AMERGE
-
naratriptan hydrochloride tablet, film coated
SmithKline Beecham Corporation
----------
AMERGE®(naratriptan hydrochloride)
Tablets
DESCRIPTION
AMERGE Tablets contain naratriptan as the hydrochloride, which is a selective 5-hydroxytryptamine1 receptor subtype agonist. Naratriptan hydrochloride is chemically designated as N-methyl-3-(1-methyl-4-piperidinyl)-1H-indole-5-ethanesulfonamide monohydrochloride, and it has the following structure:
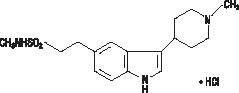
The empirical formula is C17H25N3O2S•HCl, representing a molecular weight of 371.93. Naratriptan hydrochloride is a white to pale yellow powder that is readily soluble in water. Each AMERGE Tablet for oral administration contains 1.11 or 2.78 mg of naratriptan hydrochloride equivalent to 1 or 2.5 mg of naratriptan, respectively. Each tablet also contains the inactive ingredients croscarmellose sodium; hypromellose; lactose; magnesium stearate; microcrystalline cellulose; triacetin; and titanium dioxide, iron oxide yellow (2.5-mg tablet only), and indigo carmine aluminum lake (FD&C Blue No. 2) (2.5-mg tablet only) for coloring.
CLINICAL PHARMACOLOGY
Mechanism of Action
Naratriptan binds with high affinity to 5-HT1D and 5-HT1B receptors and has no significant affinity or pharmacological activity at 5-HT2-4 receptor subtypes or at adrenergic α1, α2, or β; dopaminergic D1 or D2; muscarinic; or benzodiazepine receptors.
The therapeutic activity of naratriptan in migraine is generally attributed to its agonist activity at 5-HT1D/1B receptors. Two current theories have been proposed to explain the efficacy of 5-HT1D/1B receptor agonists in migraine. One theory suggests that activation of 5-HT1D/1B receptors located on intracranial blood vessels, including those on the arteriovenous anastomoses, leads to vasoconstriction, which is correlated with the relief of migraine headache. The other hypothesis suggests that activation of 5-HT1D/1B receptors on sensory nerve endings in the trigeminal system results in the inhibition of pro-inflammatory neuropeptide release.
In the anesthetized dog, naratriptan has been shown to reduce the carotid arterial blood flow with little or no effect on arterial blood pressure or total peripheral resistance. While the effect on blood flow was selective for the carotid arterial bed, increases in vascular resistance of up to 30% were seen in the coronary arterial bed. Naratriptan has also been shown to inhibit trigeminal nerve activity in rat and cat. In 10 human subjects with suspected coronary artery disease (CAD) undergoing coronary artery catheterization, there was a 1% to 10% reduction in coronary artery diameter following subcutaneous injection of 1.5 mg of naratriptan.
Pharmacokinetics
Naratriptan tablets are well absorbed, with about 70% oral bioavailability. Following administration of a 2.5-mg tablet orally, the peak concentrations are obtained in 2 to 3 hours. After administration of 1- or 2.5-mg tablets, the Cmax is somewhat (about 50%) higher in women (not corrected for milligram-per-kilogram dose) than in men. During a migraine attack, absorption was slower, with a Tmax of 3 to 4 hours. Food does not affect the pharmacokinetics of naratriptan. Naratriptan displays linear kinetics over the therapeutic dose range.
The steady-state volume of distribution of naratriptan is 170 L. Plasma protein binding is 28% to 31% over the concentration range of 50 to 1,000 ng/mL.
Naratriptan is predominantly eliminated in urine, with 50% of the dose recovered unchanged and 30% as metabolites in urine. In vitro, naratriptan is metabolized by a wide range of cytochrome P450 isoenzymes into a number of inactive metabolites.
The mean elimination half-life of naratriptan is 6 hours. The systemic clearance of naratriptan is 6.6 mL/min/kg. The renal clearance (220 mL/min) exceeds glomerular filtration rate, indicating active tubular secretion. Repeat administration of naratriptan tablets does not result in drug accumulation.
Special Populations
Age
A small decrease in clearance (approximately 26%) was observed in healthy elderly subjects (65 to 77 years) compared to younger patients, resulting in slightly higher exposure (see PRECAUTIONS).
Race
The effect of race on the pharmacokinetics of naratriptan has not been examined.
Renal Impairment
Clearance of naratriptan was reduced by 50% in patients with moderate renal impairment (creatinine clearance, 18 to 39 mL/min) compared to the normal group. Decrease in clearances resulted in an increase of mean half-life from 6 hours (healthy) to 11 hours (range, 7 to 20 hours). The mean Cmax increased by approximately 40%. The effects of severe renal impairment (creatinine clearance, ≤15 mL/min) on the pharmacokinetics of naratriptan has not been assessed (see CONTRAINDICATIONS and DOSAGE AND ADMINISTRATION).
Hepatic Impairment
Clearance of naratriptan was decreased by 30% in patients with moderate hepatic impairment (Child-Pugh grade A or B). This resulted in an approximately 40% increase in the half-life (range, 8 to 16 hours). The effects of severe hepatic impairment (Child-Pugh grade C) on the pharmacokinetics of naratriptan have not been assessed (see CONTRAINDICATIONS and DOSAGE AND ADMINISTRATION).
Drug Interactions
In normal volunteers, coadministration of single doses of naratriptan tablets and alcohol did not result in substantial modification of naratriptan pharmacokinetic parameters.
From population pharmacokinetic analyses, coadministration of naratriptan and fluoxetine, beta-blockers, or tricyclic antidepressants did not affect the clearance of naratriptan.
Naratriptan does not inhibit monoamine oxidase (MAO) enzymes and is a poor inhibitor of P450; metabolic interactions between naratriptan and drugs metabolized by P450 or MAO are therefore unlikely.
Oral Contraceptives
Oral contraceptives reduced clearance by 32% and volume of distribution by 22%, resulting in slightly higher concentrations of naratriptan. Hormone replacement therapy had no effect on pharmacokinetics in older female patients.
