failure was more frequently reported in patients receiving a total daily dose of 400 mg although there were also cases reported among those receiving lower total daily doses.
Calcium channel blockers can have negative inotropic effects which may be additive to those of itraconazole. In addition, itraconazole can inhibit the metabolism of calcium channel blockers. Therefore, caution should be used when co-administering itraconazole and calcium channel blockers due to an increased risk of CHF. Concomitant administration of SPORANOX® and nisoldipine is contraindicated.
Cases of CHF, peripheral edema, and pulmonary edema have been reported in the post-marketing period among patients being treated for onychomycosis and/or systemic fungal infections. (See CLINICAL PHARMACOLOGY: Special Populations, CONTRAINDICATIONS, PRECAUTIONS: Drug Interactions, and ADVERSE REACTIONS: Post-marketing Experience for more information.)
PRECAUTIONS
General:
SPORANOX® (itraconazole) Capsules should be administered after a full meal. (See CLINICAL PHARMACOLOGY: Pharmacokinetics and Metabolism.)
Under fasted conditions, itraconazole absorption was decreased in the presence of decreased gastric acidity. The absorption of itraconazole may be decreased with the concomitant administration of antacids or gastric acid secretion suppressors. Studies conducted under fasted conditions demonstrated that administration with 8 ounces of a cola beverage resulted in increased absorption of itraconazole in AIDS patients with relative or absolute achlorhydria. This increase relative to the effects of a full meal is unknown. (See CLINICAL PHARMACOLOGY: Pharmacokinetics and Metabolism.)
Hepatotoxicity:
Rare cases of serious hepatotoxicity have been observed with Sporanox® treatment, including some cases within the first week. In patients with elevated or abnormal liver enzymes or active liver disease, or who have experienced liver toxicity with other drugs, treatment with Sporanox® is strongly discouraged unless there is a serious or life threatening situation where the expected benefit exceeds the risk. Liver function monitoring should be done in patients with pre-existing hepatic function abnormalities or those who have experienced liver toxicity with other medications and should be considered in all patients receiving Sporanox®. Treatment should be stopped immediately and liver function testing should be conducted in patients who develop signs and symptoms suggestive of liver dysfunction.
Neuropathy:
If neuropathy occurs that may be attributable to Sporanox® capsules, the treatment should be discontinued.
Hearing Loss:
Transient or permanent hearing loss has been reported in patients receiving treatment with itraconazole. Several of these reports included concurrent administration of quinidine which is contraindicated (see BOX WARNING: Drug Interactions; CONTRAINDICATIONS: Drug Interactions and PRECAUTIONS: Drug Interactions). The hearing loss usually resolves when treatment is stopped, but can persist in some patients.
Information for Patients:
- The topical effects of mucosal exposure may be different between the SPORANOX® Capsules and Oral Solution. Only the Oral Solution has been demonstrated effective for oral and/or esophageal candidiasis. SPORANOX® Capsules should not be used interchangeably with SPORANOX® Oral Solution.
- Instruct patients to take SPORANOX® Capsules with a full meal.
- Instruct patients about the signs and symptoms of congestive heart failure, and if these signs or symptoms occur during SPORANOX® administration, they should discontinue SPORANOX® and contact their healthcare provider immediately.
- Instruct patients to stop SPORANOX® treatment immediately and contact their healthcare provider if any signs and symptoms suggestive of liver dysfunction develop. Such signs and symptoms may include unusual fatigue, anorexia, nausea and/or vomiting, jaundice, dark urine, or pale stools.
- Instruct patients to contact their physician before taking any concomitant medications with itraconazole to ensure there are no potential drug interactions.
- Instruct patients that hearing loss can occur with the use of itraconazole. The hearing loss usually resolves when treatment is stopped, but can persist in some patients. Advise patients to discontinue therapy and inform their physicians if any hearing loss symptoms occur.
Drug Interactions:
Itraconazole and its major metabolite, hydroxyitraconazole, are inhibitors of CYP3A4. Therefore, the following drug interactions may occur (see Table 1 below and the following drug class subheadings that follow):
- SPORANOX® may decrease the elimination of drugs metabolized by CYP3A4, resulting in increased plasma concentrations of these drugs when they are administered with SPORANOX®. These elevated plasma concentrations may increase or prolong both therapeutic and adverse effects of these drugs. Whenever possible, plasma concentrations of these drugs should be monitored, and dosage adjustments made after concomitant SPORANOX® therapy is initiated. When appropriate, clinical monitoring for signs or symptoms of increased or prolonged pharmacologic effects is advised. Upon discontinuation, depending on the dose and duration of treatment, itraconazole plasma concentrations decline gradually (especially in patients with hepatic cirrhosis or in those receiving CYP3A4 inhibitors). This is particularly important when initiating therapy with drugs whose metabolism is affected by itraconazole.
- Inducers of CYP3A4 may decrease the plasma concentrations of itraconazole. SPORANOX® may not be effective in patients concomitantly taking SPORANOX® and one of these drugs. Therefore, administration of these drugs with SPORANOX® is not recommended.
