INVANZ
-
ertapenem sodium injection, powder, lyophilized, for solution
Merck & Co., Inc.
----------
INVANZ®(ERTAPENEM FOR INJECTION)
To reduce the development of drug-resistant bacteria and maintain the effectiveness of INVANZ and other antibacterial drugs, INVANZ should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria.
For Intravenous or Intramuscular Use
DESCRIPTION
INVANZ1 (Ertapenem for Injection) is a sterile, synthetic, parenteral, 1-β methyl-carbapenem that is structurally related to beta-lactam antibiotics.
Chemically, INVANZ is described as [4R-[3(3S *,5S*),4α,5β,6β(R *)]]-3-[[5-[[(3- carboxyphenyl)amino]carbonyl]-3-pyrrolidinyl]thio]-6-(1-hydroxyethyl)-4-methyl-7-oxo-1- azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid monosodium salt. Its molecular weight is 497.50. The empirical formula is C22H24N3O7SNa, and its structural formula is:
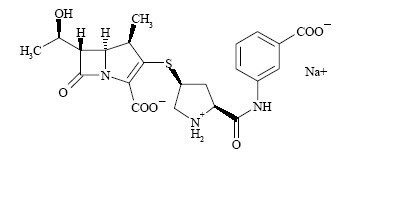
Ertapenem sodium is a white to off-white hygroscopic, weakly crystalline powder. It is soluble in water and 0.9% sodium chloride solution, practically insoluble in ethanol, and insoluble in isopropyl acetate and tetrahydrofuran.
INVANZ is supplied as sterile lyophilized powder for intravenous infusion after reconstitution with appropriate diluent (see DOSAGE AND ADMINISTRATION, PREPARATION OF SOLUTION) and transfer to 50 mL 0.9% Sodium Chloride Injection or for intramuscular injection following reconstitution with 1% lidocaine hydrochloride. Each vial contains 1.046 grams ertapenem sodium, equivalent to 1 gram ertapenem. The sodium content is approximately 137 mg (approximately 6.0 mEq).
Each vial of INVANZ contains the following inactive ingredients: 175 mg sodium bicarbonate and sodium hydroxide to adjust pH to 7.5.
- 1
-
Registered trademark of MERCK & CO., Inc. COPYRIGHT © 2001, 2003-2007 MERCK & CO., Inc. All rights reserved
CLINICAL PHARMACOLOGY
Pharmacokinetics
Average plasma concentrations (mcg/mL) of ertapenem following a single 30-minute infusion of a 1 g intravenous (IV) dose and administration of a single 1 g intramuscular (IM) dose in healthy young adults are presented in Table 1.
| Average Plasma Concentrations (mcg/mL) | |||||||||
| Dose/Route | 0.5 hr | 1 hr | 2 hr | 4 hr | 6 hr | 8 hr | 12 hr | 18 hr | 24 hr |
|
|||||||||
| 1 g IV* | 155 | 115 | 83 | 48 | 31 | 20 | 9 | 3 | 1 |
| 1 g IM | 33 | 53 | 67 | 57 | 40 | 27 | 13 | 4 | 2 |
The area under the plasma concentration-time curve (AUC) of ertapenem in adults increased less-than dose-proportional based on total ertapenem concentrations over the 0.5 to 2 g dose range, whereas the AUC increased greater-than dose-proportional based on unbound ertapenem concentrations. Ertapenem exhibits non-linear pharmacokinetics due to concentration-dependent plasma protein binding at the proposed therapeutic dose. (See CLINICAL PHARMACOLOGY, Distribution.)
There is no accumulation of ertapenem following multiple IV or IM 1 g daily doses in healthy adults.
Average plasma concentrations (mcg/mL) of ertapenem in pediatric patients are presented in Table 2.
| Age Group | Dose | Average Plasma Concentrations (mcg/mL) | |||||||
| 0.5 hr | 1 hr | 2 hr | 4 hr | 6 hr | 8 hr | 12 hr | 24 hr | ||
| 3 to 23 months | 15 mg/kg† | 103.8 | 57.3 | 43.6 | 23.7 | 13.5 | 8.2 | 2.5 | - |
| 20 mg/kg† | 126.8 | 87.6 | 58.7 | 28.4 | - | 12.0 | 3.4 | 0.4 | |
| 40 mg/kg‡ | 199.1 | 144.1 | 95.7 | 58.0 | - | 20.2 | 7.7 | 0.6 | |
| 2 to 12 years | 15 mg/kg† | 113.2 | 63.9 | 42.1 | 21.9 | 12.8 | 7.6 | 3.0 | - |
| 20 mg/kg† | 147.6 | 97.6 | 63.2 | 34.5 | - | 12.3 | 4.9 | 0.5 | |
| 40 mg/kg‡ | 241.7 | 152.7 | 96.3 | 55.6 | - | 18.8 | 7.2 | 0.6 | |
| 13 to 17 years | 20 mg/kg† | 170.4 | 98.3 | 67.8 | 40.4 | - | 16.0 | 7.0 | 1.1 |
| 1 g§ | 155.9 | 110.9 | 74.8 | - | 24.0 | - | 6.2 | - | |
| 40 mg/kg‡ | 255.0 | 188.7 | 127.9 | 76.2 | - | 31.0 | 15.3 | 2.1 | |
Absorption
Ertapenem, reconstituted with 1% lidocaine HCl injection, USP (in saline without epinephrine), is almost completely absorbed following intramuscular (IM) administration at the recommended dose of 1 g. The mean bioavailability is approximately 90%. Following 1 g daily IM administration, mean peak plasma concentrations (Cmax) are achieved in approximately 2.3 hours (Tmax).
