COPEGUS
-
ribavirin tablet, film coated
Hoffmann-La Roche Inc
----------
COPEGUS®(ribavirin, USP)
TABLETS
COPEGUS (ribavirin) monotherapy is not effective for the treatment of chronic hepatitis C virus infection and should not be used alone for this indication (see WARNINGS).
The primary clinical toxicity of ribavirin is hemolytic anemia. The anemia associated with ribavirin therapy may result in worsening of cardiac disease that has led to fatal and nonfatal myocardial infarctions. Patients with a history of significant or unstable cardiac disease should not be treated with ribavirin (see WARNINGS, ADVERSE REACTIONS, and DOSAGE AND ADMINISTRATION).
Significant teratogenic and/or embryocidal effects have been demonstrated in all animal species exposed to ribavirin. In addition, ribavirin has a multiple dose half-life of 12 days, and it may persist in non-plasma compartments for as long as 6 months. Ribavirin therapy is contraindicated in women who are pregnant and in the male partners of women who are pregnant. Extreme care must be taken to avoid pregnancy during therapy and for 6 months after completion of therapy in both female patients and in female partners of male patients who are taking ribavirin therapy. At least two reliable forms of effective contraception must be utilized during treatment and during the 6-month posttreatment follow-up period (see CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS: Information for Patients, and Pregnancy: Category X).
DESCRIPTION
COPEGUS, the Hoffmann-La Roche brand name for ribavirin, is a nucleoside analogue with antiviral activity. The chemical name of ribavirin is 1-β-D-ribofuranosyl-1H-1,2,4-triazole-3-carboxamide and has the following structural formula:
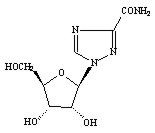
The empirical formula of ribavirin is C8H12N4O5 and the molecular weight is 244.2. Ribavirin is a white to off-white powder. It is freely soluble in water and slightly soluble in anhydrous alcohol.
COPEGUS (ribavirin) is available as a light pink to pink colored, flat, oval-shaped, film-coated tablet for oral administration. Each tablet contains 200 mg of ribavirin and the following inactive ingredients: pregelatinized starch, microcrystalline cellulose, sodium starch glycolate, cornstarch, and magnesium stearate. The coating of the tablet contains Chromatone-P® or Opadry® Pink (made by using hydroxypropyl methyl cellulose, talc, titanium dioxide, synthetic yellow iron oxide, and synthetic red iron oxide), ethyl cellulose (ECD-30), and triacetin.
Mechanism of Action
Ribavirin is a synthetic nucleoside analogue. The mechanism by which the combination of ribavirin and an interferon product exerts its effects against the hepatitis C virus has not been fully established.
CLINICAL PHARMACOLOGY
Pharmacokinetics
Multiple dose ribavirin pharmacokinetic data are available for HCV patients who received ribavirin in combination with peginterferon alfa-2a. Following administration of 1200 mg/day with food for 12 weeks mean±SD (n=39; body weight >75 kg) AUC0-12hr was 25,361±7110 ng∙hr/mL and Cmax was 2748±818 ng/mL. The average time to reach Cmax was 2 hours. Trough ribavirin plasma concentrations following 12 weeks of dosing with food were 1662±545 ng/mL in HCV infected patients who received 800 mg/day (n=89), and 2112±810 ng/mL in patients who received 1200 mg/day (n=75; body weight >75 kg).
The terminal half-life of ribavirin following administration of a single oral dose of COPEGUS is about 120 to 170 hours. The total apparent clearance following administration of a single oral dose of COPEGUS is about 26 L/h. There is extensive accumulation of ribavirin after multiple dosing (twice daily) such that the Cmax at steady state was four-fold higher than that of a single dose.
Effect of Food on Absorption of Ribavirin
Bioavailability of a single oral dose of ribavirin was increased by co-administration with a high-fat meal. The absorption was slowed (Tmax was doubled) and the AUC0-192h and Cmax increased by 42% and 66%, respectively, when COPEGUS was taken with a high-fat meal compared with fasting conditions (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Elimination and Metabolism
The contribution of renal and hepatic pathways to ribavirin elimination after administration of COPEGUS is not known. In vitro studies indicate that ribavirin is not a substrate of CYP450 enzymes.
Special Populations
Race
A pharmacokinetic study in 42 subjects demonstrated there is no clinically significant difference in ribavirin pharmacokinetics among Black (n=14), Hispanic (n=13) and Caucasian (n=15) subjects.
Renal Dysfunction
The pharmacokinetics of ribavirin following administration of COPEGUS have not been studied in patients with renal impairment and there are limited data from clinical trials on administration of COPEGUS in patients with creatinine clearance <50 mL/min. Therefore, patients with creatinine clearance <50 mL/min should not be treated with COPEGUS (see WARNINGS and DOSAGE AND ADMINISTRATION).
Hepatic Impairment
The effect of hepatic impairment on the pharmacokinetics of ribavirin following administration of COPEGUS has not been evaluated. The clinical trials of COPEGUS were restricted to patients with Child-Pugh class A disease.
Pediatric Patients
Pharmacokinetic evaluations in pediatric patients have not been performed.
Elderly Patients
Pharmacokinetic evaluations in elderly patients have not been performed.
Gender
Ribavirin pharmacokinetics, when corrected for weight, are similar in male and female patients.
Drug Interactions
In vitro studies indicate that ribavirin does not inhibit CYP450 enzymes.
Nucleoside Analogues
In vitro data indicate ribavirin reduces phosphorylation of lamivudine, stavudine, and zidovudine. However, no pharmacokinetic (e.g., plasma concentrations or intracellular triphosphorylated active metabolite concentrations) or pharmacodynamic (e.g., loss of HIV/HCV virologic suppression) interaction was observed when ribavirin and lamivudine (n=18), stavudine (n=10), or zidovudine (n=6) were co-administered as part of a multi-drug regimen to HCV/HIV coinfected patients (see PRECAUTIONS: Drug Interactions).
In vitro, didanosine or its active metabolite (dideoxyadenosine 5'-triphosphate) is increased when didanosine is co-administered with ribavirin, which could cause or worsen clinical toxicities (see PRECAUTIONS: Drug Interactions).
Drugs Metabolized by Cytochrome P450
There was no effect on the pharmacokinetics of representative drugs metabolized by CYP 2C9, CYP 2C19, CYP 2D6 or CYP 3A4.
Treatment with PEGASYS® once weekly for 4 weeks in healthy subjects was associated with an inhibition of P450 1A2 and a 25% increase in theophylline AUC (see PRECAUTIONS: Drug Interactions).
CLINICAL STUDIES
HCV Patients
The safety and effectiveness of PEGASYS in combination with COPEGUS for the treatment of hepatitis C virus infection were assessed in two randomized controlled clinical trials. All patients were adults, had compensated liver disease, detectable hepatitis C virus, liver biopsy diagnosis of chronic hepatitis, and were previously untreated with interferon. Approximately 20% of patients in both studies had compensated cirrhosis (Child-Pugh class A). Patients coinfected with HIV were excluded from these studies.