Smoking increased the clearance of naratriptan by 30%.
CLINICAL TRIALS
The efficacy of AMERGE Tablets in the acute treatment of migraine headaches was evaluated in 6 randomized, double-blind, placebo-controlled studies of which 4 used the recommended dosing regimen and were conducted as outpatient trials. Three of these studies enrolled adult patients who were predominantly female (86%) and Caucasian (96%) with a mean age of 41 (range, 18 to 65). One study enrolled adolescents with a mean age of 14 (range, 12 to 17). In the adolescent study, 54% of the patients were female and 89% were Caucasian. In all studies, patients were instructed to treat at least 1 moderate to severe headache. Headache response, defined as a reduction in headache severity from moderate or severe pain to mild or no pain, was assessed up to 4 hours after dosing. Associated symptoms such as nausea, vomiting, photophobia, and phonophobia were also assessed. Maintenance of response was assessed for up to 24 hours postdose. A second dose of AMERGE Tablets or other medication was allowed 4 to 24 hours after the initial treatment for recurrent headache. The frequency and time to use of these additional treatments were also determined.
In all 3 trials in adults utilizing the recommended dosage regimen and outpatient use, the percentage of patients achieving headache response 4 hours after treatment, the primary outcome measure, was significantly greater among patients receiving AMERGE compared to those who received placebo. In all studies, response to 2.5 mg was numerically greater than response to 1 mg and in the largest of the 3 studies, there was a statistically significant greater percentage of patients with headache response at 4 hours in the 2.5-mg group compared to the 1-mg group. The results are summarized in Table 1.
| Placebo |
AMERGE 1.0 mg |
AMERGE 2.5 mg |
|
| Study 1 | 34% (n = 122) | 50%* (n = 117) | 60%* (n = 127) |
| Study 2 | 27% (n = 104) | 52%* (n = 208) | 66%*† (n = 199) |
| Study 3 | 32% (n = 169) | 54%* (n = 166) | 65%* (n = 167) |
*p<0.05 in comparison with placebo.
†p<0.05 in comparison with 1 mg.
In the single study in adolescents, there were no statistically significant differences between any of the treatment groups. The headache response rates at 4 hours (n) were 65% (n = 74), 67% (n = 78), and 64% (n = 70) for placebo, 1-mg, and 2.5-mg groups, respectively.
Comparisons of drug performance based upon results obtained in different clinical trials are never reliable. Because studies are conducted at different times, with different samples of patients, by different investigators, employing different criteria and/or different interpretations of the same criteria, under different conditions (dose, dosing regimen, etc.), quantitative estimates of treatment response and the timing of response may be expected to vary considerably from study to study.
The estimated probability of achieving an initial headache response in adults over the 4 hours following treatment is depicted in Figure 1.
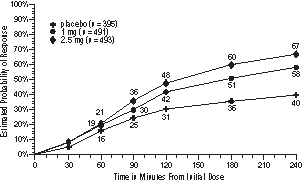
Figure 1. Estimated Probability of Achieving Initial Headache Response Within 4 Hours*
*The figure shows the probability over time of obtaining headache response (no or mild pain) following treatment with AMERGE Tablets. The averages displayed are based on pooled data from the 3 controlled clinical trials providing evidence of efficacy (Studies 1, 2, and 3). In this Kaplan-Meier plot, patients not achieving response within 240 minutes were censored at 240 minutes.
For patients with migraine-associated nausea, photophobia, and phonophobia at baseline, there was a lower incidence of these symptoms 4 hours following administration of 1- and 2.5-mg AMERGE Tablets compared to placebo.
Four to 24 hours following the initial dose of study treatment, patients were allowed to use additional treatment for pain relief in the form of a second dose of study treatment or other medication. The estimated probability of patients taking a second dose or other medication for migraine over the 24 hours following the initial dose of study treatment is summarized in Figure 2.
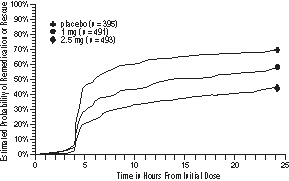
Figure 2. Estimated Probability of Patients Taking a Second Dose of AMERGE Tablets or Other Medication for Migraine Over the 24 Hours Following the Initial Dose of Study Treatment*
*Kaplan-Meier plot based on data obtained in the 3 controlled clinical trials (Studies 1, 2, and 3) providing evidence of efficacy with patients not using additional treatments censored at 24 hours. The plot also includes patients who had no response to the initial dose. Remedication was discouraged prior to 4 hours postdose.
There is no evidence that doses of 5 mg provide a greater effect than 2.5 mg. There was no evidence to suggest that treatment with AMERGE was associated with an increase in the severity or frequency of migraine attacks. The efficacy of AMERGE Tablets was unaffected by presence of aura; gender, age, or weight of the patient; oral contraceptive use; or concomitant use of common migraine prophylactic drugs (e.g., beta-blockers, calcium channel blockers, tricyclic antidepressants). There was insufficient data to assess the impact of race on efficacy.
INDICATIONS AND USAGE
AMERGE Tablets are indicated for the acute treatment of migraine attacks with or without aura in adults.
AMERGE Tablets are not intended for the prophylactic therapy of migraine or for use in the management of hemiplegic or basilar migraine (see CONTRAINDICATIONS). Safety and effectiveness of AMERGE Tablets have not been established for cluster headache, which is present in an older, predominantly male population.
CONTRAINDICATIONS
AMERGE Tablets should not be given to patients with history, symptoms, or signs of ischemic cardiac, cerebrovascular, or peripheral vascular syndromes. In addition, patients with other significant underlying cardiovascular diseases should not receive AMERGE Tablets. Ischemic cardiac syndromes include, but are not limited to, angina pectoris of any type (e.g., stable angina of effort and vasospastic forms of angina such as the Prinzmetal variant), all forms of myocardial infarction, and silent myocardial ischemia. Cerebrovascular syndromes include, but are not limited to, strokes of any type as well as transient ischemic attacks. Peripheral vascular disease includes, but is not limited to, ischemic bowel disease (see WARNINGS).