Distribution
Ertapenem is highly bound to human plasma proteins, primarily albumin. In healthy young adults, the protein binding of ertapenem decreases as plasma concentrations increase, from approximately 95% bound at an approximate plasma concentration of <100 micrograms (mcg)/mL to approximately 85% bound at an approximate plasma concentration of 300 mcg/mL.
The apparent volume of distribution at steady state (Vss) of ertapenem in adults is approximately 0.12 liter/kg, approximately 0.2 liter/kg in pediatric patients 3 months to 12 years of age and approximately 0.16 liter/kg in pediatric patients 13 to 17 years of age.
The concentrations of ertapenem achieved in suction-induced skin blister fluid at each sampling point on the third day of 1 g once daily IV doses are presented in Table 3. The ratio of AUC0-24 in skin blister fluid/AUC0-24 in plasma is 0.61.
| 0.5 hr | 1 hr | 2 hr | 4 hr | 8 hr | 12 hr | 24 hr |
| 7 | 12 | 17 | 24 | 24 | 21 | 8 |
The concentration of ertapenem in breast milk from 5 lactating women with pelvic infections (5 to 14 days postpartum) was measured at random time points daily for 5 consecutive days following the last 1 g dose of intravenous therapy (3-10 days of therapy). The concentration of ertapenem in breast milk within 24 hours of the last dose of therapy in all 5 women ranged from <0.13 (lower limit of quantitation) to 0.38 mcg/mL; peak concentrations were not assessed. By day 5 after discontinuation of therapy, the level of ertapenem was undetectable in the breast milk of 4 women and below the lower limit of quantitation (<0.13 mcg/mL) in 1 woman.
Metabolism
In healthy young adults, after infusion of 1 g IV radiolabeled ertapenem, the plasma radioactivity consists predominantly (94%) of ertapenem. The major metabolite of ertapenem is the inactive ring-opened derivative formed by hydrolysis of the beta-lactam ring.
In vitro studies in human liver microsomes indicate that ertapenem does not inhibit metabolism mediated by any of the following cytochrome p450 (CYP) isoforms: 1A2, 2C9, 2C19, 2D6, 2E1 and 3A4. (See PRECAUTIONS, Drug Interactions.)
In vitro studies indicate that ertapenem does not inhibit P-glycoprotein-mediated transport of digoxin or vinblastine and that ertapenem is not a substrate for P-glycoprotein-mediated transport. (See PRECAUTIONS, Drug Interactions.)
Elimination
Ertapenem is eliminated primarily by the kidneys. The mean plasma half-life in healthy young adults is approximately 4 hours and the plasma clearance is approximately 1.8 L/hour. The mean plasma half-life in pediatric patients 13 to 17 years of age is approximately 4 hours and approximately 2.5 hours in pediatric patients 3 months to 12 years of age.
Following the administration of 1 g IV radiolabeled ertapenem to healthy young adults, approximately 80% is recovered in urine and 10% in feces. Of the 80% recovered in urine, approximately 38% is excreted as unchanged drug and approximately 37% as the ring-opened metabolite.
In healthy young adults given a 1 g IV dose, the mean percentage of the administered dose excreted in urine was 17.4% during 0-2 hours postdose, 5.4% during 4-6 hours postdose, and 2.4% during 12-24 hours postdose.
Special Populations
Renal Insufficiency
Total and unbound fractions of ertapenem pharmacokinetics were investigated in 26 adult subjects (31 to 80 years of age) with varying degrees of renal impairment. Following a single 1 g IV dose of ertapenem, the unbound AUC increased 1.5-fold and 2.3-fold in subjects with mild renal insufficiency (CLCR 60-90 mL/min/1.73 m2) and moderate renal insufficiency (CLCR 31-59 mL/min/1.73 m2), respectively, compared with healthy young subjects (25 to 45 years of age). No dosage adjustment is necessary in patients with CLCR≥31 mL/min/1.73 m2. The unbound AUC increased 4.4-fold and 7.6-fold in subjects with advanced renal insufficiency (CLCR 5-30 mL/min/1.73 m2) and end-stage renal insufficiency (CLCR<10 mL/min/1.73 m2), respectively, compared with healthy young subjects. The effects of renal insufficiency on AUC of total drug were of smaller magnitude. The recommended dose of ertapenem in adult patients with CLCR≤30 mL/min/1.73 m2 is 0.5 grams every 24 hours. Following a single 1 g IV dose given immediately prior to a 4 hour hemodialysis session in 5 adult patients with end-stage renal insufficiency, approximately 30% of the dose was recovered in the dialysate. A supplementary dose of 150 mg is recommended if ertapenem is administered within 6 hours prior to hemodialysis. (See DOSAGE AND ADMINISTRATION.) There are no data in pediatric patients with renal insufficiency.