In Study NV15801 (described as Study 4 in the PEGASYS Package Insert), patients were randomized to receive either PEGASYS 180 µg sc once weekly (qw) with an oral placebo, PEGASYS 180 µg qw with COPEGUS 1000 mg po (body weight <75 kg) or 1200 mg po (body weight≥75 kg) or REBETRON™ (interferon alfa-2b 3 MIU sc tiw plus ribavirin 1000 mg or 1200 mg po). All patients received 48 weeks of therapy followed by 24 weeks of treatment-free follow-up. COPEGUS or placebo treatment assignment was blinded. Sustained virological response was defined as undetectable (<50 IU/mL) HCV RNA on or after study week 68. PEGASYS in combination with COPEGUS resulted in a higher SVR compared to PEGASYS alone or interferon alfa-2b and ribavirin (Table 1). In all treatment arms, patients with viral genotype 1, regardless of viral load, had a lower response rate to PEGASYS in combination with COPEGUS compared to patients with other viral genotypes.
| Interferon alfa-2b+ Ribavirin 1000 mg or 1200 mg | PEGASYS + placebo | PEGASYS + COPEGUS 1000 mg or 1200 mg |
|
|---|---|---|---|
| Difference in overall treatment response (PEGASYS/COPEGUS– Interferon alfa-2b/ribavirin) was 9% (95% CI 2.3, 15.3). | |||
|
|||
| All patients | 197/444 (44%) | 65/224 (29%) | 241/453 (53%) |
| Genotype 1 | 103/285 (36%) | 29/145 (20%) | 132/298 (44%) |
| Genotypes 2-6 | 94/159 (59%) | 36/79 (46%) | 109/155 (70%) |
In Study NV15942 (described as Study 5 in the PEGASYS Package Insert), all patients received PEGASYS 180 µg sc qw and were randomized to treatment for either 24 or 48 weeks and to a COPEGUS dose of either 800 mg or 1000 mg/1200 mg (for body weight<75 kg/≥75 kg). Assignment to the four treatment arms was stratified by viral genotype and baseline HCV viral titer. Patients with genotype 1 and high viral titer (defined as >2 × 106 HCV RNA copies/mL serum) were preferentially assigned to treatment for 48 weeks.
HCV Genotypes
HCV 1 and 4 - Irrespective of baseline viral titer, treatment for 48 weeks with PEGASYS and 1000 mg or 1200 mg of COPEGUS resulted in higher SVR (defined as undetectable HCV RNA at the end of the 24-week treatment-free follow-up period) compared to shorter treatment (24 weeks) and/or 800 mg COPEGUS.
HCV 2 and 3 - Irrespective of baseline viral titer, treatment for 24 weeks with PEGASYS and 800 mg of COPEGUS resulted in a similar SVR compared to longer treatment (48 weeks) and/or 1000 mg or 1200 mg of COPEGUS (see Table 2).
The numbers of patients with genotype 5 and 6 were too few to allow for meaningful assessment.
| 24 Weeks Treatment | 48 Weeks Treatment | |||
|---|---|---|---|---|
| PEGASYS + COPEGUS 800 mg (N=207) | PEGASYS + COPEGUS 1000 mg or 1200 mg† (N=280) | PEGASYS + COPEGUS 800 mg (N=361) | PEGASYS + COPEGUS 1000 mg or 1200 mg† (N=436) |
|
| Genotype 1 | 29/101 (29%) | 48/118 (41%) | 99/250 (40%) | 138/271 (51%) |
| Genotypes 2, 3 | 79/96 (82%) | 116/144 (81%) | 75/99 (76%) | 117/153 (76%) |
| Genotype 4 | 0/5 (0%) | 7/12 (58%) | 5/8 (63%) | 9/11 (82%) |
Other Treatment Response Predictors
Treatment response rates are lower in patients with poor prognostic factors receiving pegylated interferon alpha therapy. In studies NV15801 and NV15942, treatment response rates were lower in patients older than 40 years (50% vs. 66%), in patients with cirrhosis (47% vs. 59%), in patients weighing over 85 kg (49% vs. 60%), and in patients with genotype 1 with high vs. low viral load (43% vs. 56%). African-American patients had lower response rates compared to Caucasians.
Paired liver biopsies were performed on approximately 20% of patients in studies NV15801 and NV15942. Modest reductions in inflammation compared to baseline were seen in all treatment groups.
In studies NV15801 and NV15942, lack of early virologic response by 12 weeks (defined as HCV RNA undetectable or >2log10 lower than baseline) was grounds for discontinuation of treatment. Of patients who lacked an early viral response by 12 weeks and completed a recommended course of therapy despite a protocol-defined option to discontinue therapy, 5/39 (13%) achieved an SVR. Of patients who lacked an early viral response by 24 weeks, 19 completed a full course of therapy and none achieved an SVR.
CHC and Coinfection with HIV (CHC/HIV): Study NR15961
In Study NR15961 (described as Study 6 in the PEGASYS Package Insert), patients with CHC/HIV were randomized to receive either PEGASYS 180 µg sc once weekly (qw) plus an oral placebo, PEGASYS 180 µg qw plus COPEGUS 800 mg po daily or ROFERON®-A (interferon alfa-2a), 3 MIU sc tiw plus COPEGUS 800 mg po daily. All patients received 48 weeks of therapy and sustained virologic response (SVR) was assessed at 24 weeks of treatment-free follow-up. COPEGUS or placebo treatment assignment was blinded in the PEGASYS treatment arms. All patients were adults, had compensated liver disease, detectable hepatitis C virus, liver biopsy diagnosis of chronic hepatitis C, and were previously untreated with interferon. Patients also had CD4+ cell count ≥200 cells/µL or CD4+ cell count ≥100 cells/µL but <200 cells/µL and HIV-1 RNA <5000 copies/mL, and stable status of HIV. Approximately 15% of patients in the study had cirrhosis. Results are shown in Table 3.
| ROFERON-A + COPEGUS 800 mg (N=289) | PEGASYS + Placebo (N=289) | PEGASYS + COPEGUS 800 mg (N=290) |
|
|---|---|---|---|
|
|||
| All patients | 33 (11%)* | 58 (20%)* | 116 (40%)* |
| Genotype 1 | 12/171 (7%) | 24/175 (14%) | 51/176 (29%) |
| Genotypes 2, 3 | 18/89 (20%) | 32/90 (36%) | 59/95 (62%) |
Treatment response rates are lower in CHC/HIV patients with poor prognostic factors (including HCV genotype 1, HCV RNA >800,000 IU/mL, and cirrhosis) receiving pegylated interferon alpha therapy. Geographic region is not a prognostic factor for response. However, poor prognostic factors occur more frequently in the US population than in the non-US population.
Of the patients who did not demonstrate either undetectable HCV RNA or at least a 2log10 reduction from baseline in HCV RNA titer by 12 weeks of PEGASYS and COPEGUS combination therapy, 2% (2/85) achieved an SVR.
In CHC patients with HIV coinfection who received 48 weeks of PEGASYS alone or in combination with COPEGUS treatment, mean and median HIV RNA titers did not increase above baseline during treatment or 24 weeks posttreatment.