- Other inhibitors of CYP3A4 may increase the plasma concentrations of itraconazole. Patients who must take SPORANOX® concomitantly with one of these drugs should be monitored closely for signs or symptoms of increased or prolonged pharmacologic effects of SPORANOX®.
| 1 This list is not all-inclusive. 2 Contraindicated with SPORANOX® based on clinical and/or pharmacokinetics studies. (See WARNINGS and below.) 3 For information on parenterally administered midazolam, see the Benzodiazepine paragraph below. |
|
| Drug plasma concentration increased by itraconazole | |
| Antiarrhythmics | digoxin, dofetilide,2 quinidine,2 disopyramide |
| Anticonvulsants | carbamazepine |
| Antimycobacterials | rifabutin |
| Antineoplastics | busulfan, docetaxel, vinca alkaloids |
| Antipsychotics | pimozide2 |
| Benzodiazepines | alprazolam, diazepam, midazolam,2, 3 triazolam2 |
| Calcium Channel Blockers | dihydropyridines (including nisoldipine2), verapamil |
| Gastrointestinal Motility Agents | cisapride2 |
| HMG CoA-Reductase Inhibitors | atorvastatin, cerivastatin, lovastatin,2 simvastatin2 |
| Immunosuppressants | cyclosporine, tacrolimus, sirolimus |
| Oral Hypoglycemics | oral hypoglycemics |
| Protease Inhibitors | indinavir, ritonavir, saquinavir |
| Other | levacetylmethadol (levomethadyl),2 ergot alkaloids,2 halofantrine, alfentanil, buspirone, methylprednisolone, budesonide, dexamethasone, fluticasone, trimetrexate, warfarin, cilostazol, eletriptan, fentanyl |
| Decrease plasma concentration of itraconazole | |
| Anticonvulsants | carbamazepine, phenobarbital, phenytoin |
| Antimycobacterials | isoniazid, rifabutin, rifampin |
| Gastric Acid Suppressors/Neutralizers | antacids, H2-receptor antagonists, proton pump inhibitors |
| Non-nucleoside Reverse Transcriptase Inhibitors | nevirapine |
| Increase plasma concentration of itraconazole | |
| Macrolide Antibiotics | clarithromycin, erythromycin |
| Protease Inhibitors | indinavir, ritonavir |
Antiarrhythmics:
The class IA antiarrhythmic quinidine and class III antiarrhythmic dofetilide are known to prolong the QT interval. Coadministration of quinidine or dofetilide with SPORANOX® may increase plasma concentrations of quinidine or dofetilide which could result in serious cardiovascular events. Therefore, concomitant administration of SPORANOX® and quinidine or dofetilide is contraindicated. (See BOX WARNING, CONTRAINDICATIONS, and WARNINGS.)
The class IA antiarrhythmic disopyramide has the potential to increase the QT interval at high plasma concentrations. Caution is advised when SPORANOX® and disopyramide are administered concomitantly.
Concomitant administration of digoxin and SPORANOX® has led to increased plasma concentrations of digoxin via inhibition of P-glycoprotein.
Anticonvulsants:
Reduced plasma concentrations of itraconazole were reported when SPORANOX® was administered concomitantly with phenytoin. Carbamazepine, phenobarbital, and phenytoin are all inducers of CYP3A4. Although interactions with carbamazepine and phenobarbital have not been studied, concomitant administration of SPORANOX® and these drugs would be expected to result in decreased plasma concentrations of itraconazole. In addition, in vivo studies have demonstrated an increase in plasma carbamazepine concentrations in subjects concomitantly receiving ketoconazole. Although there are no data regarding the effect of itraconazole on carbamazepine metabolism, because of the similarities between ketoconazole and itraconazole, concomitant administration of SPORANOX® and carbamazepine may inhibit the metabolism of carbamazepine.
Antimycobacterials:
Drug interaction studies have demonstrated that plasma concentrations of azole antifungal agents and their metabolites, including itraconazole and hydroxyitraconazole, were significantly decreased when these agents were given concomitantly with rifabutin or rifampin. In vivo data suggest that rifabutin is metabolized in part by CYP3A4. SPORANOX® may inhibit the metabolism of rifabutin. Although no formal study data are available for isoniazid, similar effects should be anticipated. Therefore, the efficacy of SPORANOX® could be substantially reduced if given concomitantly with one of these agents. Coadministration is not recommended.
Antineoplastics:
SPORANOX® may inhibit the metabolism of busulfan, docetaxel, and vinca alkaloids.
Antipsychotics:
Pimozide is known to prolong the QT interval and is partially metabolized by CYP3A4. Coadministration of pimozide with SPORANOX® could result in serious cardiovascular events. Therefore, concomitant administration of SPORANOX® and pimozide is contraindicated. (See BOX WARNING, CONTRAINDICATIONS, and WARNINGS.)
Benzodiazepines:
Concomitant administration of SPORANOX® and alprazolam, diazepam, oral midazolam, or triazolam could lead to increased plasma concentrations of these benzodiazepines. Increased plasma concentrations could potentiate and prolong hypnotic and sedative effects. Concomitant administration of SPORANOX® and oral midazolam or triazolam is contraindicated. (See CONTRAINDICATIONS and WARNINGS.) If midazolam is administered parenterally, special precaution and patient monitoring is required since the sedative effect may be prolonged.
Calcium Channel Blockers:
Edema has been reported in patients concomitantly receiving SPORANOX® and dihydropyridine calcium channel blockers. Appropriate dosage adjustment may be necessary.
Calcium channel blockers can have a negative inotropic effect which may be additive to those of itraconazole; itraconazole can inhibit the metabolism of calcium channel blockers such as dihydropyridines (e.g., nifedipine and felodipine) and verapamil. Therefore, caution should be used when co-administering itraconazole and calcium channel blockers due to an increased risk of CHF. Concomitant administration of SPORANOX® and nisoldipine results in clinically significant increases in nisoldipine plasma concentrations, which cannot be managed by dosage reduction, therefore the concomitant administration of SPORANOX® and nisoldipine is contraindicated. (See CLINICAL PHARMACOLOGY: Special Populations, CONTRAINDICATIONS, WARNINGS, and ADVERSE REACTIONS: Post-marketing Experience for more information).