Hepatic Insufficiency
The pharmacokinetics of ertapenem in patients with hepatic insufficiency have not been established. However, ertapenem does not appear to undergo hepatic metabolism based on in vitro studies and approximately 10% of an administered dose is recovered in the feces. (See PRECAUTIONS and DOSAGE AND ADMINISTRATION.)
Gender
The effect of gender on the pharmacokinetics of ertapenem was evaluated in healthy male (n=8) and healthy female (n=8) subjects. The differences observed could be attributed to body size when body weight was taken into consideration. No dose adjustment is recommended based on gender.
Geriatric Patients
The impact of age on the pharmacokinetics of ertapenem was evaluated in healthy male (n=7) and healthy female (n=7) subjects ≥65 years of age. The total and unbound AUC increased 37% and 67%, respectively, in elderly adults relative to young adults. These changes were attributed to age-related changes in creatinine clearance. No dosage adjustment is necessary for elderly patients with normal (for their age) renal function.
Pediatric Patients
Plasma concentrations of ertapenem are comparable in pediatric patients 13 to 17 years of age and adults following a 1 g once daily IV dose.
Following the 20 mg/kg dose (up to a maximum dose of 1 g), the pharmacokinetic parameter values in patients 13 to 17 years of age (N=6) were generally comparable to those in healthy young adults.
Plasma concentrations at the midpoint of the dosing interval following a single 15 mg/kg IV dose of ertapenem in patients 3 months to 12 years of age are comparable to plasma concentrations at the midpoint of the dosing interval following a 1 g once daily IV dose in adults (see Pharmacokinetics.) The plasma clearance (mL/min/kg) of ertapenem in patients 3 months to 12 years of age is approximately 2-fold higher as compared to that in adults. At the 15 mg/kg dose, the AUC value (doubled to model a twice daily dosing regimen, i.e., 30 mg/kg/day exposure) in patients 3 months to 12 years of age was comparable to the AUC value in young healthy adults receiving a 1 g IV dose of ertapenem.
Microbiology
Ertapenem has in vitro activity against gram-positive and gram-negative aerobic and anaerobic bacteria. The bactericidal activity of ertapenem results from the inhibition of cell wall synthesis and is mediated through ertapenem binding to penicillin binding proteins (PBPs). In Escherichia coli, it has strong affinity toward PBPs 1a, 1b, 2, 3, 4 and 5 with preference for PBPs 2 and 3. Ertapenem is stable against hydrolysis by a variety of beta-lactamases, including penicillinases, and cephalosporinases and extended spectrum beta-lactamases. Ertapenem is hydrolyzed by metallo-beta-lactamases.
Ertapenem has been shown to be active against most isolates of the following microorganisms in vitro and in clinical infections. (See INDICATIONS AND USAGE):
Aerobic and facultative gram-positive microorganisms:
Staphylococcus aureus (methicillin susceptible isolates only)
Streptococcus agalactiae
Streptococcus pneumoniae (penicillin susceptible isolates only)
Streptococcus pyogenes
Note: Methicillin-resistant staphylococci and Enterococcus spp. are resistant to ertapenem.
Aerobic and facultative gram-negative microorganisms:
Escherichia coli
Haemophilus influenzae (Beta-lactamase negative isolates only)
Klebsiella pneumoniae
Moraxella catarrhalis
Proteus mirabilis
Anaerobic microorganisms:
Bacteroides fragilis
Bacteroides distasonis
Bacteroides ovatus
Bacteroides thetaiotaomicron
Bacteroides uniformis
Clostridium clostridioforme
Eubacterium lentum
Peptostreptococcus species
Porphyromonas asaccharolytica
Prevotella bivia
The following in vitro data are available, but their clinical significance is unknown.
At least 90% of the following microorganisms exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for ertapenem; however, the safety and effectiveness of ertapenem in treating clinical infections due to these microorganisms have not been established in adequate and well-controlled clinical studies:
Aerobic and facultative gram-positive microorganisms:
Staphylococcus epidermidis (methicillin susceptible isolates only)
Streptococcus pneumoniae (penicillin-intermediate isolates only)
Aerobic and facultative gram-negative microorganisms:
Citrobacter freundii
Citrobacter koseri
Enterobacter aerogenes
Enterobacter cloacae
Haemophilus influenzae (Beta-lactamase positive isolates)
Haemophilus parainfluenzae
Klebsiella oxytoca (excluding ESBL producing isolates)
Morganella morganii
Proteus vulgaris
Providencia rettgeri
Providencia stuartii
Serratia marcescens
Anaerobic microorganisms:
Bacteroides vulgatus
Clostridium perfringens
Fusobacterium spp.
