oxytrol (Oxybutynin) patch
[Watson Pharma]
DESCRIPTION
OXYTROL, oxybutynin transdermal system, is designed to deliver oxybutynin continuously and consistently over a 3- to 4-day interval after application to intact skin. OXYTROL is available as a 39 cm2 system containing 36 mg of oxybutynin. OXYTROL has a nominal in vivo delivery rate of 3.9 mg oxybutynin per day through skin of average permeability (interindividual variation in skin permeability is approximately 20%).
Oxybutynin is an antispasmodic, anticholinergic agent. Oxybutynin is administered as a racemate of R- and S-isomers. Chemically, oxybutynin is d, l (racemic) 4-diethylamino-2-butynyl phenylcyclohexylglycolate. The empirical formula of oxybutynin is C22H31NO3. Its structural formula is:
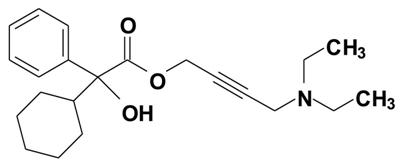
Oxybutynin is a white powder with a molecular weight of 357. It is soluble in alcohol, but relatively insoluble in water.
Transdermal System Components
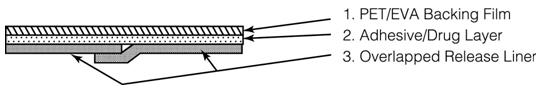
OXYTROL is a matrix-type transdermal system composed of three layers as illustrated in Figure 1 below. Layer 1 (Backing Film) is a thin flexible polyester/ethylene-vinyl acetate film that provides the matrix system with occlusivity and physical integrity and protects the adhesive/drug layer. Layer 2 (Adhesive/Drug Layer) is a cast film of acrylic adhesive containing oxybutynin and triacetin, USP. Layer 3 (Release Liner) is two overlapped siliconized polyester strips that are peeled off and discarded by the patient prior to applying the matrix system.
Figure 1: Side and top views of the OXYTROL system.
(Not to scale)
Side View
Top View
CLINICAL PHARMACOLOGY
The free base form of oxybutynin is pharmacologically equivalent to oxybutynin hydrochloride. Oxybutynin acts as a competitive antagonist of acetylcholine at postganglionic muscarinic receptors, resulting in relaxation of bladder smooth muscle. In patients with conditions characterized by involuntary detrusor contractions, cystometric studies have demonstrated that oxybutynin increases maximum urinary bladder capacity and increases the volume to first detrusor contraction. Oxybutynin thus decreases urinary urgency and the frequency of both incontinence episodes and voluntary urination.
Oxybutynin is a racemic (50:50) mixture of R- and S-isomers. Antimuscarinic activity resides predominantly in the R-isomer. The active metabolite, N-desethyloxybutynin, has pharmacological activity on the human detrusor muscle that is similar to that of oxybutynin in in vitro studies.
Pharmacokinetics
Absorption
Oxybutynin is transported across intact skin and into the systemic circulation by passive diffusion across the stratum corneum. The average daily dose of oxybutynin absorbed from the 39 cm2 OXYTROL system is 3.9 mg. The average (SD) nominal dose, 0.10 (0.02) mg oxybutynin per cm2 surface area, was obtained from analysis of residual oxybutynin content of systems worn over a continuous 4-day period during 303 separate occasions in 76 healthy volunteers. Following application of the first OXYTROL 3.9 mg/day system, oxybutynin plasma concentration increases for approximately 24 to 48 hours, reaching average maximum concentrations of 3 to 4 ng/mL. Thereafter, steady concentrations are maintained for up to 96 hours. Absorption of oxybutynin is bioequivalent when OXYTROL is applied to the abdomen, buttocks, or hip. Average plasma concentrations measured during a randomized, crossover study of the three recommended application sites in 24 healthy men and women are shown in Figure 2.