INDICATIONS AND USAGE
COPEGUS in combination with PEGASYS (peginterferon alfa-2a) is indicated for the treatment of adults with chronic hepatitis C virus infection who have compensated liver disease and have not been previously treated with interferon alpha. Patients in whom efficacy was demonstrated included patients with compensated liver disease and histological evidence of cirrhosis (Child-Pugh class A) and patients with HIV disease that is clinically stable (e.g., antiretroviral therapy not required or receiving stable antiretroviral therapy).
CONTRAINDICATIONS
COPEGUS (ribavirin) is contraindicated in:
- Patients with known hypersensitivity to COPEGUS or to any component of the tablet.
- Women who are pregnant.
- Men whose female partners are pregnant.
- Patients with hemoglobinopathies (e.g., thalassemia major or sickle-cell anemia).
COPEGUS and PEGASYS combination therapy is contraindicated in patients with:
- Autoimmune hepatitis.
- Hepatic decompensation (Child-Pugh score greater than 6; class B and C) in cirrhotic CHC monoinfected patients before or during treatment.
- Hepatic decompensation with Child-Pugh score greater than or equal to 6 in cirrhotic CHC patients coinfected with HIV before or during treatment.
WARNINGS
COPEGUS must not be used alone because ribavirin monotherapy is not effective for the treatment of chronic hepatitis C virus infection. The safety and efficacy of COPEGUS have only been established when used together with PEGASYS (pegylated interferon alfa-2a, recombinant).
COPEGUS and PEGASYS should be discontinued in patients who develop evidence of hepatic decompensation during treatment.
There are significant adverse events caused by COPEGUS/PEGASYS therapy, including severe depression and suicidal ideation, hemolytic anemia, suppression of bone marrow function, autoimmune and infectious disorders, pulmonary dysfunction, pancreatitis, and diabetes. The PEGASYS Package Insert and MEDICATION GUIDE should be reviewed in their entirety prior to initiation of combination treatment for additional safety information.
General
Treatment with COPEGUS and PEGASYS should be administered under the guidance of a qualified physician and may lead to moderate to severe adverse experiences requiring dose reduction, temporary dose cessation or discontinuation of therapy.
Pregnancy
Ribavirin may cause birth defects and/or death of the exposed fetus. Extreme care must be taken to avoid pregnancy in female patients and in female partners of male patients. Ribavirin has demonstrated significant teratogenic and/or embryocidal effects in all animal species in which adequate studies have been conducted. These effects occurred at doses as low as one twentieth of the recommended human dose of ribavirin. COPEGUS THERAPY SHOULD NOT BE STARTED UNLESS A REPORT OF A NEGATIVE PREGNANCY TEST HAS BEEN OBTAINED IMMEDIATELY PRIOR TO PLANNED INITIATION OF THERAPY. Patients should be instructed to use at least two forms of effective contraception during treatment and for 6 months after treatment has been stopped. Pregnancy testing should occur monthly during COPEGUS therapy and for 6 months after therapy has stopped (see CONTRAINDICATIONS and PRECAUTIONS: Information for Patients and Pregnancy: Category X).
Anemia
The primary toxicity of ribavirin is hemolytic anemia (hemoglobin <10 g/dL), which was observed in approximately 13% of all COPEGUS and PEGASYS treated patients in clinical trials (see PRECAUTIONS: Laboratory Tests). The anemia associated with COPEGUS occurs within 1 to 2 weeks of initiation of therapy. BECAUSE THE INITIAL DROP IN HEMOGLOBIN MAY BE SIGNIFICANT, IT IS ADVISED THAT HEMOGLOBIN OR HEMATOCRIT BE OBTAINED PRETREATMENT AND AT WEEK 2 AND WEEK 4 OF THERAPY OR MORE FREQUENTLY IF CLINICALLY INDICATED. Patients should then be followed as clinically appropriate.
Fatal and nonfatal myocardial infarctions have been reported in patients with anemia caused by ribavirin. Patients should be assessed for underlying cardiac disease before initiation of ribavirin therapy. Patients with pre-existing cardiac disease should have electrocardiograms administered before treatment, and should be appropriately monitored during therapy. If there is any deterioration of cardiovascular status, therapy should be suspended or discontinued (see DOSAGE AND ADMINISTRATION: COPEGUS Dosage Modification Guidelines). Because cardiac disease may be worsened by drug induced anemia, patients with a history of significant or unstable cardiac disease should not use COPEGUS (see ADVERSE REACTIONS).
Hepatic Failure
Chronic hepatitis C (CHC) patients with cirrhosis may be at risk of hepatic decompensation and death when treated with alpha interferons, including PEGASYS. Cirrhotic CHC patients coinfected with HIV receiving highly active antiretroviral therapy (HAART) and interferon alfa-2a with or without ribavirin appear to be at increased risk for the development of hepatic decompensation compared to patients not receiving HAART. In Study NR15961 (described as Study 6 in the PEGASYS Package Insert), among 129 CHC/HIV cirrhotic patients receiving HAART, 14 (11%) of these patients across all treatment arms developed hepatic decompensation resulting in 6 deaths. All 14 patients were on NRTIs, including stavudine, didanosine, abacavir, zidovudine, and lamivudine. These small numbers of patients do not permit discrimination between specific NRTIs or the associated risk. During treatment, patients' clinical status and hepatic function should be closely monitored, and PEGASYS treatment should be immediately discontinued if decompensation (Child-Pugh score ≥6) is observed (see CONTRAINDICATIONS).
Hypersensitivity
Severe acute hypersensitivity reactions (e.g., urticaria, angioedema, bronchoconstriction, and anaphylaxis) have been rarely observed during alpha interferon and ribavirin therapy. If such reaction occurs, therapy with PEGASYS and COPEGUS should be discontinued and appropriate medical therapy immediately instituted. Serious skin reactions including vesiculobullous eruptions, reactions in the spectrum of Stevens Johnson Syndrome (erythema multiforme major) with varying degrees of skin and mucosal involvement and exfoliative dermatitis (erythroderma) have been rarely reported in patients receiving PEGASYS with and without ribavirin. Patients developing signs or symptoms of severe skin reactions must discontinue therapy. (see ADVERSE REACTIONS: Postmarketing Experience).
Pulmonary
Pulmonary symptoms, including dyspnea, pulmonary infiltrates, pneumonitis and occasional cases of fatal pneumonia, have been reported during therapy with ribavirin and interferon. In addition, sarcoidosis or the exacerbation of sarcoidosis has been reported. If there is evidence of pulmonary infiltrates or pulmonary function impairment, the patient should be closely monitored and, if appropriate, combination COPEGUS/PEGASYS treatment should be discontinued.
Other
COPEGUS and PEGASYS therapy should be suspended in patients with signs and symptoms of pancreatitis, and discontinued in patients with confirmed pancreatitis.
COPEGUS should not be used in patients with creatinine clearance <50 mL/min (see CLINICAL PHARMACOLOGY: Special Populations).
COPEGUS must be discontinued immediately and appropriate medical therapy instituted if an acute hypersensitivity reaction (e.g., urticaria, angioedema, bronchoconstriction, anaphylaxis) develops. Transient rashes do not necessitate interruption of treatment.