Gastric Acid Suppressors/Neutralizers:
Reduced plasma concentrations of itraconazole were reported when SPORANOX® Capsules were administered concomitantly with H2-receptor antagonists. Studies have shown that absorption of itraconazole is impaired when gastric acid production is decreased. Therefore, SPORANOX® should be administered with a cola beverage if the patient has achlorhydria or is taking H2-receptor antagonists or other gastric acid suppressors. Antacids should be administered at least 1 hour before or 2 hours after administration of SPORANOX® Capsules. In a clinical study, when SPORANOX® Capsules were administered with omeprazole (a proton pump inhibitor), the bioavailability of itraconazole was significantly reduced.
Gastrointestinal Motility Agents:
Coadministration of SPORANOX® with cisapride can elevate plasma cisapride concentrations which could result in serious cardiovascular events. Therefore, concomitant administration of SPORANOX® with cisapride is contraindicated. (See BOX WARNING, CONTRAINDICATIONS, and WARNINGS.)
HMG CoA-Reductase Inhibitors:
Human pharmacokinetic data suggest that SPORANOX® inhibits the metabolism of atorvastatin, cerivastatin, lovastatin, and simvastatin, which may increase the risk of skeletal muscle toxicity, including rhabdomyolysis. Concomitant administration of SPORANOX® with HMG CoA-reductase inhibitors, such as lovastatin and simvastatin, is contraindicated. (See CONTRAINDICATIONS and WARNINGS.)
Immunosuppressants:
Concomitant administration of SPORANOX® and cyclosporine or tacrolimus has led to increased plasma concentrations of these immunosuppressants. Concomitant administration of SPORANOX® and sirolimus could increase plasma concentrations of sirolimus.
Macrolide Antibiotics:
Erythromycin and clarithromycin are known inhibitors of CYP3A4 (See Table 1) and may increase plasma concentrations of itraconazole. In a small pharmacokinetic study involving HIV infected patients, clarithromycin was shown to increase plasma concentrations of itraconazole. Similarly, following administration of 1 gram of erythromycin ethyl succinate and 200 mg itraconazole as single doses, the mean Cmax and AUC 0-∞ of itraconazole increased by 44% (90% CI: 119-175%) and 36% (90% CI: 108-171%), respectively.
Non-nucleoside Reverse Transcriptase Inhibitors:
Nevirapine is an inducer of CYP3A4. In vivo studies have shown that nevirapine induces the metabolism of ketoconazole, significantly reducing the bioavailability of ketoconazole. Studies involving nevirapine and itraconazole have not been conducted. However, because of the similarities between ketoconazole and itraconazole, concomitant administration of SPORANOX® and nevirapine is not recommended.
In a clinical study, when 8 HIV-infected subjects were treated concomitantly with SPORANOX® Capsules 100 mg twice daily and the nucleoside reverse transcriptase inhibitor zidovudine 8 ± 0.4 mg/kg/day, the pharmacokinetics of zidovudine were not affected. Other nucleoside reverse transcriptase inhibitors have not been studied.
Oral Hypoglycemic Agents:
Severe hypoglycemia has been reported in patients concomitantly receiving azole antifungal agents and oral hypoglycemic agents. Blood glucose concentrations should be carefully monitored when SPORANOX® and oral hypoglycemic agents are coadministered.
Polyenes:
Prior treatment with itraconazole, like other azoles, may reduce or inhibit the activity of polyenes such as amphotericin B. However, the clinical significance of this drug effect has not been clearly defined.
Protease Inhibitors:
Concomitant administration of SPORANOX® and protease inhibitors metabolized by CYP3A4, such as indinavir, ritonavir, and saquinavir, may increase plasma concentrations of these protease inhibitors. In addition, concomitant administration of SPORANOX® and indinavir and ritonavir (but not saquinavir) may increase plasma concentrations of itraconazole. Caution is advised when SPORANOX® and protease inhibitors must be given concomitantly.
Other:
- Levacetylmethadol (levomethadyl) is known to prolong the QT interval and is metabolized by CYP3A4. Co-administration of levacetylmethadol with SPORANOX® could result in serious cardiovascular events. Therefore, concomitant administration of SPORANOX® and levacetylmethadol is contraindicated.
- Elevated concentrations of ergot alkaloids can cause ergotism, ie. a risk for vasospasm potentially leading to cerebral ischemia and/or ischemia of the extremities. Concomitant administration of ergot alkaloids such as dihydroergotamine, ergometrine (ergonovine), ergotamine and methylergometrine (methylergonovine) with SPORANOX® is contraindicated.
- Halofantrine has the potential to prolong the QT interval at high plasma concentrations. Caution is advised when SPORANOX® and halofantrine are administered concomitantly.
- In vitro data suggest that alfentanil is metabolized by CYP3A4. Administration with SPORANOX® may increase plasma concentrations of alfentanil.
- Human pharmacokinetic data suggest that concomitant administration of SPORANOX® and buspirone results in significant increases in plasma concentrations of buspirone.
- SPORANOX® may inhibit the metabolism of certain glucocorticosteroids such as budesonide, dexamethasone, fluticasone and methylprednisolone.
- In vitro data suggest that trimetrexate is extensively metabolized by CYP3A4. In vitro animal models have demonstrated that ketoconazole potently inhibits the metabolism of trimetrexate. Although there are no data regarding the effect of itraconazole on trimetrexate metabolism, because of the similarities between ketoconazole and itraconazole, concomitant administration of SPORANOX® and trimetrexate may inhibit the metabolism of trimetrexate.
- SPORANOX® enhances the anticoagulant effect of coumarin-like drugs, such as warfarin.
- Cilostazol and eletriptan are CYP3A4 metabolized drugs that should be used with caution when co-administered with SPORANOX®.
- Fentanyl plasma concentrations could be increased or prolonged by concomitant use of SPORANOX® and may cause potentially fatal respiratory depression.