Because AMERGE Tablets may increase blood pressure, they should not be given to patients with uncontrolled hypertension (see WARNINGS).
AMERGE Tablets are contraindicated in patients with severe renal impairment (creatinine clearance, <15 mL/min) (see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
AMERGE Tablets are contraindicated in patients with severe hepatic impairment (Child-Pugh grade C) (see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
AMERGE Tablets should not be administered to patients with hemiplegic or basilar migraine.
AMERGE Tablets should not be used within 24 hours of treatment with another 5-HT1 agonist, an ergotamine-containing or ergot-type medication like dihydroergotamine or methysergide.
AMERGE Tablets are contraindicated in patients with hypersensitivity to naratriptan or any of the components.
WARNINGS
AMERGE Tablets should only be used where a clear diagnosis of migraine has been established.
Risk of Myocardial Ischemia and/or Infarction and Other Adverse Cardiac Events
Because of the potential of this class of compounds (5-HT1B/1D agonists) to cause coronary vasospasm, naratriptan should not be given to patients with documented ischemic or vasospastic coronary artery disease (CAD) (see CONTRAINDICATIONS). It is strongly recommended that 5-HT1 agonists (including naratriptan) not be given to patients in whom unrecognized CAD is predicted by the presence of risk factors (e.g., hypertension, hypercholesterolemia, smoker, obesity, diabetes, strong family history of CAD, female with surgical or physiological menopause, or male over 40 years of age) unless a cardiovascular evaluation provides satisfactory clinical evidence that the patient is reasonably free of coronary artery and ischemic myocardial disease or other significant underlying cardiovascular disease. The sensitivity of cardiac diagnostic procedures to detect cardiovascular disease or predisposition to coronary artery vasospasm is modest, at best. If, during the cardiovascular evaluation, the patient’s medical history, electrocardiographic, or other investigations reveal findings indicative of, or consistent with, coronary artery vasospasm or myocardial ischemia, naratriptan should not be administered (see CONTRAINDICATIONS).
For patients with risk factors predictive of CAD, who are determined to have a satisfactory cardiovascular evaluation, it is strongly recommended that administration of the first dose of naratriptan take place in the setting of a physician’s office or similar medically staffed and equipped facility. Because cardiac ischemia can occur in the absence of clinical symptoms, consideration should be given to obtaining on the first occasion of use an electrocardiogram (ECG) during the interval immediately following administration of AMERGE Tablets, in these patients with risk factors.
It is recommended that patients who are intermittent long-term users of 5-HT1 agonists, including AMERGE Tablets, and who have or acquire risk factors predictive of CAD, as described above, undergo periodic cardiovascular evaluation as they continue to use AMERGE Tablets.
The systematic approach described above is intended to reduce the likelihood that patients with unrecognized cardiovascular disease will be inadvertently exposed to naratriptan.
Cardiac Events and Fatalities Associated With 5-HT1 Agonists
Naratriptan can cause coronary artery vasospasm (see CLINICAL PHARMACOLOGY). Serious adverse cardiac events, including acute myocardial infarction, life-threatening disturbances of cardiac rhythm, and death have been reported within a few hours following the administration of 5-HT1 agonists. Considering the extent of use of 5-HT1 agonists in patients with migraine, the incidence of these events is extremely low.
Premarketing Experience With AMERGE Tablets
Among approximately 3,500 patients with migraine who participated in premarketing clinical trials of naratriptan tablets, 4 patients treated with single oral doses of naratriptan ranging from 1 to 10 mg experienced asymptomatic ischemic ECG changes with at least 1, who took 7.5 mg, likely due to coronary vasospasm.
Cerebrovascular Events and Fatalities With 5-HT1 Agonists
Cerebral hemorrhage, subarachnoid hemorrhage, stroke, and other cerebrovascular events have been reported in patients treated with 5-HT1 agonists, and some have resulted in fatalities. In a number of cases, it appears possible that the cerebrovascular events were primary, the agonist having been administered in the incorrect belief that the symptoms experienced were a consequence of migraine, when they were not. It should be noted that patients with migraine may be at increased risk of certain cerebrovascular events (e.g., stroke, hemorrhage, transient ischemic attack).
Other Vasospasm-Related Events
5-HT1 agonists may cause vasospastic reactions other than coronary artery spasm. Both peripheral vascular ischemia and colonic ischemia with abdominal pain and bloody diarrhea have been reported with naratriptan.
Serotonin Syndrome
The development of a potentially life-threatening serotonin syndrome may occur with triptans, including treatment with AMERGE, particularly during combined use with selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs). If concomitant treatment with naratriptan and an SSRI (e.g., fluoxetine, paroxetine, sertraline, fluvoxamine, citalopram, escitalopram) or SNRI (e.g., venlafaxine, duloxetine) is clinically warranted, careful observation of the patient is advised, particularly during treatment initiation and dose increases. Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g., hyperreflexia, incoordination), and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea).
Increase in Blood Pressure
In healthy volunteers, dose-related increases in systemic blood pressure have been observed after administration of up to 20 mg of oral naratriptan. At the recommended doses, the elevations are generally small, although an increase of systolic pressure of 32 mmHg was seen in 1 patient following a single 2.5-mg dose. The effect may be more pronounced in the elderly and hypertensive patients. A patient who was mildly hypertensive (the baseline blood pressure was 150/98) experienced a significant increase in blood pressure to 204/144 mmHg 225 minutes after administration of a 10-mg oral dose. Significant elevation in blood pressure, including hypertensive crisis, has been reported on rare occasions in patients receiving 5-HT1 agonists with and without a history of hypertension. Naratriptan is contraindicated in patients with uncontrolled hypertension (see CONTRAINDICATIONS).
An 18% increase in mean pulmonary artery pressure and an 8% increase in mean aortic pressure was seen following dosing with 1.5 mg of subcutaneous naratriptan in a study evaluating 10 subjects with suspected CAD undergoing cardiac catheterization.