PRECAUTIONS
The safety and efficacy of COPEGUS and PEGASYS therapy for the treatment of adenovirus, RSV, parainfluenza or influenza infections have not been established. COPEGUS should not be used for these indications. Ribavirin for inhalation has a separate package insert, which should be consulted if ribavirin inhalation therapy is being considered.
The safety and efficacy of COPEGUS and PEGASYS therapy have not been established in liver or other organ transplant patients, patients with decompensated liver disease due to hepatitis C virus infection, patients who are non-responders to interferon therapy or patients coinfected with HBV or HIV and a CD4+ cell count <100 cells/µL.
Information for Patients
Patients must be informed that ribavirin may cause birth defects and/or death of the exposed fetus. COPEGUS therapy must not be used by women who are pregnant or by men whose female partners are pregnant. Extreme care must be taken to avoid pregnancy in female patients and in female partners of male patients taking COPEGUS therapy and for 6 months posttherapy. COPEGUS therapy should not be initiated until a report of a negative pregnancy test has been obtained immediately prior to initiation of therapy. Patients must perform a pregnancy test monthly during therapy and for 6 months posttherapy.
Female patients of childbearing potential and male patients with female partners of childbearing potential must be advised of the teratogenic/embryocidal risks and must be instructed to practice effective contraception during COPEGUS therapy and for 6 months posttherapy. Patients should be advised to notify the healthcare provider immediately in the event of a pregnancy (see CONTRAINDICATIONS and WARNINGS).
The most common adverse event associated with ribavirin is anemia, which may be severe (see ADVERSE REACTIONS). Patients should be advised that laboratory evaluations are required prior to starting COPEGUS therapy and periodically thereafter (see Laboratory Tests). It is advised that patients be well hydrated, especially during the initial stages of treatment.
Patients who develop dizziness, confusion, somnolence, and fatigue should be cautioned to avoid driving or operating machinery.
Patients should be informed regarding the potential benefits and risks attendant to the use of COPEGUS. Instructions on appropriate use should be given, including review of the contents of the enclosed MEDICATION GUIDE, which is not a disclosure of all or possible adverse effects.
Patients should be advised to take COPEGUS with food.
Laboratory Tests
Before beginning COPEGUS therapy, standard hematological and biochemical laboratory tests must be conducted for all patients. Pregnancy screening for women of childbearing potential must be done.
After initiation of therapy, hematological tests should be performed at 2 weeks and 4 weeks and biochemical tests should be performed at 4 weeks. Additional testing should be performed periodically during therapy. Monthly pregnancy testing should be done during combination therapy and for 6 months after discontinuing therapy.
The entrance criteria used for the clinical studies of COPEGUS and PEGASYS combination therapy may be considered as a guideline to acceptable baseline values for initiation of treatment:
- Platelet count ≥90,000 cells/mm3 (as low as 75,000 cells/mm3 in patients with cirrhosis or 70,000 cells/mm3 in patients with CHC and HIV)
- Absolute neutrophil count (ANC) ≥1500 cells/mm3
- TSH and T4 within normal limits or adequately controlled thyroid function
- ECG (see WARNINGS)
- CD4+ cell count ≥200 cells/µL or CD4+ cell count≥100 cells/µL but <200 cells/µL and HIV-1 RNA<5000 copies/mL in patients coinfected with HIV
- Hemoglobin ≥12 g/dL for women and ≥13 g/dL for men in CHC monoinfected patients
- Hemoglobin ≥11 g/dL for women and ≥12 g/dL for men in patients with CHC and HIV
The maximum drop in hemoglobin usually occurred during the first 8 weeks of initiation of COPEGUS therapy. Because of this initial acute drop in hemoglobin, it is advised that a complete blood count should be obtained pretreatment and at week 2 and week 4 of therapy or more frequently if clinically indicated. Additional testing should be performed periodically during therapy. Patients should then be followed as clinically appropriate.
Drug Interactions
Results from a pharmacokinetic sub-study demonstrated no pharmacokinetic interaction between PEGASYS (peginterferon alfa-2a) and ribavirin.
Nucleoside Analogues
NRTIs
In Study NR15961 among the CHC/HIV coinfected cirrhotic patients receiving NRTIs cases of hepatic decompensation (some fatal) were observed (see WARNINGS: Hepatic Failure).
Patients receiving PEGASYS/COPEGUS and NRTIs should be closely monitored for treatment associated toxicities. Physicians should refer to prescribing information for the respective NRTIs for guidance regarding toxicity management. In addition, dose reduction or discontinuation of PEGASYS, COPEGUS or both should also be considered if worsening toxicities are observed (see WARNINGS, PRECAUTIONS, and DOSAGE AND ADMINISTRATION: Dose Modifications).
Didanosine
Co-administration of COPEGUS and didanosine is not recommended. Reports of fatal hepatic failure, as well as peripheral neuropathy, pancreatitis, and symptomatic hyperlactatemia/lactic acidosis have been reported in clinical trials (see CLINICAL PHARMACOLOGY: Drug Interactions).
Zidovudine
In Study NR15961, patients who were administered zidovudine in combination with PEGASYS/COPEGUS developed severe neutropenia (ANC <500) and severe anemia (hemoglobin <8 g/dL) more frequently than similar patients not receiving zidovudine (neutropenia 15% vs. 9%) (anemia 5% vs. 1%).
Lamivudine, Stavudine, and Zidovudine
In vitro studies have shown ribavirin can reduce the phosphorylation of pyrimidine nucleoside analogs such as lamivudine, stavudine, and zidovudine. No evidence of a pharmacokinetic or pharmacodynamic interaction was seen when ribavirin was co-administered with lamivudine, stavudine, and/or zidovudine in HIV/HCV coinfected patients (see CLINICAL PHARMACOLOGY: Drug Interactions).
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a p53 (+/-) mouse carcinogenicity study and a rat 2-year carcinogenicity study at doses up to the maximum tolerated doses of 100 mg/kg/day and 60 mg/kg/day, respectively, ribavirin was not oncogenic. On a body surface area basis, these doses are approximately 0.5 and 0.6 times the maximum recommended human 24-hour dose of ribavirin.
Mutagenesis
Ribavirin demonstrated mutagenic activity in the in vitro mouse lymphoma assay. No clastogenic activity was observed in an in vivo mouse micronucleus assay at doses up to 2000 mg/kg. However, results from studies published in the literature show clastogenic activity in the in vivo mouse micronucleus assay at oral doses up to 2000 mg/kg. A dominant lethal assay in rats was negative, indicating that if mutations occurred in rats they were not transmitted through male gametes. However, potential carcinogenic risk to humans cannot be excluded.
Impairment of Fertility
In a fertility study in rats, ribavirin showed a marginal reduction in sperm counts at the dose of 100 mg/kg/day with no effect on fertility. Upon cessation of treatment, total recovery occurred after 1 spermatogenesis cycle. Abnormalities in sperm were observed in studies in mice designed to evaluate the time course and reversibility of ribavirin-induced testicular degeneration at doses of 15 to 150 mg/kg/day (approximately 0.1 to 0.8 times the maximum recommended human 24-hour dose of ribavirin) administered for 3 to 6 months. Upon cessation of treatment, essentially total recovery from ribavirin-induced testicular toxicity was apparent within 1 or 2 spermatogenic cycles.