Carcinogenesis, Mutagenesis, and Impairment of Fertility:
Itraconazole showed no evidence of carcinogenicity potential in mice treated orally for 23 months at dosage levels up to 80 mg/kg/day (approximately 10x the maximum recommended human dose [MRHD]). Male rats treated with 25 mg/kg/day (3.1x MRHD) had a slightly increased incidence of soft tissue sarcoma. These sarcomas may have been a consequence of hypercholesterolemia, which is a response of rats, but not dogs or humans, to chronic itraconazole administration. Female rats treated with 50 mg/kg/day (6.25x MRHD) had an increased incidence of squamous cell carcinoma of the lung (2/50) as compared to the untreated group. Although the occurrence of squamous cell carcinoma in the lung is extremely uncommon in untreated rats, the increase in this study was not statistically significant.
Itraconazole produced no mutagenic effects when assayed in DNA repair test (unscheduled DNA synthesis) in primary rat hepatocytes, in Ames tests with Salmonella typhimurium (6 strains) and Escherichia coli, in the mouse lymphoma gene mutation tests, in a sex-linked recessive lethal mutation (Drosophila melanogaster) test, in chromosome aberration tests in human lymphocytes, in a cell transformation test with C3H/10T½ C18 mouse embryo fibroblasts cells, in a dominant lethal mutation test in male and female mice, and in micronucleus tests in mice and rats.
Itraconazole did not affect the fertility of male or female rats treated orally with dosage levels of up to 40 mg/kg/day (5x MRHD), even though parental toxicity was present at this dosage level. More severe signs of parental toxicity, including death, were present in the next higher dosage level, 160 mg/kg/day (20x MRHD).
Pregnancy: Teratogenic effects. Pregnancy Category C:
Itraconazole was found to cause a dose-related increase in maternal toxicity, embryotoxicity, and teratogenicity in rats at dosage levels of approximately 40-160 mg/kg/day (5-20x MRHD), and in mice at dosage levels of approximately 80 mg/kg/day (10x MRHD). In rats, the teratogenicity consisted of major skeletal defects; in mice, it consisted of encephaloceles and/or macroglossia.
There are no studies in pregnant women. SPORANOX® should be used for the treatment of systemic fungal infections in pregnancy only if the benefit outweighs the potential risk.
SPORANOX® should not be administered for the treatment of onychomycosis to pregnant patients or to women contemplating pregnancy. SPORANOX® should not be administered to women of childbearing potential for the treatment of onychomycosis unless they are using effective measures to prevent pregnancy and they begin therapy on the second or third day following the onset of menses. Effective contraception should be continued throughout SPORANOX® therapy and for 2 months following the end of treatment.
During post-marketing experience, cases of congenital abnormalities have been reported. (See ADVERSE REACTIONS, Post-marketing Experience.)
Nursing Mothers:
Itraconazole is excreted in human milk; therefore, the expected benefits of SPORANOX® therapy for the mother should be weighed against the potential risk from exposure of itraconazole to the infant. The U.S. Public Health Service Centers for Disease Control and Prevention advises HIV-infected women not to breast-feed to avoid potential transmission of HIV to uninfected infants.
Pediatric Use:
The efficacy and safety of SPORANOX® have not been established in pediatric patients. No pharmacokinetic data on SPORANOX® Capsules are available in children. A small number of patients ages 3 to 16 years have been treated with 100 mg/day of itraconazole capsules for systemic fungal infections, and no serious unexpected adverse events have been reported. SPORANOX® Oral Solution (5 mg/kg/day) has been administered to pediatric patients (N=26; ages 6 months to 12 years) for 2 weeks and no serious unexpected adverse events were reported.
The long-term effects of itraconazole on bone growth in children are unknown. In three toxicology studies using rats, itraconazole induced bone defects at dosage levels as low as 20 mg/kg/day (2.5x MRHD). The induced defects included reduced bone plate activity, thinning of the zona compacta of the large bones, and increased bone fragility. At a dosage level of 80 mg/kg/day (10x MRHD) over 1 year or 160 mg/kg/day (20x MRHD) for 6 months, itraconazole induced small tooth pulp with hypocellular appearance in some rats. No such bone toxicity has been reported in adult patients.
Geriatric Use:
Transient or permanent hearing loss has been reported in elderly patients receiving treatment with itraconazole. Several of these reports included concurrent administration of quinidine which is contraindicated (see BOX WARNING: Drug Interactions, CONTRAINDICATIONS: Drug Interactions and PRECAUTIONS: Drug Interactions). Itraconazole should be used with care in elderly patients (see PRECAUTIONS).
HIV-Infected Patients:
Because hypochlorhydria has been reported in HIV-infected individuals, the absorption of itraconazole in these patients may be decreased.
Renal Impairment:
Limited data are available on the use of oral itraconazole in patients with renal impairment. Caution should be exercised when this drug is administered in this patient population. (See CLINICAL PHARMACOLOGY: Special Populations and DOSAGE AND ADMINISTRATION.)
Hepatic Impairment:
Limited data are available on the use of oral itraconazole in patients with hepatic impairment. Caution should be exercised when this drug is administered in this patient population. (See CLINICAL PHARMACOLOGY: Special Populations and DOSAGE AND ADMINISTRATION.)
ADVERSE REACTIONS
SPORANOX® has been associated with rare cases of serious hepatotoxicity, including liver failure and death. Some of these cases had neither pre-existing liver disease nor a serious underlying medical condition. If clinical signs or symptoms develop that are consistent with liver disease, treatment should be discontinued and liver function testing performed. The risks and benefits of SPORANOX® use should be reassessed. (See WARNINGS: Hepatic Effects and PRECAUTIONS: Hepatotoxicity and Information for Patients.)