Susceptibility Test Methods:
When available, the results of in vitro susceptibility tests should be provided to the physician as periodic reports which describe the susceptibility profile of nosocomial and community-acquired pathogens. These reports should aid the physician in selecting the most effective antimicrobial.
Dilution Techniques:
Quantitative methods are used to determine antimicrobial minimum inhibitory concentrations (MICs). These MICs provide estimates of the susceptibility of bacteria to antimicrobial compounds. The MICs should be determined using a standardized procedure. Standardized procedures are based on a broth dilution method(1,2) or equivalent with standardized inoculum concentrations and standardized concentrations of ertapenem powder. The MIC values should be interpreted according to criteria provided in Table 4.
Diffusion Techniques:
Quantitative methods that require measurement of zone diameters also provide reproducible estimates of the susceptibility of bacteria to antimicrobial compounds. One such standardized procedure(2,3) requires the use of standardized inoculum concentrations. This procedure uses paper disks impregnated with 10-µg ertapenem to test the susceptibility of microorganisms to ertapenem. The disk diffusion interpretive criteria should be interpreted according to criteria provided in Table 4.
Anaerobic Techniques:
For anaerobic bacteria, the susceptibility to ertapenem as MICs can be determined by standardized test methods(4). The MIC values obtained should be interpreted according to criteria provided in Table 4.
| Pathogen | Minimum Inhibitory Concentrations*
MIC (μg/mL) | Disk Diffusion*
Zone Diameter (mm) |
||||
| S | I | R | S | I | R | |
|
||||||
|
Enterobacteriaceae and Staphylococcus spp. | ≤2.0 | 4.0 | ≥8.0 | ≥19 | 16-18 | ≤15 |
| Haemophilus spp. | ≤0.5 | - | - | ≥19 | - | - |
| Streptococcus pneumoniae†,‡ | ≤1.0 | - | - | ≥19 | - | - |
| Streptococcus spp. other than Streptococcus pneumoniae§,¶ | ≤1.0 | - | - | ≥19 | - | - |
| Anaerobes | ≤4.0 | 8.0 | ≥16.0 | - | - | - |
Note: Staphylococcus spp. can be considered susceptible to ertapenem if the penicillin MIC is ≤0.12 µg/mL. If the penicillin MIC is >0.12 µg/mL, then test oxacillin. Staphylococcus aureus can be considered susceptible to ertapenem if the oxacillin MIC is ≤2.0 µg/mL and resistant to ertapenem if the oxacillin MIC is ≥4.0 µg/mL. Coagulase negative staphylococci can be considered susceptible to ertapenem if the oxacillin MIC is ≤0.25 µg/mL and resistant to ertapenem if the oxacillin MIC ≥0.5 µg/mL.
Staphylococcus spp. can be considered susceptible to ertapenem if the penicillin (10 U disk) zone is ≥29 mm. If the penicillin zone is ≤28 mm, then test oxacillin by disk diffusion (1 µg disk). Staphylococcus aureus can be considered susceptible to ertapenem if the oxacillin (1 µg disk) zone is ≥13 mm and resistant to ertapenem if the oxacillin zone is ≤10 mm. Coagulase negative staphylococci can be considered susceptible to ertapenem if the oxacillin zone is ≥18 mm and resistant to ertapenem if the oxacillin (1 µg disk) zone is ≤17 mm.
A report of “Susceptible” indicates that the pathogen is likely to be inhibited if the antimicrobial compound in blood reaches the concentrations usually achievable. A report of “Intermediate” indicates that the result should be considered equivocal, and, if the microorganism is not fully susceptible to alternative, clinically feasible drugs, the test should be repeated. This category implies possible clinical applicability in body sites where the drug is physiologically concentrated or in situations where high dosage of drug can be used. This category also provides a buffer zone which prevents small uncontrolled technical factors from causing major discrepancies in interpretation. A report of “Resistant” indicates that the pathogen is not likely to be inhibited if the antimicrobial compound in the blood reaches the concentrations usually achievable; other therapy should be selected.
Quality Control
Standardized susceptibility test procedures require the use of laboratory control microorganisms to control the technical aspects of the laboratory procedures (1,2,3,4). Quality control microorganisms are specific strains of organisms with intrinsic biological properties. QC strains are very stable strains which will give a standard and repeatable susceptibility pattern. The specific strains used for microbiological quality control are not clinically significant. Standard ertapenem powder should provide the following range of values noted in Table 5.