Female patients of childbearing potential and male patients with female partners of childbearing potential should not receive COPEGUS unless the patient and his/her partner are using effective contraception (two reliable forms). Based on a multiple dose half-life (t1/2) of ribavirin of 12 days, effective contraception must be utilized for 6 months posttherapy (ie, 15 half-lives of clearance for ribavirin).
No reproductive toxicology studies have been performed using PEGASYS in combination with COPEGUS. However, peginterferon alfa-2a and ribavirin when administered separately, each has adverse effects on reproduction. It should be assumed that the effects produced by either agent alone would also be caused by the combination of the two agents.
Pregnancy
Pregnancy: Category X
(see CONTRAINDICATIONS)
Ribavirin produced significant embryocidal and/or teratogenic effects in all animal species in which adequate studies have been conducted. Malformations of the skull, palate, eye, jaw, limbs, skeleton, and gastrointestinal tract were noted. The incidence and severity of teratogenic effects increased with escalation of the drug dose. Survival of fetuses and offspring was reduced.
In conventional embryotoxicity/teratogenicity studies in rats and rabbits, observed no-effect dose levels were well below those for proposed clinical use (0.3 mg/kg/day for both the rat and rabbit; approximately 0.06 times the recommended human 24-hour dose of ribavirin). No maternal toxicity or effects on offspring were observed in a peri/postnatal toxicity study in rats dosed orally at up to 1 mg/kg/day (approximately 0.01 times the maximum recommended human 24-hour dose of ribavirin).
Treatment and Posttreatment: Potential Risk to the Fetus
Ribavirin is known to accumulate in intracellular components from where it is cleared very slowly. It is not known whether ribavirin is contained in sperm, and if so, will exert a potential teratogenic effect upon fertilization of the ova. In a study in rats, it was concluded that dominant lethality was not induced by ribavirin at doses up to 200 mg/kg for 5 days (up to 1.7 times the maximum recommended human dose of ribavirin). However, because of the potential human teratogenic effects of ribavirin, male patients should be advised to take every precaution to avoid risk of pregnancy for their female partners.
COPEGUS should not be used by pregnant women or by men whose female partners are pregnant. Female patients of childbearing potential and male patients with female partners of childbearing potential should not receive COPEGUS unless the patient and his/her partner are using effective contraception (two reliable forms) during therapy and for 6 months posttherapy.
Ribavirin Pregnancy Registry
A Ribavirin Pregnancy Registry has been established to monitor maternal-fetal outcomes of pregnancies of female patients and female partners of male patients exposed to ribavirin during treatment and for 6 months following cessation of treatment. Healthcare providers and patients are encouraged to report such cases by calling 1-800-593-2214.
Animal Toxicology
Long-term study in the mouse and rat (18 to 24 months; dose 20 to 75, and 10 to 40 mg/kg/day, respectively, approximately 0.1 to 0.4 times the maximum human daily dose of ribavirin) have demonstrated a relationship between chronic ribavirin exposure and an increased incidence of vascular lesions (microscopic hemorrhages) in mice. In rats, retinal degeneration occurred in controls, but the incidence was increased in ribavirin-treated rats.
Nursing Mothers
It is not known whether ribavirin is excreted in human milk. Because many drugs are excreted in human milk and to avoid any potential for serious adverse reactions in nursing infants from ribavirin, a decision should be made either to discontinue nursing or therapy with COPEGUS, based on the importance of the therapy to the mother.
Pediatric Use
Safety and effectiveness of COPEGUS have not been established in patients below the age of 18.
Geriatric Use
Clinical studies of COPEGUS and PEGASYS did not include sufficient numbers of subjects aged 65 or over to determine whether they respond differently from younger subjects. Specific pharmacokinetic evaluations for ribavirin in the elderly have not been performed. The risk of toxic reactions to this drug may be greater in patients with impaired renal function. COPEGUS should not be administered to patients with creatinine clearance <50 mL/min. (see CLINICAL PHARMACOLOGY: Special Populations).
Effect of Gender
No clinically significant differences in the pharmacokinetics of ribavirin were observed between male and female subjects.
ADVERSE REACTIONS
PEGASYS in combination with COPEGUS causes a broad variety of serious adverse reactions (see BOXED WARNING and WARNINGS).
The most common life-threatening or fatal events induced or aggravated by PEGASYS and COPEGUS were depression, suicide, relapse of drug abuse/overdose, and bacterial infections, each occurring at a frequency of <1%. Hepatic decompensation occurred in 2% (10/574) of CHC/HIV patients (see WARNINGS: Hepatic Failure).
In all studies, one or more serious adverse reactions occurred in 10% of CHC monoinfected patients and in 19% of CHC/HIV patients receiving PEGASYS alone or in combination with COPEGUS. The most common serious adverse event (3% in CHC and 5% in CHC/HIV) was bacterial infection (e.g., sepsis, osteomyelitis, endocarditis, pyelonephritis, pneumonia). Other SAEs occurred at a frequency of <1% and included: suicide, suicidal ideation, psychosis, aggression, anxiety, drug abuse and drug overdose, angina, hepatic dysfunction, fatty liver, cholangitis, arrhythmia, diabetes mellitus, autoimmune phenomena (e.g., hyperthyroidism, hypothyroidism, sarcoidosis, systemic lupus erythematosus, rheumatoid arthritis), peripheral neuropathy, aplastic anemia, peptic ulcer, gastrointestinal bleeding, pancreatitis, colitis, corneal ulcer, pulmonary embolism, coma, myositis, cerebral hemorrhage, thrombotic thrombocytopenic purpura, psychotic disorder, and hallucination.
Nearly all patients in clinical trials experienced one or more adverse events. The most commonly reported adverse reactions were psychiatric reactions, including depression, insomnia, irritability, anxiety, and flu-like symptoms such as fatigue, pyrexia, myalgia, headache and rigors. Other common reactions were anorexia, nausea and vomiting, diarrhea, arthralgias, injection site reactions, alopecia, and pruritus.
Ten percent of CHC monoinfected patients receiving 48 weeks of therapy with PEGASYS in combination with COPEGUS discontinued therapy; 16% of CHC/HIV coinfected patients discontinued therapy. The most common reasons for discontinuation of therapy were psychiatric, flu-like syndrome (e.g., lethargy, fatigue, headache), dermatologic and gastrointestinal disorders and laboratory abnormalities (thrombocytopenia, neutropenia, and anemia).
Overall 39% of patients with CHC or CHC/HIV required modification of PEGASYS and/or COPEGUS therapy. The most common reason for dose modification of PEGASYS in CHC and CHC/HIV patients was for laboratory abnormalities; neutropenia (20% and 27%, respectively) and thrombocytopenia (4% and 6%, respectively). The most common reason for dose modification of COPEGUS in CHC and CHC/HIV patients was anemia (22% and 16%, respectively).