Adverse Events in the Treatment of Systemic Fungal Infections
Adverse event data were derived from 602 patients treated for systemic fungal disease in U.S. clinical trials who were immunocompromised or receiving multiple concomitant medications. Treatment was discontinued in 10.5% of patients due to adverse events. The median duration before discontinuation of therapy was 81 days (range: 2 to 776 days). The table lists adverse events reported by at least 1% of patients.
| Body System/Adverse Event | Incidence (%) (N=602) |
|---|---|
| * Rash tends to occur more frequently in immunocompromised patients receiving immunosuppressive medications. | |
| Gastrointestinal | |
| Nausea | 11 |
| Vomiting | 5 |
| Diarrhea | 3 |
| Abdominal Pain | 2 |
| Anorexia | 1 |
| Body as a Whole | |
| Edema | 4 |
| Fatigue | 3 |
| Fever | 3 |
| Malaise | 1 |
| Skin and Appendages | |
| Rash* | 9 |
| Pruritus | 3 |
| Central/Peripheral Nervous System | |
| Headache | 4 |
| Dizziness | 2 |
| Psychiatric | |
| Libido Decreased | 1 |
| Somnolence | 1 |
| Cardiovascular | |
| Hypertension | 3 |
| Metabolic/Nutritional | |
| Hypokalemia | 2 |
| Urinary System | |
| Albuminuria | 1 |
| Liver and Biliary System | |
| Hepatic Function Abnormal | 3 |
| Reproductive System, Male | |
| Impotence | 1 |
Adverse events infrequently reported in all studies included constipation, gastritis, depression, insomnia, tinnitus, menstrual disorder, adrenal insufficiency, gynecomastia, and male breast pain.
Adverse Events Reported in Toenail Onychomycosis Clinical Trials
Patients in these trials were on a continuous dosing regimen of 200 mg once daily for 12 consecutive weeks.
The following adverse events led to temporary or permanent discontinuation of therapy.
| Adverse Event | Incidence (%) Itraconazole (N=112) |
|---|---|
| Elevated Liver Enzymes (greater than twice the upper limit of normal) | 4 |
| Gastrointestinal Disorders | 4 |
| Rash | 3 |
| Hypertension | 2 |
| Orthostatic Hypotension | 1 |
| Headache | 1 |
| Malaise | 1 |
| Myalgia | 1 |
| Vasculitis | 1 |
| Vertigo | 1 |
The following adverse events occurred with an incidence of greater than or equal to 1% (N=112): headache: 10%; rhinitis: 9%; upper respiratory tract infection: 8%; sinusitis, injury: 7%; diarrhea, dyspepsia, flatulence, abdominal pain, dizziness, rash: 4%; cystitis, urinary tract infection, liver function abnormality, myalgia, nausea: 3%; appetite increased, constipation, gastritis, gastroenteritis, pharyngitis, asthenia, fever, pain, tremor, herpes zoster, abnormal dreaming: 2%.
Adverse Events Reported in Fingernail Onychomycosis Clinical Trials
Patients in these trials were on a pulse regimen consisting of two 1-week treatment periods of 200 mg twice daily, separated by a 3-week period without drug.
The following adverse events led to temporary or permanent discontinuation of therapy.
| Adverse Event | Incidence (%) Itraconazole (N=37) |
|---|---|
| Rash/Pruritus | 3 |
| Hypertriglyceridemia | 3 |
The following adverse events occurred with an incidence of greater than or equal to 1% (N=37): headache: 8%; pruritus, nausea, rhinitis: 5%; rash, bursitis, anxiety, depression, constipation, abdominal pain, dyspepsia, ulcerative stomatitis, gingivitis, hypertriglyceridemia, sinusitis, fatigue, malaise, pain, injury: 3%.
Post-marketing Experience
Adverse drug reactions that have been identified during post-approval use of SPORANOX® (all formulations) are listed in the table below. Because these reactions are reported voluntarily from a population of uncertain size, reliably estimating their frequency or establishing a causal relationship to drug exposure is not always possible.
| Blood and lymphatic system disorders: | Leukopenia, neutropenia, thrombocytopenia |
| Immune system disorders: | Anaphylaxis; anaphylactic, anaphylactoid and allergic reactions; serum sickness; angioneurotic edema |
| Metabolism and nutrition disorders: | Hypertriglyceridemia, hypokalemia |
| Nervous system disorders: | Peripheral neuropathy, paresthesia, hypoesthesia, headache, dizziness |
| Eye disorders: | Visual disturbances, including vision blurred and diplopia |
| Ear and labyrinth disorder: | Transient or permanent hearing loss, tinnitus |
| Cardiac disorders: | Congestive heart failure |
| Respiratory, thoracic and mediastinal disorders: | Pulmonary edema |
| Gastrointestinal disorders: | Pancreatitis, abdominal pain, vomiting, dyspepsia, nausea, diarrhea, constipation, dysgeusia |
| Hepato-biliary disorders: | Serious hepatotoxicity (including some cases of fatal acute liver failure), hepatitis, reversible increases in hepatic enzymes |
| Skin and subcutaneous tissue disorders: | Toxic epidermal necrolysis, Stevens-Johnson syndrome, exfoliative dermatitis, leukocytoclastic vasculitis, erythema multiforme, alopecia, photosensitivity, rash, urticaria, pruritus |
| Musculoskeletal and connective tissue disorders: | Myalgia, arthralgia |
| Renal and urinary disorders: | Urinary incontinence, pollakiuria |
| Reproductive system and breast disorders: | Menstrual disorders, erectile dysfunction |
| General disorders and administration site conditions: | Peripheral edema, pyrexia |
There is limited information on the use of SPORANOX® during pregnancy. Cases of congenital abnormalities including skeletal, genitourinary tract, cardiovascular and ophthalmic malformations as well as chromosomal and multiple malformations have been reported during post-marketing experience. A causal relationship with SPORANOX® has not been established. (See CLINICAL PHARMACOLOGY: Special Populations, CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS: Drug Interactions for more information.)