| Microorganism | Minimum Inhibitory Concentrations MIC Range (µg/mL) | Disk Diffusion Zone Diameter (mm) |
| Escherichia coli ATCC 25922 | 0.004-0.016 | 29-36 |
| Haemophilus influenzae ATCC 49766 | 0.016-0.06 | 27-33 |
| Staphylococcus aureus ATCC 29213 | 0.06-0.25 | - |
| Staphylococcus aureus ATCC 25923 | - | 24-31 |
| Streptococcus pneumoniae ATCC 49619 | 0.03-0.25 | 28-35 |
| Bacteroides fragilis ATCC 25285 |
0.06-0.5 * 0.06-0.25† | - |
| Bacteroides thetaiotaomicron ATCC 29741 |
0.5-2.0 * 0.25-1.0 † | - |
| Eubacterium lentum ATCC 43055 |
0.5-4.0 * 0.5-2.0 † | - |
INDICATIONS AND USAGE
Treatment
INVANZ is indicated for the treatment of patients with the following moderate to severe infections caused by susceptible isolates of the designated microorganisms. (See DOSAGE AND ADMINISTRATION):
Complicated Intra-abdominal Infections due to Escherichia coli, Clostridium clostridioforme, Eubacterium lentum, Peptostreptococcus species, Bacteroides fragilis, Bacteroides distasonis, Bacteroides ovatus, Bacteroides thetaiotaomicron, or Bacteroides uniformis.
Complicated Skin and Skin Structure Infections, including diabetic foot infections without osteomyelitis due to Staphylococcus aureus (methicillin susceptible isolates only), Streptococcus agalactiae, Streptococcus pyogenes, Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, Bacteroides fragilis, Peptostreptococcus species, Porphyromonas asaccharolytica, or Prevotella bivia. INVANZ has not been studied in diabetic foot infections with concomitant osteomyelitis (see CLINICAL STUDIES).
Community Acquired Pneumonia due to Streptococcus pneumoniae (penicillin susceptible isolates only) including cases with concurrent bacteremia, Haemophilus influenzae (beta-lactamase negative isolates only), or Moraxella catarrhalis.
Complicated Urinary Tract Infections including pyelonephritis due to Escherichia coli, including cases with concurrent bacteremia, or Klebsiella pneumoniae.
Acute Pelvic Infections including postpartum endomyometritis, septic abortion and post surgical gynecologic infections due to Streptococcus agalactiae, Escherichia coli, Bacteroides fragilis, Porphyromonas asaccharolytica, Peptostreptococcus species, or Prevotella bivia.
Prevention
INVANZ is indicated in adults for the prophylaxis of surgical site infection following elective colorectal surgery.
Appropriate specimens for bacteriological examination should be obtained in order to isolate and identify the causative organisms and to determine their susceptibility to ertapenem. Therapy with INVANZ (ertapenem) may be initiated empirically before results of these tests are known; once results become available, antimicrobial therapy should be adjusted accordingly.
To reduce the development of drug-resistant bacteria and maintain the effectiveness of INVANZ and other antibacterial drugs, INVANZ should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
CONTRAINDICATIONS
INVANZ is contraindicated in patients with known hypersensitivity to any component of this product or to other drugs in the same class or in patients who have demonstrated anaphylactic reactions to beta-lactams.
Due to the use of lidocaine HCl as a diluent, INVANZ administered intramuscularly is contraindicated in patients with a known hypersensitivity to local anesthetics of the amide type. (Refer to the prescribing information for lidocaine HCl.)
WARNINGS
SERIOUS AND OCCASIONALLY FATAL HYPERSENSITIVITY (ANAPHYLACTIC) REACTIONS HAVE BEEN REPORTED IN PATIENTS RECEIVING THERAPY WITH BETA-LACTAMS. THESE REACTIONS ARE MORE LIKELY TO OCCUR IN INDIVIDUALS WITH A HISTORY OF SENSITIVITY TO MULTIPLE ALLERGENS. THERE HAVE BEEN REPORTS OF INDIVIDUALS WITH A HISTORY OF PENICILLIN HYPERSENSITIVITY WHO HAVE EXPERIENCED SEVERE HYPERSENSITIVITY REACTIONS WHEN TREATED WITH ANOTHER BETA-LACTAM. BEFORE INITIATING THERAPY WITH INVANZ, CAREFUL INQUIRY SHOULD BE MADE CONCERNING PREVIOUS HYPERSENSITIVITY REACTIONS TO PENICILLINS, CEPHALOSPORINS, OTHER BETA-LACTAMS AND OTHER ALLERGENS. IF AN ALLERGIC REACTION TO INVANZ OCCURS, DISCONTINUE THE DRUG IMMEDIATELY. SERIOUS ANAPHYLACTIC REACTIONS REQUIRE IMMEDIATE EMERGENCY TREATMENT WITH EPINEPHRINE, OXYGEN, INTRAVENOUS STEROIDS, AND AIRWAY MANAGEMENT, INCLUDING INTUBATION. OTHER THERAPY MAY ALSO BE ADMINISTERED AS INDICATED.
Seizure Potential
Seizures and other CNS adverse experiences have been reported during treatment with INVANZ. (See PRECAUTIONS and ADVERSE REACTIONS.)