PEGASYS dose was reduced in 12% of patients receiving 1000 mg to 1200 mg COPEGUS for 48 weeks and in 7% of patients receiving 800 mg COPEGUS for 24 weeks. COPEGUS dose was reduced in 21% of patients receiving 1000 mg to 1200 mg COPEGUS for 48 weeks and in 12% of patients receiving 800 mg COPEGUS for 24 weeks.
Chronic hepatitis C monoinfected patients treated for 24 weeks with PEGASYS and 800 mg COPEGUS were observed to have lower incidence of serious adverse events (3% vs. 10%), hemoglobin <10g/dL (3% vs. 15%), dose modification of PEGASYS (30% vs. 36%) and COPEGUS (19% vs. 38%), and of withdrawal from treatment (5% vs. 15%) compared to patients treated for 48 weeks with PEGASYS and 1000 mg or 1200 mg COPEGUS. On the other hand, the overall incidence of adverse events appeared to be similar in the two treatment groups.
Because clinical trials are conducted under widely varying and controlled conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug. Also, the adverse event rates listed here may not predict the rates observed in a broader patient population in clinical practice.
| CHC Combination Therapy Study NV15801 | ||
|---|---|---|
| Body System | PEGASYS 180 µg + 1000 mg or 1200 mg COPEGUS 48 week | Intron A + 1000 mg or 1200 mg REBETOL® 48 week |
| N=451 | N=443 | |
| % | % | |
| Application Site Disorders | ||
| Injection site reaction | 23 | 16 |
| Endocrine Disorders | ||
| Hypothyroidism | 4 | 5 |
| Flu-like Symptoms and Signs | ||
| Fatigue/Asthenia | 65 | 68 |
| Pyrexia | 41 | 55 |
| Rigors | 25 | 37 |
| Pain | 10 | 9 |
| Gastrointestinal | ||
| Nausea/Vomiting | 25 | 29 |
| Diarrhea | 11 | 10 |
| Abdominal pain | 8 | 9 |
| Dry mouth | 4 | 7 |
| Dyspepsia | 6 | 5 |
| Hematologic† | ||
| Lymphopenia | 14 | 12 |
| Anemia | 11 | 11 |
| Neutropenia | 27 | 8 |
| Thrombocytopenia | 5 | <1 |
| Metabolic and Nutritional | ||
| Anorexia | 24 | 26 |
| Weight decrease | 10 | 10 |
| Musculoskeletal, Connective Tissue and Bone | ||
| Myalgia | 40 | 49 |
| Arthralgia | 22 | 23 |
| Back pain | 5 | 5 |
| Neurological | ||
| Headache | 43 | 49 |
| Dizziness (excluding vertigo) | 14 | 14 |
| Memory impairment | 6 | 5 |
| Psychiatric | ||
| Irritability/Anxiety/Nervousness | 33 | 38 |
| Insomnia | 30 | 37 |
| Depression | 20 | 28 |
| Concentration impairment | 10 | 13 |
| Mood alteration | 5 | 6 |
| Resistance Mechanism Disorders | ||
| Overall | 12 | 10 |
| Respiratory, Thoracic and Mediastinal | ||
| Dyspnea | 13 | 14 |
| Cough | 10 | 7 |
| Dyspnea exertional | 4 | 7 |
| Skin and Subcutaneous Tissue | ||
| Alopecia | 28 | 33 |
| Pruritus | 19 | 18 |
| Dermatitis | 16 | 13 |
| Dry skin | 10 | 13 |
| Rash | 8 | 5 |
| Sweating increased | 6 | 5 |
| Eczema | 5 | 4 |
| Visual Disorders | ||
| Vision blurred | 5 | 2 |
Common Adverse Reactions in CHC With HIV Coinfection
The adverse event profile of coinfected patients treated with PEGASYS and COPEGUS in Study NR15961 was generally similar to that shown for monoinfected patients in Study NV15801 (Table 4). Events occurring more frequently in coinfected patients were neutropenia (40%), anemia (14%), thrombocytopenia (8%), weight decrease (16%), and mood alteration (9%).
Laboratory Test Values
Anemia due to hemolysis is the most significant toxicity of ribavirin therapy. Anemia (hemoglobin <10 g/dL) was observed in 13% of all COPEGUS and PEGASYS combination-treated patients in clinical trials. The maximum drop in hemoglobin occurred during the first 8 weeks of initiation of ribavirin therapy (see DOSAGE AND ADMINISTRATION: Dose Modifications).
The following adverse reactions have been identified and reported during post-approval use of PEGASYS therapy: dehydration, hearing impairment, hearing loss, serious skin reactions (see WARNINGS: Hypersensitivity), and serous retinal detachment.
OVERDOSAGE
No cases of overdose with COPEGUS have been reported in clinical trials. Hypocalcemia and hypomagnesemia have been observed in persons administered greater than the recommended dosage of ribavirin. In most of these cases, ribavirin was administered intravenously at dosages up to and in some cases exceeding four times the recommended maximum oral daily dose.
DOSAGE AND ADMINISTRATION
CHC Monoinfection
The recommended dose of COPEGUS tablets is provided in Table 5. The recommended duration of treatment for patients previously untreated with ribavirin and interferon is 24 to 48 weeks.
The daily dose of COPEGUS is 800 mg to 1200 mg administered orally in two divided doses. The dose should be individualized to the patient depending on baseline disease characteristics (e.g., genotype), response to therapy, and tolerability of the regimen (see Table 5).
In the pivotal clinical trials, patients were instructed to take COPEGUS with food; therefore, patients are advised to take COPEGUS with food.
| Genotype | PEGASYS Dose | COPEGUS Dose | Duration |
|---|---|---|---|
| Genotypes non-1 showed no increased response to treatment beyond 24 weeks (see Table 2). Data on genotypes 5 and 6 are insufficient for dosing recommendations. | |||
| Genotypes 1, 4 | 180 µg | <75 kg = 1000 mg | 48 weeks |
| ≥75 kg = 1200 mg | 48 weeks | ||
| Genotypes 2, 3 | 180 µg | 800 mg | 24 weeks |
CHC with HIV Coinfection
The recommended dose for hepatitis C in HCV/HIV coinfected patients is PEGASYS 180 µg sc once weekly and COPEGUS 800 mg po daily for a total of 48 weeks, regardless of genotype.
Dose Modifications
If severe adverse reactions or laboratory abnormalities develop during combination COPEGUS/PEGASYS therapy, the dose should be modified or discontinued, if appropriate, until the adverse reactions abate. If intolerance persists after dose adjustment, COPEGUS/PEGASYS therapy should be discontinued.