Hypersensitivity
Hypersensitivity (anaphylaxis/anaphylactoid) reactions may occur in patients receiving naratriptan. Such reactions can be life threatening or fatal. In general, hypersensitivity reactions to drugs are more likely to occur in individuals with a history of sensitivity to multiple allergens (see CONTRAINDICATIONS).
PRECAUTIONS
General
Chest discomfort (including pain, pressure, heaviness, tightness) has been reported after administration of 5-HT1 agonists, including AMERGE Tablets. These events have not been associated with arrhythmias or ischemic ECG changes in clinical trials with AMERGE Tablets. Because naratriptan may cause coronary artery vasospasm, patients who experience signs or symptoms suggestive of angina following naratriptan should be evaluated for the presence of CAD or a predisposition to Prinzmetal variant angina before receiving additional doses of naratriptan, and should be monitored electrocardiographically if dosing is resumed and similar symptoms recur. Similarly, patients who experience other symptoms or signs suggestive of decreased arterial flow, such as ischemic bowel syndrome or Raynaud syndrome following naratriptan administration should be evaluated for atherosclerosis or predisposition to vasospasm (see CONTRAINDICATIONS and WARNINGS).
AMERGE Tablets should also be administered with caution to patients with diseases that may alter the absorption, metabolism, or excretion of drugs, such as impaired renal or hepatic function (see CLINICAL PHARMACOLOGY, CONTRAINDICATIONS, and DOSAGE AND ADMINISTRATION).
Care should be taken to exclude other potentially serious neurological conditions before treating headache in patients not previously diagnosed with migraine or who experience a headache that is atypical for them. There have been rare reports where patients received 5-HT1 agonists for severe headaches that were subsequently shown to have been secondary to an evolving neurologic lesion (see WARNINGS).
For a given attack, if a patient has no response to the first dose of AMERGE, the diagnosis of migraine should be reconsidered before administration of a second dose.
Overuse of acute migraine treatments has been associated with the exacerbation of headache (medication overuse headache) in susceptible patients. Withdrawal of the treatment may be necessary.
Binding to Melanin-Containing Tissues
In rats treated with a single oral dose (10 mg/kg) of radiolabeled naratriptan, the elimination half-life of radioactivity from the eye was 90 days, suggesting that naratriptan and/or its metabolites may bind to the melanin of the eye. Because there could be accumulation in melanin-rich tissues over time, this raises the possibility that naratriptan could cause toxicity in these tissues after extended use. Although no systematic monitoring of ophthalmologic function was undertaken in clinical trials, and no specific recommendations for ophthalmologic monitoring are offered, prescribers should be aware of the possibility of long-term ophthalmologic effects.
Changes in the Precorneal Tear Film
Dogs receiving oral naratriptan showed transient changes in the precorneal tear film. Corneal stippling was seen at the lowest dose tested, 1 mg/kg/day, and occurred intermittently from day 1 throughout the first 2 to 3 weeks of treatment. Although a no-effect dose was not established, the exposure at the lowest dose tested was approximately 5 times the human exposure after a 5-mg oral dose.
Information for Patients
See PATIENT INFORMATION at the end of this labeling for the text of the separate leaflet provided for patients.
Patients should be cautioned about the risk of serotonin syndrome with the use of naratriptan or other triptans, especially during combined use with SSRIs or SNRIs.
Laboratory Tests
No specific laboratory tests are recommended for monitoring patients prior to and/or after treatment with AMERGE Tablets.
Drug Interactions
Selective Serotonin Reuptake Inhibitors/Serotonin Norepinephrine Reuptake Inhibitors and Serotonin Syndrome
Cases of life-threatening serotonin syndrome have been reported during combined use of SSRIs or SNRIs and triptans (see WARNINGS).
Ergot-Containing Drugs
Ergot-containing drugs have been reported to cause prolonged vasospastic reactions. Because there is a theoretical basis that these effects may be additive, use of ergotamine-containing or ergot-type medications (like dihydroergotamine or methysergide) and naratriptan within 24 hours is contraindicated (see CONTRAINDICATIONS).
Other 5-HT1 Agonists
The administration of naratriptan with other 5-HT1 agonists has not been evaluated in migraine patients. Because their vasospastic effects may be additive, coadministration of naratriptan and other 5-HT1 agonists within 24 hours of each other is not recommended (see CONTRAINDICATIONS).
Drug/Laboratory Test Interactions
AMERGE Tablets are not known to interfere with commonly employed clinical laboratory tests.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Lifetime carcinogenicity studies, 104 weeks in duration, were carried out in mice and rats by oral gavage. There was no evidence of an increase in tumors related to naratriptan administration in mice receiving up to 200 mg/kg/day. That dose was associated with a plasma area-under-the-curve (AUC) exposure that was 110 times the exposure in humans receiving the maximum recommended daily dose of 5 mg. Two rat studies were conducted, 1 using a standard diet and the other a nitrite-supplemented diet (naratriptan can be nitrosated in vitro to form a mutagenic product that has been detected in the stomachs of rats fed a high nitrite diet). Doses of 5, 20, and 90 mg/kg were associated with week 13 AUC exposures that in the standard diet study were 7, 40, and 236 times, respectively, and in the nitrite-supplemented diet study were 7, 29, and 180 times, respectively, the exposure attained in humans given the maximum recommended daily dose of 5 mg. In both studies, there was an increase in the incidence of thyroid follicular hyperplasia in high-dose males and females and in thyroid follicular adenomas in high-dose males. In the standard diet study only, there was also an increase in the incidence of benign c-cell adenomas in the thyroid of high-dose males and females. The exposures achieved at the no-effect dose for thyroid tumors were 40 (standard diet) and 29 (nitrite-supplemented diet) times the exposure achieved in humans receiving the maximum recommended daily dose of 5 mg. In the nitrite-supplemented diet study only, the incidence of benign lymphocytic thymoma was increased in all treated groups of females. It was not determined if the nitrosated product is systemically absorbed. However, no changes were seen in the stomachs of rats in that study.