OVERDOSAGE
Itraconazole is not removed by dialysis. In the event of accidental overdosage, supportive measures, including gastric lavage with sodium bicarbonate, should be employed.
Limited data exist on the outcomes of patients ingesting high doses of itraconazole. In patients taking either 1000 mg of SPORANOX® (itraconazole) Oral Solution or up to 3000 mg of SPORANOX® (itraconazole) Capsules, the adverse event profile was similar to that observed at recommended doses.
DOSAGE AND ADMINISTRATION
SPORANOX® (itraconazole) Capsules should be taken with a full meal to ensure maximal absorption.
SPORANOX® Capsules is a different preparation than SPORANOX® Oral Solution and should not be used interchangeably.
Treatment of Blastomycosis and Histoplasmosis:
The recommended dose is 200 mg once daily (2 capsules). If there is no obvious improvement, or there is evidence of progressive fungal disease, the dose should be increased in 100-mg increments to a maximum of 400 mg daily. Doses above 200 mg/day should be given in two divided doses.
Treatment of Aspergillosis:
A daily dose of 200 to 400 mg is recommended.
Treatment in Life-Threatening Situations:
In life-threatening situations, a loading dose should be used whether given as oral capsules or intravenously.
- IV Injection: the recommended intravenous dose is 200 mg b.i.d. for four consecutive doses, followed by 200 mg once daily thereafter. Each intravenous dose should be infused over 1 hour. The safety and efficacy of SPORANOX® Injection administered for greater than 14 days is not known. See complete prescribing information for SPORANOX® (itraconazole) Injection.
- Capsules: although clinical studies did not provide for a loading dose, it is recommended, based on pharmacokinetic data, that a loading dose of 200 mg (2 capsules) three times daily (600 mg/day) be given for the first 3 days of treatment.
Treatment should be continued for a minimum of three months and until clinical parameters and laboratory tests indicate that the active fungal infection has subsided. An inadequate period of treatment may lead to recurrence of active infection.
SPORANOX® Capsules and SPORANOX® Oral Solution should not be used interchangeably. Only the oral solution has been demonstrated effective for oral and/or esophageal candidiasis.
Treatment of Onychomycosis:
Toenails with or without fingernail involvement: The recommended dose is 200 mg (2 capsules) once daily for 12 consecutive weeks.
Treatment of Onychomycosis:
Fingernails only: The recommended dosing regimen is 2 treatment pulses, each consisting of 200 mg (2 capsules) b.i.d. (400 mg/day) for 1 week. The pulses are separated by a 3-week period without SPORANOX®.
Use in Patients with Renal Impairment:
Limited data are available on the use of oral itraconazole in patients with renal impairment. Caution should be exercised when this drug is administered in this patient population. (See CLINICAL PHARMACOLOGY: Special Populations and PRECAUTIONS for further information.)
Use in Patients with Hepatic Impairment:
Limited data are available on the use of oral itraconazole in patients with hepatic impairment. Caution should be exercised when this drug is administered in this patient population. (See CLINICAL PHARMACOLOGY: Special Populations, WARNINGS, and PRECAUTIONS.)
HOW SUPPLIED
SPORANOX® (itraconazole) Capsules are available containing 100 mg of itraconazole, with a blue opaque cap and pink transparent body, imprinted with “JANSSEN” and “SPORANOX 100.” The capsules are supplied in unit-dose blister packs of 3 × 10 capsules (NDC 50458-290-01), bottles of 30 capsules (NDC 50458-290-04) and in the PulsePak® containing 7 blister packs × 4 capsules each (NDC 50458-290-28).
Store at controlled room temperature 15°-25°C (59°-77°F). Protect from light and moisture.
Keep out of reach of children.
© Ortho-McNeil-Janssen Pharmaceuticals, Inc. 2001
U.S. Patent Nos. 4,267,179; 5,633,015
Revised November 2009
Capsule contents manufactured by:
Janssen Pharmaceutica N.V.
Olen, Belgium
Manufactured by:
JOLLC, Gurabo, Puerto Rico 00778
Manufactured for:
PriCara, Division of Ortho-McNeil-Janssen Pharmaceuticals, Inc.
Raritan, NJ 08869
PATIENT INFORMATION
This summary contains important information about SPORANOX® (SPOR-ah-nox). This information is for patients who have been prescribed SPORANOX® to treat fungal nail infections. If your doctor prescribed SPORANOX® for medical problems other than fungal nail infections, ask your doctor if there is any information in this summary that does not apply to you. Read this information carefully each time you start to use SPORANOX®. This information does not take the place of discussion between you and your doctor. Only your doctor can decide if SPORANOX® is the right treatment for you. If you do not understand some of this information or have any questions, talk with your doctor or pharmacist.
WHAT IS THE MOST IMPORTANT INFORMATION I SHOULD KNOW ABOUT SPORANOX®?
SPORANOX® is used to treat fungal nail infections. However, SPORANOX® is not for everyone. Do not take SPORANOX® for fungal nail infections if you have had heart failure, including congestive heart failure. You should not take SPORANOX® if you are taking certain medicines that could lead to serious or life-threatening medical problems. (See “Who Should Not Take SPORANOX®?” below.)
If you have had heart, lung, liver, kidney or other serious health problems, ask your doctor if it is safe for you to take SPORANOX®.
WHAT HAPPENS IF I HAVE A FUNGAL NAIL INFECTION?
Anyone can have a fungal nail infection, but it is usually found in adults. When a fungus infects the tip or sides of a nail, the infected part of the nail may turn yellow or brown. If not treated, the fungus may spread under the nail towards the cuticle. If the fungus spreads, more of the nail may change color, may become thick or brittle, and the tip of the nail may become raised. In some patients, this can cause pain and discomfort.