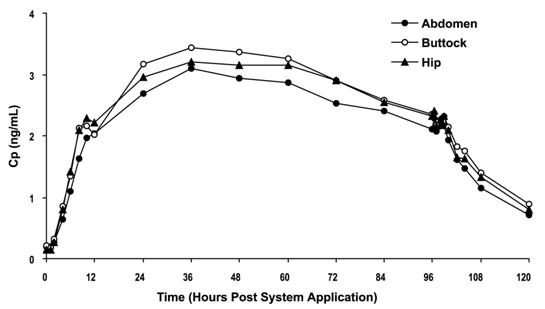
Figure 2: Average plasma oxybutynin concentrations (Cp) in 24 healthy male and female volunteers during single-dose application of OXYTROL 3.9 mg/day to the abdomen, buttock, and hip (System removal at 96 hours).
Steady-state conditions are reached during the second OXYTROL application. Average steady-state plasma concentrations were 3.1 ng/mL for oxybutynin and 3.8 ng/mL for N-desethyloxybutynin (Figure 3). Table 1 provides a summary of pharmacokinetic parameters of oxybutynin in healthy volunteers after single and multiple applications of OXYTROL.
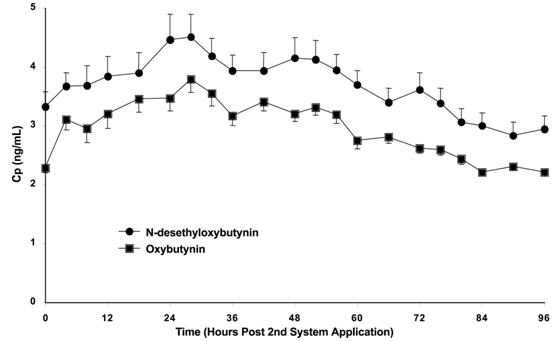
Figure 3: Average (SEM) steady-state oxybutynin and N-desethyloxybutynin plasma concentrations (Cp) measured in 13 healthy volunteers following the second transdermal system application in a multiple-dose, randomized, crossover study.
| Dosing | Oxybutynin | |||
|---|---|---|---|---|
| Cmax (SD) | Tmax1 | Cavg (SD) | AUC (SD) | |
| (ng/mL) | (hr) | (ng/mL) | (ng/mLxh) | |
| 1 Tmax given as median | ||||
| 2 AUCinf | ||||
| 3 AUC0-96 | ||||
| 4 AUC0-84 | ||||
| Single | 3.0 (0.8) | 48 | — | 245 (59) 2 |
| 3.4(1.1) | 36 | — | 279 (99) 2 | |
| Multiple | 6.6(2.4) | 10 | 4.2(1.1) | 408 (108) 3 |
| 4.2(1.0) | 28 | 3.1(0.7) | 259 (57) 4 | |
Distribution
Oxybutynin is widely distributed in body tissues following systemic absorption. The volume of distribution was estimated to be 193 L after intravenous administration of 5 mg oxybutynin chloride.
Metabolism
Oxybutynin is metabolized primarily by the cytochrome P450 enzyme systems, particularly CYP3A4, found mostly in the liver and gut wall. Metabolites include phenylcyclohexylglycolic acid, which is pharmacologically inactive, and N-desethyloxybutynin, which is pharmacologically active.
After oral administration of oxybutynin, pre-systemic first-pass metabolism results in an oral bioavailability of approximately 6% and higher plasma concentration of the N-desethyl metabolite compared to oxybutynin (see Figure 4). The plasma concentration AUC ratio of N-desethyl metabolite to parent compound following a single 5 mg oral dose of oxybutynin chloride was 11.9:1.
Transdermal administration of oxybutynin bypasses the first-pass gastrointestinal and hepatic metabolism, reducing the formation of the N-desethyl metabolite (see Figure 4). Only small amounts of CYP3A4 are found in skin, limiting pre-systemic metabolism during transdermal absorption. The resulting plasma concentration AUC ratio of N-desethyl metabolite to parent compound following multiple OXYTROL applications was 1.3:1.