Mutagenesis
Naratriptan was not mutagenic when tested in 2 gene mutation assays, the Ames test and the in vitro thymidine locus mouse lymphoma assay. It was not clastogenic in 2 cytogenetics assays, the in vitro human lymphocyte assay and the in vivo mouse micronucleus assay. Naratriptan can be nitrosated in vitro to form a mutagenic product (WHO nitrosation assay) that has been detected in the stomachs of rats fed a nitrite-supplemented diet.
Impairment of Fertility
In a reproductive toxicity study in which male and female rats were dosed prior to and throughout the mating period with 10, 60, 170, or 340 mg/kg/day (plasma exposures [AUC] approximately 11, 70, 230, and 470 times, respectively, the human exposure at the maximum recommended daily dose [MRDD] of 5 mg), there was a treatment-related decrease in the number of females exhibiting normal estrous cycles at doses of 170 mg/kg/day or greater and an increase in preimplantation loss at 60 mg/kg/day or greater. In high-dose group males, testicular/epididymal atrophy accompanied by spermatozoa depletion reduced mating success and may have contributed to the observed preimplantation loss. The exposures achieved at the no-effect doses for preimplantation loss, anestrus, and testicular effects were approximately 11, 70, and 230 times, respectively, the exposures in humans receiving the MRDD.
In a study in which rats were dosed orally with 10, 60, or 340 mg/kg/day for 6 months, changes in the female reproductive tract including atrophic or cystic ovaries and anestrus were seen at the high dose. The exposure at the no-effect dose of 60 mg/kg was approximately 85 times the exposure in humans receiving the MRDD.
Pregnancy
Pregnancy Category C. There are no adequate and well-controlled studies in pregnant women; therefore, naratriptan should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
To monitor fetal outcomes of pregnant women exposed to AMERGE, GlaxoSmithKline maintains a Naratriptan Pregnancy Registry. Healthcare providers are encouraged to register patients by calling (800) 336-2176.
In reproductive toxicity studies in rats and rabbits, oral administration of naratriptan was associated with developmental toxicity (embryolethality, fetal abnormalities, pup mortality, offspring growth retardation) at doses producing maternal plasma drug exposures as low as 11 and 2.5 times, respectively, the exposure in humans receiving the MRDD of 5 mg.
When pregnant rats were administered naratriptan during the period of organogenesis at doses of 10, 60, or 340 mg/kg/day, there was a dose-related increase in embryonic death, with a statistically significant difference at the highest dose, and incidences of fetal structural variations (incomplete/irregular ossification of skull bones, sternebrae, ribs) were increased at all doses. The maternal plasma exposures (AUC) at these doses were approximately 11, 70, and 470 times the exposure in humans at the MRDD. The high dose was maternally toxic, as evidenced by decreased maternal body weight gain during gestation. A no-effect dose for developmental toxicity in rats exposed during organogenesis was not established.
When doses of 1, 5, or 30 mg/kg/day were given to pregnant Dutch rabbits throughout organogenesis, the incidence of a specific fetal skeletal malformation (fused sternebrae) was increased at the high dose, and increased incidences of embryonic death and fetal variations (major blood vessel variations, supernumerary ribs, incomplete skeletal ossification) were observed at all doses (4, 20, and 120 times, respectively, the MRDD on a body surface area basis). Maternal toxicity (decreased body weight gain) was evident at the high dose in this study. In a similar study in New Zealand White rabbits (1, 5, or 30 mg/kg/day throughout organogenesis), decreased fetal weights and increased incidences of fetal skeletal variations were observed at all doses (maternal exposures equivalent to 2.5, 19, and 140 times exposure in humans receiving the MRDD), while maternal body weight gain was reduced at 5 mg/kg or greater. A no-effect dose for developmental toxicity in rabbits exposed during organogenesis was not established.
When female rats were treated with 10, 60, or 340 mg/kg/day during late gestation and lactation, offspring behavioral impairment (tremors) and decreased offspring viability and growth were observed at doses of 60 mg/kg or greater, while maternal toxicity occurred only at the highest dose. Maternal exposures at the no-effect dose for developmental effects in this study were approximately 11 times the exposure in humans receiving the MRDD.
Nursing Mothers
Naratriptan-related material is excreted in the milk of rats. Therefore, caution should be exercised when considering the administration of AMERGE Tablets to a nursing woman.
Pediatric Use
Safety and effectiveness of AMERGE Tablets in pediatric patients (younger than 18 years) have not been established.
One randomized, placebo-controlled clinical trial evaluating oral naratriptan (0.25 to 2.5 mg) in pediatric patients aged 12 to 17 years evaluated a total of 300 adolescent migraineurs. This study did not establish the efficacy of oral naratriptan compared to placebo in the treatment of migraine in adolescents (see CLINICAL TRIALS). Adverse events observed in this clinical trial were similar in nature to those reported in clinical trials in adults.
Geriatric Use
The use of AMERGE Tablets in elderly patients is not recommended.
Naratriptan is known to be substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in elderly patients who have reduced renal function. In addition, elderly patients are more likely to have decreased hepatic function; they are at higher risk for CAD; and blood pressure increases may be more pronounced in the elderly. Clinical studies of AMERGE Tablets did not include patients over 65 years of age.
ADVERSE REACTIONS
Serious cardiac events, including some that have been fatal, have occurred following the use of 5-HT1 agonists. These events are extremely rare and most have been reported in patients with risk factors predictive of CAD. Events reported have included coronary artery vasospasm, transient myocardial ischemia, myocardial infarction, ventricular tachycardia, and ventricular fibrillation (see CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS).
Incidence in Controlled Clinical Trials
The most common adverse events were paresthesias, dizziness, drowsiness, malaise/fatigue, and throat/neck symptoms, which occurred at a rate of 2% and at least 2 times placebo rate. Since patients treated only 1 to 3 headaches in the controlled clinical trials, the opportunity for discontinuation of therapy in response to an adverse event was limited. In a long-term, open-label study where patients were allowed to treat multiple migraine attacks for up to 1 year, 15 patients (3.6%) discontinued treatment due to adverse events.