COPEGUS should be administered with caution to patients with pre-existing cardiac disease (see Table 6). Patients should be assessed before commencement of therapy and should be appropriately monitored during therapy. If there is any deterioration of cardiovascular status, therapy should be stopped (see WARNINGS).
| Laboratory Values | Reduce Only COPEGUS Dose to 600 mg/day* if: | Discontinue COPEGUS if: |
|---|---|---|
|
||
| Hemoglobin in patients with no cardiac disease | <10 g/dL | <8.5 g/dL |
| Hemoglobin in patients with history of stable cardiac disease | ≥2 g/dL decrease in hemoglobin during any 4 week period treatment | <12 g/dL despite 4 weeks at reduced dose |
Once COPEGUS has been withheld due to either a laboratory abnormality or clinical manifestation, an attempt may be made to restart COPEGUS at 600 mg daily and further increase the dose to 800 mg daily depending upon the physician's judgment. However, it is not recommended that COPEGUS be increased to its original assigned dose (1000 mg to 1200 mg).
Renal Impairment
COPEGUS should not be used in patients with creatinine clearance <50 mL/min (see WARNINGS and CLINICAL PHARMACOLOGY: Special Populations).
HOW SUPPLIED
COPEGUS® (ribavirin) is available as tablets for oral administration. Each tablet contains 200 mg of ribavirin and is light pink to pink colored, flat, oval-shaped, film-coated, and engraved with RIB 200 on one side and ROCHE on the other side. They are packaged as bottle of 168 tablets (NDC 0004-0086-94).
Storage Conditions
Store the COPEGUS® Tablets bottle at 25°C (77°F); excursions are permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature]. Keep bottle tightly closed.
REBETRON™ is a trademark of Schering Corporation.
PI Revised: April 2009
MEDICATION GUIDE
COPEGUS®
(ribavirin, USP)
TABLETS
Read this Medication Guide carefully before you start taking COPEGUS (Co-PEG-UHS) and read the Medication Guide each time you get more COPEGUS. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about COPEGUS?
- 1.
-
COPEGUS, a form of ribavirin, may cause birth defects or death of an unborn child. Therefore, if you are pregnant or your partner is pregnant or plans to become pregnant, do not take COPEGUS. Female patients and female partners of male patients being treated with COPEGUS must not become pregnant during treatment and for 6 months after treatment has stopped.
During this time you must have pregnancy tests that show you are not pregnant. You must also use 2 effective forms of birth control during therapy and for 6 months after stopping therapy. Male patients should use a condom with spermicide as one of the two forms.
If pregnancy occurs, report the pregnancy to your healthcare provider right away. (See "What should I avoid while taking COPEGUS?".)
If you or a female sexual partner becomes pregnant, you should tell your healthcare provider. There is a Ribavirin Pregnancy Registry that collects information about pregnancy outcomes of female patients and female partners of male patients exposed to ribavirin. You or your healthcare provider are encouraged to contact the Registry at 1-800-593-2214.
- 2.
- COPEGUS can cause a dangerous drop in your red blood cell count. COPEGUS can cause anemia, which is a decrease in the number of red blood cells. This can be dangerous, especially if you have heart or breathing problems. This may cause a worsening of heart (cardiovascular) or circulatory problems. Some patients may get chest pain and rarely, a heart attack. Patients with a history of heart disease have the highest chance of this. Tell your healthcare provider, before taking COPEGUS if you have or have ever had any heart or breathing problems. Your healthcare provider should check your red blood cell count before you start treatment with COPEGUS and often during the first 4 weeks of treatment. Your red blood cell count may be done more often if you have any heart or breathing problems.
- 3.
- Do not take COPEGUS alone to treat hepatitis C virus infection. COPEGUS does not treat hepatitis C virus infections by itself. COPEGUS should be used in combination with PEGASYS® (peginterferon alfa-2a) to treat continuing (chronic) hepatitis C virus infections. You should read the Medication Guide for PEGASYS because it has additional important information about treatment that is not covered in this Medication Guide. Your healthcare provider or pharmacist should give you a copy of the PEGASYS Medication Guide.
What is COPEGUS?
COPEGUS is the antiviral medicine ribavirin. It is used in combination with a medicine called PEGASYS (peginterferon alfa-2a) to treat some adults with chronic hepatitis C whose liver still works normally, and who have not been treated before with a medicine called an interferon alpha. It is not known how COPEGUS and PEGASYS work together to fight hepatitis C virus infections.
It is not known if treatment with COPEGUS and PEGASYS combination therapy can cure hepatitis C or if it can prevent liver damage (cirrhosis), liver failure or liver cancer that is caused by hepatitis C virus infections. It is not known if treatment with COPEGUS and PEGASYS combination therapy will prevent an infected person from spreading the hepatitis C virus to another person.
Treatment with COPEGUS has not been studied in children under 18 years of age.
Who should not take COPEGUS?
Do not use COPEGUS if:
- You are a female and you are pregnant or plan to become pregnant during treatment or during the 6 months after your treatment has ended. (See "What is the most important information I should know about COPEGUS?" and "What should I avoid while taking COPEGUS?".)
- You are a male patient with a female sexual partner who is pregnant or plans to become pregnant at any time while you are being treated with COPEGUS or during the 6 months after your treatment has ended. (See "What is the most important information I should know about COPEGUS?" and "What should I avoid while taking COPEGUS?".)
- You are breast feeding. We do not know if COPEGUS can pass through your milk and if it can harm your baby. You will need to choose either to breast-feed or take COPEGUS, but not both.
- You have a liver disease called autoimmune hepatitis (hepatitis caused by your immune system attacking your liver).
- You have unstable or severe liver disease.
- You are allergic to any of the ingredients in COPEGUS. The active ingredient in COPEGUS is ribavirin. See the end of this Medication Guide for a list of all the ingredients in COPEGUS.
Tell your healthcare provider before starting treatment with COPEGUS in combination with PEGASYS (see also the PEGASYS Medication Guide) if you have any of the following medical conditions:
- mental health problems, such as depression or anxiety: COPEGUS and PEGASYS combination therapy may make them worse. Tell your healthcare provider if you are being treated or had treatment in the past for any mental problems, including depression, thoughts of ending your life (suicidal thoughts) or a feeling of loss of contact with reality, such as hearing voices or seeing things that are not there (psychosis). Tell your healthcare provider if you take any medicines for these problems.
- high blood pressure, heart problems or have had a heart attack. COPEGUS may worsen heart problems such as high blood pressure, increased heart rate, and chest pain. Tell your healthcare provider if you have or had a heart problem. Patients who have had certain heart problems should not take COPEGUS.
- blood disorders, including anemia (low red blood cell count), thalassemia (Mediterranean anemia) and sickle-cell anemia. COPEGUS can reduce the number of red blood cells you have. This may make you feel dizzy or weak and could worsen any heart problems you might have.
- kidney problems. If your kidneys do not work properly, you may have worse side effects from COPEGUS treatment and require a lower dose.
- liver problems (other than hepatitis C virus infection).
- organ transplant, and you are taking medicine that keeps your body from rejecting your transplant (suppresses your immune system).
- thyroid disease. COPEGUS and PEGASYS combination therapy may make your thyroid disease worse or harder to treat. COPEGUS and PEGASYS treatment may be stopped if you develop thyroid problems that cannot be controlled by medicine.
- have or had drug or alcohol addiction or abuse.
- cancer.
- infection with hepatitis B virus.
- diabetes. COPEGUS and PEGASYS combination therapy may make your diabetes worse or harder to treat.