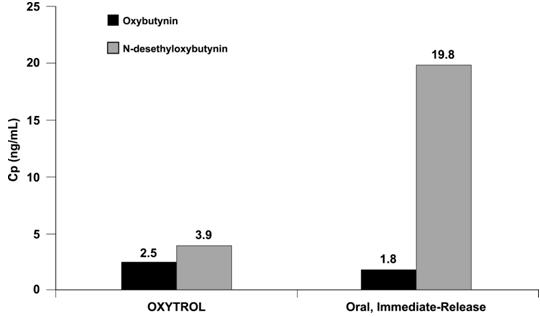
Figure 4: Average plasma concentrations (Cp) measured after a single, 96-hour application of the OXYTROL 3.9 mg/day system (AUCinf/96) and a single, 5 mg, oral immediate-release dose of oxybutynin chloride (AUCinf/8) in 16 healthy male and female volunteers.
Following intravenous administration, the elimination half-life of oxybutynin is approximately 2 hours. Following removal of OXYTROL, plasma concentrations of oxybutynin and N-desethyloxybutynin decline with an apparent half-life of approximately 7 to 8 hours.
Excretion
Oxybutynin is extensively metabolized by the liver, with less than 0.1% of the administered dose excreted unchanged in the urine. Also, less than 0.1% of the administered dose is excreted as the metabolite N-desethyloxybutynin.
Special Populations
Geriatric: The pharmacokinetics of oxybutynin and N-desethyloxybutynin were similar in all patients studied.
Pediatric: The pharmacokinetics of oxybutynin and N-desethyloxybutynin were not evaluated in individuals younger than 18 years of age. See PRECAUTIONS: Pediatric Use.
Gender: There were no significant differences in the pharmacokinetics of oxybutynin in healthy male and female volunteers following application of OXYTROL.
Race: Available data suggest that there are no significant differences in the pharmacokinetics of oxybutynin based on race in healthy volunteers following administration of OXYTROL. Japanese volunteers demonstrated a somewhat lower metabolism of oxybutynin to N-desethyloxybutynin compared to Caucasian volunteers.
Renal Insufficiency: There is no experience with the use of OXYTROL in patients with renal insufficiency.
Hepatic Insufficiency: There is no experience with the use of OXYTROL in patients with hepatic insufficiency.
Drug-Drug Interactions: See PRECAUTIONS: Drug Interactions.
Adhesion
Adhesion was periodically evaluated during the Phase 3 studies. Of the 4,746 OXYTROL evaluations in the Phase 3 trials, 20 (0.4%) were observed at clinic visits to have become completely detached and 35 (0.7%) became partially detached during routine clinic use. Similar to the pharmacokinetic studies, > 98% of the systems evaluated in the Phase 3 studies were assessed as being ≥ 75% attached and thus would be expected to perform as anticipated.
Clinical Studies
The efficacy and safety of OXYTROL were evaluated in patients with urge urinary incontinence in two Phase 3 controlled studies and one open-label extension. Study 1 was a Phase 3, placebo controlled study, comparing the safety and efficacy of OXYTROL at dose levels of 1.3, 2.6, and 3.9 mg/day to placebo in 520 patients. Open-label treatment was available for patients completing the study. Study 2 was a Phase 3 study, comparing the safety and efficacy of OXYTROL 3.9 mg/day versus active and placebo controls in 361 patients.
Study 1 was a randomized, double-blind, placebo-controlled, parallel group study of three dose levels of OXYTROL conducted in 520 patients. The 12-week double-blind treatment included OXYTROL doses of 1.3, 2.6, and 3.9 mg/day with matching placebo. An open-label, dose titration treatment extension allowed continued treatment for up to an additional 40 weeks for patients completing the double-blind period. The majority of patients were Caucasian (91%) and female (92%) with a mean age of 61 years (range, 20 to 88 years). Entry criteria required that patients have urge or mixed incontinence (with a predominance of urge), urge incontinence episodes of ≥ 10 per week, and ≥ 8 micturitions per day. The patient’s medical history and a urinary diary during the treatment-free baseline period confirmed the diagnosis of urge incontinence. Approximately 80% of patients had no prior pharmacological treatment for incontinence. Reductions in weekly incontinence episodes, urinary frequency, and urinary void volume between placebo and active treatment groups are summarized in Table 2.