Table 2 lists adverse events that occurred in 5 placebo-controlled clinical trials of approximately 1,752 exposures to placebo and AMERGE Tablets in adult migraine patients. The events cited reflect experience gained under closely monitored conditions of clinical trials in a highly selected patient population. In actual clinical practice or in other clinical trials, these frequency estimates may not apply, as the conditions of use, reporting behavior, and the kinds of patients treated may differ. Only events that occurred at a frequency of 2% or more in the group treated with AMERGE Tablets 2.5 mg and were more frequent in that group than in the placebo group are included in Table 2. From this table, it appears that many of these adverse events are dose related.
| Adverse Event Type |
Placebo (n = 498) |
AMERGE 1 mg (n = 627) |
AMERGE 2.5 mg (n = 627) |
| Atypical sensation | 1% | 2% | 4% |
| Paresthesias (all types) | <1% | 1% | 2% |
| Gastrointestinal | 5% | 6% | 7% |
| Nausea | 4% | 4% | 5% |
| Neurological | 3% | 4% | 7% |
| Dizziness | 1% | 1% | 2% |
| Drowsiness | <1% | 1% | 2% |
| Malaise/fatigue | 1% | 2% | 2% |
| Pain and pressure sensation | 2% | 2% | 4% |
| Throat/neck symptoms | 1% | 1% | 2% |
One event (vomiting) present in more than 1% of patients receiving AMERGE Tablets occurred more frequently on placebo than on naratriptan 2.5 mg.
AMERGE Tablets are generally well tolerated. Most adverse reactions were mild and transient.
The incidence of adverse events in placebo-controlled clinical trials was not affected by age or weight of the patients, duration of headache prior to treatment, presence of aura, use of prophylactic medications, or tobacco use. There was insufficient data to assess the impact of race on the incidence of adverse events.
Other Events Observed in Association With the Administration of AMERGE Tablets
In the paragraphs that follow, the frequencies of less commonly reported adverse clinical events are presented. Because the reports include events observed in open and uncontrolled studies, the role of AMERGE Tablets in their causation cannot be reliably determined. Furthermore, variability associated with adverse event reporting, the terminology used to describe adverse events, etc., limit the value of the quantitative frequency estimates provided. Event frequencies are calculated as the number of patients reporting an event divided by the total number of patients (n = 3,557) exposed to oral naratriptan doses up to 10 mg. All reported events are included except those already listed in the previous table, those too general to be informative, and those not reasonably associated with the use of the drug. Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are those occurring in at least 1/100 patients, infrequent adverse events are those occurring in 1/100 to 1/1,000 patients, and rare adverse events are those occurring in fewer than 1/1,000 patients.
Atypical Sensations
Frequent were warm/cold temperature sensations. Infrequent were feeling strange and burning/stinging sensation.
Cardiovascular
Infrequent were palpitations, increased blood pressure, tachyarrhythmias, and abnormal ECG (PR prolongation, QTc prolongation, ST/T wave abnormalities, premature ventricular contractions, atrial flutter, or atrial fibrillation), and syncope. Rare were bradycardia, varicosities, hypotension, and heart murmurs.
Ear, Nose, and Throat
Frequent were ear, nose, and throat infections. Infrequent were phonophobia, sinusitis, upper respiratory inflammation, and tinnitus. Rare were allergic rhinitis; labyrinthitis; ear, nose, and throat hemorrhage; and hearing difficulty.
Endocrine and Metabolic
Infrequent were thirst and polydipsia, dehydration, and fluid retention. Rare were hyperlipidemia, hypercholesterolemia, hypothyroidism, hyperglycemia, glycosuria and ketonuria, and parathyroid neoplasm.
Eye
Frequent was photophobia. Infrequent was blurred vision. Rare were eye pain and discomfort, sensation of eye pressure, eye hemorrhage, dry eyes, difficulty focusing, and scotoma.
Gastrointestinal
Frequent were hyposalivation and vomiting. Infrequent were dyspeptic symptoms, diarrhea, gastrointestinal discomfort and pain, gastroenteritis, and constipation. Rare were abnormal liver function tests, abnormal bilirubin levels, hemorrhoids, gastritis, esophagitis, salivary gland inflammation, oral itching and irritation, regurgitation and reflux, and gastric ulcers.
Hematological Disorders
Infrequent was increased white cells. Rare were thrombocytopenia, quantitative red cell or hemoglobin defects, anemia, and purpura.
Lower Respiratory Tract
Infrequent were bronchitis, cough, and pneumonia. Rare were tracheitis, asthma, pleuritis, and airway constriction and obstruction.
Musculoskeletal
Infrequent were muscle pain, arthralgia and articular rheumatism, muscle cramps and spasms, joint and muscle stiffness, tightness, and rigidity. Rare were bone and skeletal pain.
Neurological
Frequent was vertigo. Infrequent were tremors, cognitive function disorders, sleep disorders, and disorders of equilibrium. Rare were compressed nerve syndromes, confusion, sedation, hyperesthesia, coordination disorders, paralysis of cranial nerves, decreased consciousness, dreams, altered sense of taste, neuralgia, neuritis, aphasia, hypoesthesia, motor retardation, muscle twitching and fasciculation, psychomotor restlessness, and convulsions.
Non-Site Specific
Infrequent were chills and/or fever, descriptions of odor or taste, edema and swelling, allergies, and allergic reactions. Rare were spasms and mobility disorders.
Pain and Pressure Sensations
Frequent were pressure/tightness/heaviness sensations.
Psychiatry
Infrequent were anxiety, depressive disorders, and detachment. Rare were aggression and hostility, agitation, hallucinations, panic, and hyperactivity.
Reproduction
Rare were lumps of female reproductive tract, breast inflammation, inflammation of vagina, inflammation of fallopian tube, breast discharge, endometrium disorders, decreased libido, and lumps of breast.
Skin
Infrequent were sweating, skin rashes, pruritus, and urticaria. Rare were skin erythema, dermatitis and dermatosis, hair loss and alopecia, pruritic skin rashes, acne and folliculitis, allergic skin reactions, macular skin/rashes, skin photosensitivity, photodermatitis, skin flakiness, and dry skin.