WHAT IS SPORANOX®?
SPORANOX® is a prescription medicine used to treat fungal infections of the toenails and fingernails. It is also used to treat some types of fungal infections in other areas of your body. We do not know if SPORANOX® works in children with fungal nail infections or if it is safe for children to take.
SPORANOX® comes in the form of capsules and liquid (oral solution). The capsule and liquid forms work differently, so you should not use one in place of the other. This Patient Information discusses only the capsule form of SPORANOX®. You will get these capsules in a medicine bottle or a SPORANOX PulsePak®. The PulsePak® contains 28 capsules for treatment of your fungal nail infection.
SPORANOX® goes into your bloodstream and travels to the source of the infection underneath the nail so that it can fight the infection there. Improved nails may not be obvious for several months after the treatment period is finished because it usually takes about 6 months to grow a new fingernail and 12 months to grow a new toenail.
WHO SHOULD NOT TAKE SPORANOX®?
SPORANOX® is not for everyone. Your doctor will decide if SPORANOX® is the right treatment for you. Some patients should not take SPORANOX® because they may have certain health problems or may be taking certain medicines that could lead to serious or life-threatening medical problems.
Tell your doctor and pharmacist the name of all the prescription and non-prescription medicines you are taking, including dietary supplements and herbal remedies. Also tell your doctor about any other medical conditions you have had, especially heart, lung, liver or kidney conditions.
- have had heart failure, including congestive heart failure.
- are taking any of the medicines listed below. Dangerous or even life-threatening abnormal heartbeats could result:
- quinidine (such as Cardioquin®, Quinaglute®, Quinidex®)
- dofetilide (such as Tikosyn™)
- cisapride (such as Propulsid®)
- pimozide (such as Orap®)
- levacetylmethadol (such as Orlaam®)
- are taking any of the following medicines:
- lovastatin (such as Mevacor®, Advicor®, Altocor™)
- simvastatin (such as Zocor®)
- triazolam (such as Halcion®)
- midazolam (such as Versed®)
- nisoldipine (such as Sular®)
- ergot alkaloids (such as Migranal®, Ergonovine, Cafergot®, Methergine®)
- have ever had an allergic reaction to itraconazole or any of the other ingredients in SPORANOX® Capsules. Ask your doctor or pharmacist for a list of these ingredients.
Taking SPORANOX® with certain other medicines could lead to serious or life-threatening medical problems. For example, taking fentanyl, a strong opioid narcotic pain medicine, with SPORANOX® could cause serious side effects, including trouble breathing, that may be life-threatening. Tell your doctor and pharmacist the name of all the prescription and non-prescription medicines you are taking. Your doctor will decide if SPORANOX® is the right treatment for you.
WHAT SHOULD I KNOW ABOUT SPORANOX® AND PREGNANCY OR BREAST FEEDING?
Never take SPORANOX® if you have a fungal nail infection and are pregnant or planning to become pregnant within 2 months after you have finished your treatment.
If you are able to become pregnant, you should use effective birth control during SPORANOX® treatment and for 2 months after finishing treatment. Ask your doctor about effective types of birth control.
If you are breast-feeding, talk with your doctor about whether you should take SPORANOX®.
HOW SHOULD I TAKE SPORANOX®?
Always take SPORANOX® Capsules during or right after a full meal.
Your doctor will decide the right dose for you. Depending on your infection, you will take SPORANOX® once a day for 12 weeks, or twice a day for 1 week in a “pulse” dosing schedule. You will receive either a bottle of capsules or a PulsePak®. Do not skip any doses. Be sure to finish all your SPORANOX® as prescribed by your doctor.
If you have ever had liver problems, your doctor should do a blood test to check your condition. If you haven't had liver problems, your doctor may recommend blood tests to check the condition of your liver because patients taking SPORANOX® can develop liver problems.
If you forget to take or miss doses of SPORANOX®, ask your doctor what you should do with the missed doses.
THE SPORANOX PulsePak®
If you use the PulsePak®, you will take SPORANOX® for 1 week and then take no SPORANOX® for the next 3 weeks before repeating the 1-week treatment. This is called “pulse dosing.” The SPORANOX PulsePak® contains enough medicine for one “pulse” (1 week of treatment).
The SPORANOX PulsePak® comes with special instructions. It contains 7 pouches-one for each day of treatment. Inside each pouch is a card containing 4 capsules. Looking at the back of the card, fold it back along the dashed line and peel away the backing so that you can remove 2 capsules.
- Take 2 capsules in the morning and 2 capsules in the evening. This means you will take 4 capsules a day for 7 days. At the end of 7 days, you will have taken all of the capsules in the PulsePak® box.
- After you finish the PulsePak®, do not take any SPORANOX® for the next 3 weeks. Even though you are not taking any capsules during this time, SPORANOX® keeps working inside your nails to help fight the fungal infection.
- You will need more than one “pulse” to treat your fungal nail infection. When your doctor prescribes another pulse treatment, be sure to get your refill before the end of week 4.
|
SPORANOX® Pulse Dosing |
||||||||||||||
|
Take 2 SPORANOX® capsules twice a day for 1 week |
||||||||||||||
|
| Day1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 | Day 7 | |||||||
|
| AM | PM | AM | PM | AM | PM | AM | PM | AM | PM | AM | PM | AM | PM |
|
Week 1 | // | // | // | // | // | // | // | // | // | // | // | // | // | // |
|
Week 2 |
For the next 3 weeks, do not take any SPORANOX® capsules. Remember to get a refill before the end of Week 4 when your doctor prescribes another PulsePak®. |
|||||||||||||
|
Week 3 |
||||||||||||||
|
Week 4 |
||||||||||||||
WHAT ARE THE POSSIBLE SIDE EFFECTS OF SPORANOX®?