| Parameter | Placebo | OXYTROL 3.9 mg/day | ||
|---|---|---|---|---|
| (N=127) | (N=120) | |||
| Mean (SD) | Median | Mean (SD) | Median | |
| *Comparison significant if p < 0.05 | ||||
| **Comparison significant if p ≤ 0.0167 | ||||
| Weekly Incontinence Episodes | ||||
| Baseline | 37.7 (24.0) | 30 | 34.3 (18.2) | 31 |
| Reduction | 19.2 (21.4) | 15 | 21.0 (17.1) | 19 |
| p value vs. placebo | — | 0.0265* | ||
| Daily Urinary Frequency | ||||
| Baseline | 12.3 (3.5) | 11 | 11.8 (3.1) | 11 |
| Reduction | 1.6 (3.0) | 1 | 2.2 (2.5) | 2 |
| p value vs. placebo | — | 0.0313* | ||
| Urinary Void Volume (mL) | ||||
| Baseline | 175.9 (69.5) | 166.5 | 171.6 (65.1) | 168 |
| Increase | 10.5 (56.9) | 5.5 | 31.6 (65.6) | 26 |
| p value vs. placebo | — | 0.0009** | ||
Study 2 was a randomized, double-blind, double-dummy, study of OXYTROL 3.9 mg/day versus active and placebo controls conducted in 361 patients. The 12-week double-blind treatment included an OXYTROL dose of 3.9 mg/day, an active comparator, and placebo. The majority of patients were Caucasian (95%) and female (93%) with a mean age of 64 years (range, 18 to 89 years). Entry criteria required that all patients have urge or mixed incontinence (with a predominance of urge) and had achieved a beneficial response from the anticholinergic treatment they were using at the time of study entry. The average duration of prior pharmacological treatment was greater than 2 years. The patient’s medical history and a urinary diary during the treatment-free baseline period confirmed the diagnosis of urge incontinence. Reductions in daily incontinence episodes, urinary frequency, and urinary void volume between placebo and active treatment groups are summarized in Table 3.
| Parameter | Placebo | OXYTROL 3.9 mg/day | ||
|---|---|---|---|---|
| (N=117) | (N=121) | |||
| Mean (SD) | Median | Mean (SD) | Median | |
| *Comparison significant if p < 0.05 | ||||
| Daily Incontinence Episodes | ||||
| Baseline | 5.0 (3.2) | 4 | 4.7 (2.9) | 4 |
| Reduction | 2.1 (3.0) | 2 | 2.9 (3.0) | 3 |
| p value vs. placebo | — | 0.0137* | ||
| Daily Urinary Frequency | ||||
| Baseline | 12.3 (3.3) | 12 | 12.4 (2.9) | 12 |
| Reduction | 1.4 (2.7) | 1 | 1.9 (2.7) | 2 |
| p value vs. placebo | — | 0.1010* | ||
| Urinary Void Volume (mL) | ||||
| Baseline | 175.0 (68.0) | 171.0 | 164.8 (62.3) | 160 |
| Increase | 9.3 (63.1) | 5.5 | 32.0 (55.2) | 24 |
| p value vs. placebo | — | 0.0010* | ||
INDICATIONS AND USAGE
OXYTROL is indicated for the treatment of overactive bladder with symptoms of urge urinary incontinence, urgency, and frequency.
CONTRAINDICATIONS
OXYTROL is contraindicated in patients with urinary retention, gastric retention, or uncontrolled narrow-angle glaucoma and in patients who are at risk for these conditions. OXYTROL is also contraindicated in patients who have demonstrated hypersensitivity to oxybutynin or other components of the product.
PRECAUTIONS
General
OXYTROL should be used with caution in patients with hepatic or renal impairment.
Urinary Retention: OXYTROL should be administered with caution to patients with clinically significant bladder outflow obstruction because of the risk of urinary retention (see CONTRAINDICATIONS).
Gastrointestinal Disorders: OXYTROL should be administered with caution to patients with gastrointestinal obstructive disorders because of the risk of gastric retention (see CONTRAINDICATIONS).