Urology
Infrequent were bladder inflammation and polyuria and diuresis. Rare were urinary tract hemorrhage, urinary urgency, pyelitis, and urinary incontinence.
Observed During Clinical Practice
The following section enumerates potentially important adverse events that have occurred in clinical practice and that have been reported spontaneously to various surveillance systems. The events enumerated represent reports arising from both domestic and nondomestic use of naratriptan. These events do not include those already listed in the ADVERSE REACTIONS section above. Because the reports cite events reported spontaneously from worldwide postmarketing experience, frequency of events and the role of naratriptan in their causation cannot be reliably determined.
Cardiovascular
Angina, myocardial infarction (see WARNINGS).
Gastrointestinal
Colonic ischemia (see WARNINGS).
Lower Respiratory
Dyspnea.
Miscellaneous
Hypersensitivity, including anaphylaxis/anaphylactoid reactions, in some cases severe (e.g., circulatory collapse) (see WARNINGS).
Neurologic
Cerebral vascular accident, including transient ischemic attack, subarachnoid hemorrhage, and cerebral infarction (see WARNINGS); serotonin syndrome.
DRUG ABUSE AND DEPENDENCE
In one clinical study enrolling 12 subjects, all of whom had experience using oral opiates and other psychoactive drugs, AMERGE Tablets produced less intense subjective responses ordinarily associated with many drugs of abuse than did codeine (30 to 90 mg).
OVERDOSAGE
A patient who was mildly hypertensive experienced a significant increase in blood pressure after administration of a 10-mg dose starting at 30 minutes (baseline value of 150/98 to 204/144 mmHg 225 minutes). This event resolved after treatment with antihypertensive therapy. Oral administration of 25 mg of naratriptan in 1 healthy young male subject increased blood pressure from 120/67 mmHg pretreatment up to 191/113 mmHg at approximately 6 hours postdose and resulted in adverse events including lightheadedness, tension in the neck, tiredness, and loss of coordination. Blood pressure returned to near baseline by 8 hours after dosing without any pharmacological intervention.
Another subject experienced asymptomatic ischemic ECG changes likely due to coronary artery vasospasm approximately 2 hours following a 7.5-mg oral dose.
The elimination half-life of naratriptan is about 6 hours (see CLINICAL PHARMACOLOGY), and therefore monitoring of patients after overdose with AMERGE Tablets should continue for at least 24 hours or while symptoms or signs persist. There is no specific antidote to naratriptan. Standard supportive treatment should be applied as required. If the patient presents with chest pain or other symptoms consistent with angina pectoris, ECG monitoring should be performed for evidence of ischemia. It is unknown what effect hemodialysis or peritoneal dialysis has on the serum concentrations of naratriptan.
DOSAGE AND ADMINISTRATION
In controlled clinical trials, single doses of 1 and 2.5 mg of AMERGE Tablets taken with fluid were effective for the acute treatment of migraines in adults. A greater proportion of patients had headache response following a 2.5-mg dose than following a 1-mg dose (see CLINICAL TRIALS). Individuals may vary in response to doses of AMERGE Tablets. The choice of dose should therefore be made on an individual basis, weighing the possible benefit of the 2.5-mg dose with the potential for a greater risk of adverse events. If the headache returns or if the patient has only partial response, the dose may be repeated once after 4 hours, for a maximum dose of 5 mg in a 24-hour period. There is evidence that doses of 5 mg do not provide a greater effect than 2.5 mg.
The safety of treating, on average, more than 4 headaches in a 30-day period has not been established.
Renal Impairment
The use of AMERGE is contraindicated in patients with severe renal impairment (creatinine clearance, <15 mL/min) because of decreased clearance of the drug (see CONTRAINDICATIONS and CLINICAL PHARMACOLOGY). In patients with mild to moderate renal impairment, the maximum daily dose should not exceed 2.5 mg over a 24-hour period and a lower starting dose should be considered.
Hepatic Impairment
The use of AMERGE is contraindicated in patients with severe hepatic impairment (Child-Pugh grade C) because of decreased clearance (see CONTRAINDICATIONS and CLINICAL PHARMACOLOGY). In patients with mild or moderate hepatic impairment, the maximum daily dose should not exceed 2.5 mg over a 24-hour period and a lower starting dose should be considered (see CLINICAL PHARMACOLOGY).
HOW SUPPLIED
AMERGE Tablets 1 and 2.5 mg of naratriptan (base) as the hydrochloride. AMERGE Tablets, 1 mg, are white, D-shaped, film-coated tablets debossed with “GX CE3” on one side in blister packs of 9 tablets (NDC 0173-0561-00). AMERGE Tablets, 2.5 mg, are green, D-shaped, film-coated tablets debossed with “GX CE5” on one side in blister packs of 9 tablets (NDC 0173-0562-00).
Store at controlled room temperature, 20° to 25°C (68° to 77°F) (see USP).
PATIENT INFORMATION
The following wording is contained in a separate leaflet provided for patients.
Information for the Patient
AMERGE® (naratriptan hydrochloride) Tablets
Read this leaflet carefully before you start to take AMERGE Tablets. Keep the leaflet for reference because it gives you a summary of important information about AMERGE Tablets.
Read the leaflet that comes with each refill of your prescription because there may be new information.
This leaflet does not have all the information about AMERGE Tablets. Ask your healthcare provider for more information or advice.
What are AMERGE Tablets?
AMERGE is a kind of medicine called a triptan. You should take it only if you have a prescription.
AMERGE is used to relieve your migraine. It is not used to prevent attacks or reduce the number of attacks you have. Use AMERGE only to treat an actual migraine attack.
The decision to use AMERGE Tablets is one that you and your healthcare provider should make together, based on your personal needs and health.
Talk with your healthcare provider before taking AMERGE Tablets
-
Risk factors for heart disease to tell your healthcare provider:
Tell your healthcare provider if you have risk factors for heart disease such as:- high blood pressure
- high cholesterol
- being overweight
- diabetes
- smoking
- strong family history of heart disease
- you are postmenopausal
- you are a male over 40 years of age
If you do have risk factors for heart disease, your healthcare provider should check you for heart disease to see if AMERGE is right for you.