- past interferon treatment for hepatitis C virus infection that did not work for you.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins or herbal supplements. Some medicines can cause serious side effects if taken while you also take COPEGUS. Some medicines may affect how COPEGUS works or COPEGUS may affect how your other medicines work. Be especially sure to tell your healthcare provider if you take any medicines to treat HIV.
For more information see the PEGASYS Medication Guide.
How should I take COPEGUS?
- Your healthcare provider will determine the right dose of COPEGUS based on your weight.
- Take COPEGUS 1 time in the morning and 1 time at night (2 times a day). Take COPEGUS the same 2 times each day.
- Take COPEGUS with food.
- It is very important to follow your dosing schedule and your healthcare provider's instructions on how to take your medicines.
- Take COPEGUS for as long as it is prescribed, and do not take more than your healthcare provider prescribes.
- If you miss a dose of COPEGUS and remember the same day, take the missed dose as soon as you remember. If the whole day has passed, ask your healthcare provider what to do. Do not take 2 doses at the same time.
- Your healthcare provider may adjust your dose of COPEGUS based on blood tests that show your response to treatment and side effects you may have.
-
Females taking COPEGUS or female sexual partners of male patients taking COPEGUS must have a pregnancy test:
- before treatment begins
- every month during treatment
- for 6 months after treatment ends to make sure there is no pregnancy
It is also important not to use other ribavirin medicines without talking to your healthcare provider. Please see the PEGASYS Medication Guide for the proper use of PEGASYS injection.
What should I avoid while taking COPEGUS?
Avoid the following during COPEGUS treatment:
-
Do not get pregnant. If you or your sexual partner get pregnant during treatment with COPEGUS or in the 6 months after treatment ends, tell your healthcare provider right away. (See "What is the most important information I should know about COPEGUS?".)
Talk with your healthcare provider about birth control methods and how to avoid pregnancy. You must use extreme care to avoid pregnancy during and for 6 months after treatment in female and male patients.
- Do not take COPEGUS alone to treat your hepatitis C virus infection. COPEGUS should be used in combination with PEGASYS (peginterferon alfa-2a) to treat chronic hepatitis C virus infections. (See "What is the most important information I should know about COPEGUS?".)
- Do not breast feed. COPEGUS may pass through your milk and may harm your baby.
- Do not drink alcohol, including beer, wine, and liquor. This may make your liver disease worse.
- Do not drive or operate machinery if COPEGUS makes you feel tired, dizzy or confused.
- Do not take other medicines unless your healthcare provider knows about them. Take only medicines prescribed or approved by your healthcare provider. These include prescription and non-prescription medicines, vitamins or herbal supplements. Talk to your healthcare provider before starting any new medicine.
What are the possible side effects of COPEGUS?
The most serious possible side effects of COPEGUS are:
- Harm to unborn children. COPEGUS may cause birth defects or death of an unborn child. (For more details, see "What is the most important information I should know about COPEGUS?".)
- Anemia. Anemia is a reduction in the number of red blood cells you have. Anemia can be dangerous, especially if you have heart or breathing problems. Tell your healthcare provider right away if you feel tired, have chest pain or shortness of breath. These may be signs of low red blood cell counts.
- Liver problems. Some patients may develop worsening of liver function. Some of the symptoms may include stomach bloating, confusion, brown urine, and yellow eyes. Tell your healthcare provider immediately if any of these symptoms occur.
Call your healthcare provider right away if you have any of the following symptoms. They may be signs of a serious side effect of COPEGUS and PEGASYS treatment.
- trouble breathing
- hives or swelling
- chest pain
- severe stomach pain or low back pain
- bloody diarrhea or bloody stools (bowel movements). These may look like black tar.
- bruising or unusual bleeding
- change in your vision
- high fever (temperature greater than 100.5°F)
- you have psoriasis (a skin disease) and it gets worse
- you become very depressed or think about suicide (ending your life)
- Skin rash can occur in patients taking PEGASYS. In some patients a rash can be serious. If you develop a rash with fever, blisters, or sores in your mouth, nose or eyes or conjunctivitis (red or inflamed eyes, like “pink eye”), stop using PEGASYS and call your doctor right away
The most common side effects of COPEGUS are likely to be the same as for other ribavirin products. These are:
- feeling tired
- nausea and appetite loss
- rash and itching
- cough
These are not all the possible side effects of COPEGUS treatment. For more information, ask your doctor or pharmacist and see the PEGASYS Medication Guide. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Roche at 1-800-526-6367.
What should I know about hepatitis C infection?
Hepatitis C infection is a disease caused by a virus that infects the liver. Hepatitis C is more serious for some people than others. Most people who get hepatitis C carry the virus in their blood for the rest of their lives. Most of these people will have some liver damage, but many do not feel sick from the disease. In some people, the liver becomes badly damaged and scarred. This is called cirrhosis. Cirrhosis can cause the liver to stop working. Some people may get liver cancer or liver failure from the hepatitis C virus.
Hepatitis C virus is spread from one person to another by contact with an infected person's blood. You should talk to your healthcare provider about ways to prevent you from infecting others.
How should I store COPEGUS?
Store COPEGUS tablets at room temperature (77°F).
Please referto the PEGASYS Medication Guide for storage information about PEGASYS injection.
General information about the safe and effective use of COPEGUS
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use COPEGUS for a condition for which it was not prescribed. Do not give COPEGUS to other people, even if they have the same symptoms that you have.
This Medication Guide summarizes the most important information about COPEGUS. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about COPEGUS that is written for healthcare professionals.
What are the ingredients in COPEGUS?
Active Ingredient: ribavirin
Inactive Ingredients: The core of the tablet contains corn starch, magnesium stearate, microcrystalline cellulose, pregelatinized starch, and sodium starch glycolate. The coating of the tablet contains ethyl cellulose (200 mg tablet only), hydroxypropylmethyl cellulose, red iron oxide, talc, titanium dioxide, triacetin, and yellow iron oxide.
This Medication Guide has been approved by the US Food and Drug Administration.
MG Revised: April 2009
Distributed by:
Roche Laboratories Inc.
340 Kingsland Street
Nutley, New Jersey 07110-1199
CST_209963_PI_02112008_N(1)
Copyright© 2002-2009 by Roche Laboratories Inc. All rights reserved.
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
PRINCIPAL DISPLAY PANEL - 200 mg Tablet Bottle
NDC 0004-0086-94
Copegus®
(ribavirin, USP)
Tablets
200 mg
Each tablet contains
200 mg ribavirin.
168 tablets
Rx only
AVOID PREGNANCY WHILE
TAKING THIS MEDICATION.
READ THE MEDICATION GUIDE
FOR IMPORTANT INFORMATION.
Roche
| COPEGUS
ribavirin tablet, film coated |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA021511 | 12/03/2002 | |
| Labeler - Hoffmann-La Roche Inc (002191211) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Hoffmann-La Roche Inc | 002191211 | MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Star Lake Bioscience Co Inc | 654457563 | API MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| F. Hoffmann-La Roche Ltd | 482242971 | API MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Patheon Inc. | 243790024 | MANUFACTURE | |