Carbapenems, including ertapenem, may reduce serum valproic acid concentrations to subtherapeutic levels, resulting in loss of seizure control. Serum valproic acid concentrations should be monitored frequently after initiating carbapenem therapy. Alternative antibacterial or anticonvulsant therapy should be considered if serum valproic acid concentrations drop below the therapeutic range or a seizure occurs. (See PRECAUTIONS, Drug Interactions.)
Clostridium difficile associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including ertapenem, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of Clostridium difficile.
Clostridium difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of Clostridium difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibiotic use not directed against Clostridium difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of Clostridium difficile, and surgical evaluation should be instituted as clinically indicated.
Lidocaine HCl is the diluent for intramuscular administration of INVANZ. Refer to the prescribing information for lidocaine HCl.
PRECAUTIONS
General
During clinical investigations in adult patients treated with INVANZ (1 g once a day), seizures, irrespective of drug relationship, occurred in 0.5% of patients during study therapy plus 14-day follow-up period. (See ADVERSE REACTIONS.) These experiences have occurred most commonly in patients with CNS disorders (e.g., brain lesions or history of seizures) and/or compromised renal function. Close adherence to the recommended dosage regimen is urged, especially in patients with known factors that predispose to convulsive activity. Anticonvulsant therapy should be continued in patients with known seizure disorders. If focal tremors, myoclonus, or seizures occur, patients should be evaluated neurologically, placed on anticonvulsant therapy if not already instituted, and the dosage of INVANZ reexamined to determine whether it should be decreased or the antibiotic discontinued. Dosage adjustment of INVANZ is recommended in patients with reduced renal function. (See DOSAGE AND ADMINISTRATION.)
As with other antibiotics, prolonged use of INVANZ may result in overgrowth of non-susceptible organisms. Repeated evaluation of the patient's condition is essential. If superinfection occurs during therapy, appropriate measures should be taken.
Prescribing INVANZ in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
Caution should be taken when administering INVANZ intramuscularly to avoid inadvertent injection into a blood vessel. (See DOSAGE AND ADMINISTRATION.)
Lidocaine HCl is the diluent for intramuscular administration of INVANZ. Refer to the prescribing information for lidocaine HCl for additional precautions.
Information for Patients
Patients should be counseled that antibacterial drugs including INVANZ should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When INVANZ is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by INVANZ or other antibacterial drugs in the future.
Diarrhea is a common problem caused by antibiotics which usually ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, patients should contact their physician as soon as possible.
Laboratory Tests
While INVANZ possesses toxicity similar to the beta-lactam group of antibiotics, periodic assessment of organ system function, including renal, hepatic, and hematopoietic, is advisable during prolonged therapy.
Drug Interactions
When ertapenem is co-administered with probenecid (500 mg p.o. every 6 hours), probenecid competes for active tubular secretion and reduces the renal clearance of ertapenem. Based on total ertapenem concentrations, probenecid increased the AUC by 25% and reduced the plasma and renal clearances by 20% and 35%, respectively. The half-life increased from 4.0 to 4.8 hours. Because of the small effect on half-life, the coadministration with probenecid to extend the half-life of ertapenem is not recommended.
In vitro studies indicate that ertapenem does not inhibit P-glycoprotein-mediated transport of digoxin or vinblastine and that ertapenem is not a substrate for P-glycoprotein-mediated transport. In vitro studies in human liver microsomes indicate that ertapenem does not inhibit metabolism mediated by any of the following six cytochrome p450 (CYP) isoforms: 1A2, 2C9, 2C19, 2D6, 2E1 and 3A4. Drug interactions caused by inhibition of P-glycoprotein-mediated drug clearance or CYP-mediated drug clearance with the listed isoforms are unlikely. (See CLINICAL PHARMACOLOGY, Distribution and Metabolism.)
Other than with probenecid, no specific clinical drug interaction studies have been conducted.
A clinically significant reduction in serum valproic acid concentration has been reported in patients receiving carbapenem antibiotics and may result in loss of seizure control. Although the mechanism of this interaction is not fully understood, data from in vitro and animal studies suggest that carbapenem antibiotics may inhibit valproic acid glucuronide hydrolysis. Serum valproic acid concentrations should be monitored frequently after initiating carbapenem therapy. Alternative antibacterial or anticonvulsant therapy should be considered if serum valproic acid concentrations drop below the therapeutic range or a seizure occurs. (See WARNINGS, Seizure Potential.)
Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term studies in animals have been performed to evaluate the carcinogenic potential of ertapenem.
Ertapenem was neither mutagenic nor genotoxic in the following in vitro assays: alkaline elution/rat hepatocyte assay, chromosomal aberration assay in Chinese hamster ovary cells, and TK6 human lymphoblastoid cell mutagenesis assay; and in the in vivo mouse micronucleus assay.
In mice and rats, IV doses of up to 700 mg/kg/day (for mice, approximately 3 times the recommended human dose of 1 g based on body surface area and for rats, approximately 1.2 times the human exposure at the recommended dose of 1 g based on plasma AUCs) resulted in no effects on mating performance, fecundity, fertility, or embryonic survival.