The most common side effects that cause people to stop treatment either for a short time or completely include: skin rash, high triglyceride test results, high liver test results, and digestive system problems (such as nausea, bloating, and diarrhea).
Stop SPORANOX® and call your doctor or get medical assistance right away if you have a severe allergic reaction. Symptoms of an allergic reaction may include skin rash, itching, hives, shortness of breath or difficulty breathing, and/or swelling of the face. Very rarely, an oversensitivity to sunlight, a tingling sensation in the limbs or a severe skin disorder can occur. If any of these symptoms occur, stop taking SPORANOX® and contact your doctor.
Stop SPORANOX® and call your doctor right away if you develop shortness of breath; have unusual swelling of your feet, ankles or legs; suddenly gain weight; are unusually tired; cough up white or pink phlegm; have unusual fast heartbeats; or begin to wake up at night. In rare cases, patients taking SPORANOX® could develop serious heart problems, and these could be warning signs of heart failure.
Stop SPORANOX® and call your doctor right away if you become unusually tired; lose your appetite; or develop nausea, abdominal pain, or vomiting, a yellow color to your skin or eyes, or dark colored urine or pale stools (bowel movements). In rare cases, patients taking SPORANOX® could develop serious liver problems and these could be warning signs.
Stop SPORANOX® and call your doctor right away if you experience any hearing loss symptoms. In very rare cases, patients taking SPORANOX® have reported temporary or permanent hearing loss.
Call your doctor right away if you develop tingling or numbness in your extremities (hands or feet), if your vision gets blurry or you see double, if you hear a ringing in your ears, if you lose the ability to control your urine or urinate much more than usual.
Additional possible side effects include upset stomach, vomiting, abdominal pain, constipation, headache, fever, inflammation of the pancreas, menstrual disorders, erectile dysfunction, dizziness, muscle weakness or pain, painful joints, unpleasant taste, or hair loss. These are not all the side effects of SPORANOX®. Your doctor or pharmacist can give you a more complete list.
WHAT SHOULD I DO IF I TAKE AN OVERDOSE OF SPORANOX®?
If you think you took too much SPORANOX®, call your doctor or local poison control center, or go to the nearest hospital emergency room right away.
HOW SHOULD I STORE SPORANOX®?
Keep all medicines, including SPORANOX®, out of the reach of children.
Store SPORANOX® Capsules and the PulsePak® at room temperature in a dry place away from light.
GENERAL ADVICE ABOUT SPORANOX®
Medicines are sometimes prescribed for conditions that are not mentioned in patient information leaflets. Do not use SPORANOX® for a condition for which it was not prescribed. Do not give SPORANOX® to other people, even if they have the same symptoms you have. It may harm them.
This leaflet summarizes the most important information about SPORANOX®. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about SPORANOX® that is written for health professionals or you can call 1-800-526-7736.
This patient information has been approved by the U.S. Food and Drug Administration.
The following are registered trademarks of their respective manufacturers:
Mevacor® (Merck & Co., Inc.), Advicor® (Kos Pharmaceuticals, Inc.), Altocor™ (Andrx Laboratories), Zocor® (Merck & Co., Inc.), Halcion® (Pharmacia), Versed® (Roche Pharmaceuticals), Cardioquin® (The Purdue Frederick Company), Quinaglute® (Berlex Laboratories), Quinidex® (A.H. Robins), TikosynTM (Pfizer, Inc.), Propulsid® (Janssen Pharmaceutica Products, L.P.), Orlaam® (Roxane Laboratories), Migranal® (Xcel Pharmaceuticals), Ergonovine (PDRX Pharmaceuticals), Cafergot® (Novartis Pharmaceuticals Corporation), Methergine® (Novartis Pharmaceuticals Corporation), Orap® (Gate Pharmaceuticals), and Sular® (Sciele Pharma, Inc.)
© Ortho-McNeil-Janssen Pharmaceuticals, Inc. 2001
Printed in USA/ Revised: November 2009
TO
OPEN: Fold at line
and tear open at slit.
sporanox® 100
mg
[itraconazole]
Capsules
Each
pouch contains four 100 mg capsules,
one complete day of treatment.
Keep out of reach of children.
Capsule contents manufactured by:
Janssen
Pharmaceutica, N.V., Olen, Belgium
Manufactured
by:
JOLLC, Gurabo, Puerto Rico 00778
Manufactured
for:
Janssen®
Division
of Ortho-McNeil-Janssen
Pharmaceuticals, Inc.
Titusville,
NJ 08560
(01) 10350458290285 © OMJPI 2001 Revised 4/08 8008302

Contains 7 pouches x
4 capsules each
Each capsule contains: itraconazole 100 mg
Dosage: See accompanying product literature
Store at controlled room temperature
15º-25ºC (59º-77ºF)
Protect from light and moisture.
Keep out of reach of children.
Capsule contents manufactured by:
Janssen Pharmaceutica, N.V.
Olen, Belgium
Manufactured by: JOLLC,
Gurabo, Puerto Rico 00778
Manufactured for: Janssen,
Division of Ortho-McNeil-Janssen
Pharmaceuticals, Inc.
Titusville, NJ 08560
Janssen®
Division of Ortho-McNeil-Janssen
Pharmaceuticals, Inc.

| SPORANOX
itraconazole capsule |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020083 | 09/11/1992 | |
| Labeler - Ortho-McNeil-Janssen Pharmaceuticals Inc. (063137772) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Janssen Pharmaceutical, Ltd | 989673884 | API MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Janssen Pharmaceutica, NV | 374747970 | API MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Janssen Pharmaceutica, NV | 377360784 | MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| JOLLC | 062191882 | MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Ortho-McNeil-Janssen Pharmaceuticals, Inc | 063137772 | RELABEL | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Catalent | 806746405 | RELABEL | |