Most of the people who have taken AMERGE Tablets have not had any serious side effects. Rarely, deaths and/or serious heart problems have been reported with this kind of medicine. Usually, these deaths and/or serious heart problems happened in people with heart disease. It was not clear whether the medicine had anything to do with these deaths and/or serious heart problems.
-
Important questions to ask yourself before you take AMERGE Tablets:
If the answer to any of the following questions is YES or if you do not know the answer, then please talk with your healthcare provider before you take AMERGE Tablets.- Are you pregnant? Do you think you might be pregnant? Are you trying to become pregnant? Are you not using adequate contraception? Are you breastfeeding?
- Do you have any chest pain, heart disease, shortness of breath, or irregular heartbeats? Have you had a heart attack?
- Do you have risk factors for heart disease (see list above)?
- Have you had a stroke, a mini-stroke (also called a transient ischemic attack or TIA), or Raynaud syndrome?
- Do you have high blood pressure?
- Have you ever had to stop taking this or any other medicine because of an allergy or other problems?
- Are you taking any other migraine medicines, including other triptans such as IMITREX® (sumatriptan/sumatriptan succinate)? Are you taking any medicines containing ergotamine, dihydroergotamine, or methysergide?
- Are you taking any medicine for depression or other health problems such as a selective serotonin reuptake inhibitor (SSRI) or serotonin norepinephrine reuptake inhibitor (SNRI)? Common SSRIs are citalopram HBr (CELEXA®), escitalopram oxalate (LEXAPRO®), paroxetine (PAXIL®), fluoxetine (PROZAC®/SARAFEM®), olanzapine/fluoxetine (SYMBYAX®), sertraline (ZOLOFT®), and fluvoxamine. Common SNRIs are duloxetine (CYMBALTA®) and venlafaxine (EFFEXOR®).
- Have you had, or do you have, any disease of the kidney or liver?
- Is this headache different from your usual migraine attacks?
Remember, if you answered YES to any of the above questions, then talk with your healthcare provider about it.
Important points about AMERGE Tablets
-
The use of AMERGE Tablets during pregnancy:
Do not take AMERGE Tablets if you are pregnant, think you might be pregnant, are trying to become pregnant, or are not using adequate contraception unless you have talked with your healthcare provider about this.
-
How to take AMERGE Tablets:
For adults, the usual dose is a single tablet taken whole with liquids. You can take it at any time after the headache starts. If you need more relief because you only got a partial response or your headache came back after the first tablet, you can take a second tablet 4 hours after the first tablet, but not sooner.
For any attack, if you have no response to the first tablet, do not take a second tablet without first talking with your healthcare provider. Do not take more than a total of 2 AMERGE Tablets in any 24-hour period.
If you have kidney or liver disease, take as directed by your healthcare provider.
-
What to do if you take an overdose:
If you have taken more medicine than has been prescribed for you, contact either your healthcare provider, hospital emergency department, or nearest poison control center right away.
-
How to store your medicine:
Keep your medicine in a safe place where children cannot reach it. It may be harmful to children.
Store your medicine away from heat and light. Do not store at temperatures above 77°F (25°C).
The expiration date of your medicine is printed on the packaging. If your medicine has expired, throw it away.
If your healthcare provider decides to stop your treatment, do not keep any leftover medicine unless your healthcare provider tells you to.
Some possible side effects of AMERGE Tablets
- Some patients feel pain or tightness in the chest or throat when using AMERGE Tablets. If this happens to you, tell your healthcare provider before taking any more AMERGE Tablets. If the chest pain, tightness, or pressure is severe or does not go away, call your healthcare provider right away.
- Call your healthcare provider right away if you have sudden and/or severe abdominal pain after you take AMERGE Tablets.
- Some people may have a reaction called serotonin syndrome when they take certain kinds of medicines for depression called SSRIs or SNRIs while they are taking AMERGE Tablets. Symptoms may include confusion, hallucinations, fast heartbeat, feeling faint, fever, sweating, muscle spasm, difficulty walking, and/or diarrhea. Call your healthcare provider right away if you have any of these symptoms after taking AMERGE Tablets.
- Shortness of breath; wheeziness; heart throbbing; swelling of eyelids, face, or lips; or a skin rash, skin lumps, or hives happens rarely. If it happens to you, then tell your healthcare provider right away. Do not take any more AMERGE Tablets unless your healthcare provider tells you to.
- Some people may feel tingling, heat, flushing (redness of face lasting a short time), heaviness, or pressure after taking AMERGE Tablets. A few people may feel drowsy, dizzy, tired, or sick. If you have any of these symptoms, tell your healthcare provider at your next visit.
- If you feel unwell in any other way or have any symptoms that you do not understand, you should contact your healthcare provider right away.
October 2007 AMG:1PIL
AMERGE, IMITREX, and PAXIL are registered trademarks of GlaxoSmithKline. The other brands listed are trademarks of their respective owners and are not trademarks of GlaxoSmithKline. The makers of these brands are not affiliated with and do not endorse GlaxoSmithKline or its products.
GlaxoSmithKline
Research Triangle Park, NC 27709
©2007, GlaxoSmithKline. All rights reserved.
October 2007 AMG:1PI
Principal Display Panel
NDC 0173-0561-00
Amerge®
(NARATRIPTAN HYDROCHLORIDE)
Tablets
1 mg naratriptan
9 Tablets
Rx only
Each tablet contains 1 mg of naratripan as the hydrochloride.
Store at controlled room temperature, 20o to 25oC (68o to 77oF) (see USP).
Do not use if blisters are torn, broken, or missing.
GlaxoSmithKline
Research Triangle Park, NC 27709
Made in Singapore

| AMERGE
naratriptan hydrochloride tablet, film coated |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020763 | 02/26/1998 | |
| AMERGE
naratriptan hydrochloride tablet, film coated |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020763 | 02/26/1998 | |
| Labeler - SmithKline Beecham Corporation (167380711) |