Pregnancy
Teratogenic Effects
Pregnancy Category B
In mice and rats given IV doses of up to 700 mg/kg/day (for mice, approximately 3 times the recommended human dose of 1 g based on body surface area and for rats, approximately 1.2 times the human exposure at the recommended dose of 1 g based on plasma AUCs), there was no evidence of developmental toxicity as assessed by external, visceral, and skeletal examination of the fetuses. However, in mice given 700 mg/kg/day, slight decreases in average fetal weights and an associated decrease in the average number of ossified sacrocaudal vertebrae were observed. Ertapenem crosses the placental barrier in rats.
There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
Ertapenem is excreted in human breast milk. (See CLINICAL PHARMACOLOGY, Distribution.) Caution should be exercised when INVANZ is administered to a nursing woman. INVANZ should be administered to nursing mothers only when the expected benefit outweighs the risk.
Labor and Delivery
INVANZ has not been studied for use during labor and delivery.
Pediatric Use
Safety and effectiveness of INVANZ in pediatric patients 3 months to 17 years of age are supported by evidence from adequate and well-controlled studies in adults, pharmacokinetic data in pediatric patients, and additional data from comparator-controlled studies in pediatric patients 3 months to 17 years of age with the following infections (see INDICATIONS AND USAGE and CLINICAL STUDIES):
- Complicated Intra-abdominal Infections
- Complicated Skin and Skin Structure Infections
- Community Acquired Pneumonia
- Complicated Urinary Tract Infections
- Acute Pelvic Infections
INVANZ is not recommended in infants under 3 months of age as no data are available.
INVANZ is not recommended in the treatment of meningitis in the pediatric population due to lack of sufficient CSF penetration.
Geriatric Use
Of the 1,835 patients in Phase IIb/III studies treated with INVANZ, approximately 26 percent were 65 and over, while approximately 12 percent were 75 and over. No overall differences in safety or effectiveness were observed between these patients and younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. (See DOSAGE AND ADMINISTRATION.)
Hepatic Insufficiency
The pharmacokinetics of ertapenem in patients with hepatic insufficiency have not been established. Of the total number of patients in clinical studies, 37 patients receiving ertapenem 1 g daily and 36 patients receiving comparator drugs were considered to have Child-Pugh Class A, B, or C liver impairment. The incidence of adverse experiences in patients with hepatic impairment was similar between the ertapenem group and the comparator groups.
ANIMAL PHARMACOLOGY
In repeat-dose studies in rats, treatment-related neutropenia occurred at every dose-level tested, including the lowest dose of 2 mg/kg (approximately 2% of the human dose on a body surface area basis).
Studies in rabbits and Rhesus monkeys were inconclusive with regard to the effect on neutrophil counts.
ADVERSE REACTIONS
Adults
Clinical studies enrolled 1954 patients treated with ertapenem; in some of the clinical studies, parenteral therapy was followed by a switch to an appropriate oral antimicrobial. (See CLINICAL STUDIES.) Most adverse experiences reported in these clinical studies were described as mild to moderate in severity. Ertapenem was discontinued due to adverse experiences in 4.7% of patients. Table 6 shows the incidence of adverse experiences reported in ≥1.0% of patients in these studies. The most common drug-related adverse experiences in patients treated with INVANZ, including those who were switched to therapy with an oral antimicrobial, were diarrhea (5.5%), infused vein complication (3.7%), nausea (3.1%), headache (2.2%), vaginitis in females (2.1%), phlebitis/thrombophlebitis (1.3%), and vomiting (1.1%).