OXYTROL, like other anticholinergic drugs, may decrease gastrointestinal motility and should be used with caution in patients with conditions such as ulcerative colitis, intestinal atony, and myasthenia gravis. OXYTROL should be used with caution in patients who have gastroesophageal reflux and/or who are concurrently taking drugs (such as bisphosphonates) that can cause or exacerbate esophagitis.
Information for Patients
Patients should be informed that heat prostration (fever and heat stroke due to decreased sweating) can occur when anticholinergics such as oxybutynin are used in a hot environment. Because anticholinergic agents such as oxybutynin may produce drowsiness (somnolence) or blurred vision, patients should be advised to exercise caution. Patients should be informed that alcohol may enhance the drowsiness caused by anticholinergic agents such as oxybutynin.
OXYTROL should be applied to dry, intact skin on the abdomen, hip, or buttock. A new application site should be selected with each new system to avoid re-application to the same site within 7 days. Details on use of the system are explained in the patient information leaflet that should be dispensed with the product.
Drug Interactions
The concomitant use of oxybutynin with other anticholinergic drugs or with other agents that produce dry mouth, constipation, somnolence, and/or other anticholinergic-like effects may increase the frequency and/or severity of such effects. Anticholinergic agents may potentially alter the absorption of some concomitantly administered drugs due to anticholinergic effects on gastrointestinal motility. Pharmacokinetic studies have not been performed with patients concomitantly receiving cytochrome P450 enzyme inhibitors, such as antimycotic agents (e.g. ketoconazole, itraconazole, and miconazole) or macrolide antibiotics (e.g. erythromycin and clarithromycin). No specific drug-drug interaction studies have been performed with OXYTROL.
Carcinogenesis, Mutagenesis, Impairment of Fertility
A 24-month study in rats at dosages of oxybutynin chloride of 20, 80 and 160 mg/kg showed no evidence of carcinogenicity. These doses are approximately 6, 25 and 50 times the maximum exposure in humans taking an oral dose based on body surface area.
Oxybutynin chloride showed no increase of mutagenic activity when tested in Schizosaccharomyces pompholiciformis, Saccharomyces cerevisiae, and Salmonella typhimurium test systems. Reproduction studies with oxybutynin chloride in the mouse, rat, hamster, and rabbit showed no definite evidence of impaired fertility.
Pregnancy
Teratogenic Effects
Pregnancy Category B
Reproduction studies with oxybutynin chloride in the mouse, rat, hamster, and rabbit showed no definite evidence of impaired fertility or harm to the animal fetus. Subcutaneous administration to rats at doses up to 25 mg/kg (approximately 50 times the human exposure based on surface area) and to rabbits at doses up to 0.4 mg/kg (approximately 1 times the human exposure) revealed no evidence of harm to the fetus due to oxybutynin chloride. The safety of OXYTROL administration to women who are or who may become pregnant has not been established. Therefore, OXYTROL should not be given to pregnant women unless, in the judgment of the physician, the probable clinical benefits outweigh the possible hazards.
Nursing Mothers
It is not known whether oxybutynin is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when OXYTROL is administered to a nursing woman.
Pediatric Use
The safety and efficacy of OXYTROL in pediatric patients have not been established.
Geriatric Use
Of the total number of patients in the clinical studies of OXYTROL, 49% were 65 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in response between elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out (see CLINICAL PHARMACOLOGY, Pharmacokinetics, Special Populations: Geriatric).
ADVERSE REACTIONS
The safety of OXYTROL was evaluated in a total of 417 patients who participated in two Phase 3 clinical efficacy and safety studies and an open-label extension. Additional safety information was collected in Phase 1 and Phase 2 trials. In the two pivotal studies, a total of 246 patients received OXYTROL during the 12-week treatment periods. A total of 411 patients entered the open-label extension and of those, 65 patients and 52 patients received OXYTROL for at least 24 weeks and at least 36 weeks, respectively.
No deaths were reported during treatment. No serious adverse events related to treatment were reported.
Adverse events reported in the pivotal trials are summarized in Tables 4 and 5 below.
| Adverse Event* | Placebo | OXYTROL(3.9 mg/day) | |||
|---|---|---|---|---|---|
| (N=132) | (N=125) | ||||
| N | % | N | % | ||
| *includes adverse events judged by the investigator as possibly, probably or definitely treatment-related. | |||||
| Application site pruritus | 8 | 6.1% | 21 | 16.8% | |
| Dry mouth | 11 | 8.3% | 12 | 9.6% | |
| Application site erythema | 3 | 2.3% | 7 | 5.6% | |
| Application site vesicles | 0 | 0.0% | 4 | 3.2% | |
| Diarrhea | 3 | 2.3% | 4 | 3.2% | |
| Dysuria | 0 | 0.0% | 3 | 2.4% | |
| Adverse Event* | Placebo | OXYTROL (3.9 mg/day) | ||
|---|---|---|---|---|
| (N=117) | (N=121) | |||
| N | % | N | % | |
| *includes adverse events judged by the investigator as possibly, probably or definitely treatment-related. | ||||
| Application site pruritus | 5 | 4.3% | 17 | 14.0% |
| Application site erythema | 2 | 1.7% | 10 | 8.3% |
| Dry mouth | 2 | 1.7% | 5 | 4.1% |
| Constipation | 0 | 0.0% | 4 | 3.3% |
| Application site rash | 1 | 0.9% | 4 | 3.3% |
| Application site macules | 0 | 0.0% | 3 | 2.5% |
| Abnormal vision | 0 | 0.0% | 3 | 2.5% |
Other adverse events reported by > 1% of OXYTROL-treated patients, and judged by the investigator to be possibly, probably or definitely related to treatment include: abdominal pain, nausea, flatulence, fatigue, somnolence, headache, flushing, rash, application site burning and back pain.
Most treatment-related adverse events were described as mild or moderate in intensity. Severe application site reactions were reported by 6.4% of OXYTROL-treated patients in Study 1 and by 5.0% of OXYTROL-treated patients in Study 2.
Treatment-related adverse events that resulted in discontinuation were reported by 11.2% of OXYTROL-treated patients in Study 1 and 10.7% of OXYTROL-treated patients in Study 2. Most of these were secondary to application site reaction. In the two pivotal studies, no patient discontinued OXYTROL treatment due to dry mouth.
In the open-label extension, the most common treatment-related adverse events were: application site pruritus, application site erythema and dry mouth.
OVERDOSAGE
Plasma concentration of oxybutynin declines within 1 to 2 hours after removal of transdermal system(s). Patients should be monitored until symptoms resolve. Overdosage with oxybutynin has been associated with anticholinergic effects including CNS excitation, flushing, fever, dehydration, cardiac arrhythmia, vomiting, and urinary retention. Ingestion of 100 mg oral oxybutynin chloride in association with alcohol has been reported in a 13 year old boy who experienced memory loss, and in a 34 year old woman who developed stupor, followed by disorientation and agitation on awakening, dilated pupils, dry skin, cardiac arrhythmia, and retention of urine. Both patients recovered fully with symptomatic treatment.
DOSAGE AND ADMINISTRATION
OXYTROL should be applied to dry, intact skin on the abdomen, hip, or buttock. A new application site should be selected with each new system to avoid re-application to the same site within 7 days.
The dose of OXYTROL is one 3.9 mg/day system applied twice weekly (every 3 to 4 days).
HOW SUPPLIED
OXYTROL 3.9 mg/day (oxybutynin transdermal system). Each 39 cm2 system imprinted with OXYTROL 3.9 mg/day contains 36 mg oxybutynin for nominal delivery of 3.9 mg oxybutynin per day when dosed in a twice weekly regimen.
NDC 52544-920-08 Patient Calendar Box of 8 Systems
Storage
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F). Protect from moisture and humidity. Do not store outside the sealed pouch. Apply immediately after removal from the protective pouch. Discard used OXYTROL in household trash in a manner that prevents accidental application or ingestion by children, pets, or others.
Rx only

A subsidiary of Watson Pharmaceuticals, Inc.
Corona, CA 92880 USA
U.S. Patent Nos. 5,601,839 and 5,834,010
| OXYTROL (Oxybutynin) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
Revised: 05/2006