| INVANZ*
1 g daily | Piperacillin/ Tazobactam*
3.375 g q6h | INVANZ†
1 g daily | Ceftriaxone†
1 or 2 g daily |
|
| Adverse Events | (N=802) | (N=774) | (N=1152) | (N=942) |
|
||||
| Local: | ||||
| Extravasation | 1.9 | 1.7 | 0.7 | 1.1 |
| Infused vein complication | 7.1 | 7.9 | 5.4 | 6.7 |
| Phlebitis/thrombophlebitis | 1.9 | 2.7 | 1.6 | 2.0 |
| Systemic: | ||||
| Asthenia/fatigue | 1.2 | 0.9 | 1.2 | 1.1 |
| Death | 2.5 | 1.6 | 1.3 | 1.6 |
| Edema/swelling | 3.4 | 2.5 | 2.9 | 3.3 |
| Fever | 5.0 | 6.6 | 2.3 | 3.4 |
| Abdominal pain | 3.6 | 4.8 | 4.3 | 3.9 |
| Chest pain | 1.5 | 1.4 | 1.0 | 2.5 |
| Hypertension | 1.6 | 1.4 | 0.7 | 1.0 |
| Hypotension | 2.0 | 1.4 | 1.0 | 1.2 |
| Tachycardia | 1.6 | 1.3 | 1.3 | 0.7 |
| Acid regurgitation | 1.6 | 0.9 | 1.1 | 0.6 |
| Oral candidiasis | 0.1 | 1.3 | 1.4 | 1.9 |
| Constipation | 4.0 | 5.4 | 3.3 | 3.1 |
| Diarrhea | 10.3 | 12.1 | 9.2 | 9.8 |
| Dyspepsia | 1.1 | 0.6 | 1.0 | 1.6 |
| Nausea | 8.5 | 8.7 | 6.4 | 7.4 |
| Vomiting | 3.7 | 5.3 | 4.0 | 4.0 |
| Leg pain | 1.1 | 0.5 | 0.4 | 0.3 |
| Anxiety | 1.4 | 1.3 | 0.8 | 1.2 |
| Altered mental status‡ | 5.1 | 3.4 | 3.3 | 2.5 |
| Dizziness | 2.1 | 3.0 | 1.5 | 2.1 |
| Headache | 5.6 | 5.4 | 6.8 | 6.9 |
| Insomnia | 3.2 | 5.2 | 3.0 | 4.1 |
| Cough | 1.6 | 1.7 | 1.3 | 0.5 |
| Dyspnea | 2.6 | 1.8 | 1.0 | 2.4 |
| Pharyngitis | 0.7 | 1.4 | 1.1 | 0.6 |
| Rales/rhonchi | 1.1 | 1.0 | 0.5 | 1.0 |
| Respiratory distress | 1.0 | 0.4 | 0.2 | 0.2 |
| Erythema | 1.6 | 1.7 | 1.2 | 1.2 |
| Pruritus | 2.0 | 2.6 | 1.0 | 1.9 |
| Rash | 2.5 | 3.1 | 2.3 | 1.5 |
| Vaginitis | 1.4 | 1.0 | 3.3 | 3.7 |
In patients treated for complicated intra-abdominal infections, death occurred in 4.7% (15/316) of patients receiving ertapenem and 2.6% (8/307) of patients receiving comparator drug. These deaths occurred in patients with significant co-morbidity and/or severe baseline infections. Deaths were considered unrelated to study drugs by investigators.
In clinical studies, seizure was reported during study therapy plus 14-day follow-up period in 0.5% of patients treated with ertapenem, 0.3% of patients treated with piperacillin/tazobactam and 0% of patients treated with ceftriaxone. (See PRECAUTIONS.)
Additional adverse experiences that were reported with INVANZ with an incidence >0.1% within each body system are listed below:
Body as a whole: abdominal distention, pain, chills, septicemia, septic shock, dehydration, gout, malaise, necrosis, candidiasis, weight loss, facial edema, injection site induration, injection site pain, flank pain, and syncope;
Cardiovascular System: heart failure, hematoma, cardiac arrest, bradycardia, arrhythmia, atrial fibrillation, heart murmur, ventricular tachycardia, asystole, and subdural hemorrhage;
Digestive System: gastrointestinal hemorrhage, anorexia, flatulence, C. difficile associated diarrhea, stomatitis, dysphagia, hemorrhoids, ileus, cholelithiasis, duodenitis, esophagitis, gastritis, jaundice, mouth ulcer, pancreatitis, and pyloric stenosis;
Nervous System & Psychiatric: nervousness, seizure (see WARNINGS and PRECAUTIONS), tremor, depression, hypesthesia, spasm, paresthesia, aggressive behavior, and vertigo;
Respiratory System: pleural effusion, hypoxemia, bronchoconstriction, pharyngeal discomfort, epistaxis, pleuritic pain, asthma, hemoptysis, hiccups, and voice disturbance;
Skin & Skin Appendage: sweating, dermatitis, desquamation, flushing, and urticaria;
Special Senses: taste perversion;
Urogenital System: renal insufficiency, oliguria/anuria, vaginal pruritus, hematuria, urinary retention, bladder dysfunction, vaginal candidiasis, and vulvovaginitis.
In a clinical trial for the treatment of diabetic foot infections in which 289 adult diabetic patients were treated with ertapenem, the adverse experience profile was generally similar to that seen in previous clinical trials.
In a clinical study in adults for the prophylaxis of surgical site infection following elective colorectal surgery in which 476 patients received a 1 g dose of ertapenem 1 hour prior to surgery and were then followed for safety 14 days post surgery, the overall adverse experience profile was generally comparable to that observed for ertapenem in previous clinical trials. Table 7 shows the incidence of adverse experiences other than those previously described above for ertapenem, regardless of causality, reported in ≥1.0% of patients in this study.
|
Adverse Events | INVANZ 1 g (N= 476) |
Cefotetan 2 g(N= 476) |
| Anemia | 5.7 | 6.9 |
| Small intestinal obstruction | 2.1 | 1.9 |
| Cellulitis | 1.5 | 1.5 |
| C. difficile infection or colitis | 1.7 | 0.6 |
| Pneumonia | 2.1 | 4.0 |
| Postoperative infection | 2.3 | 4.0 |
| Urinary tract infection | 3.8 | 5.5 |
| Wound infection | 6.5 | 12.4 |
| Anastomotic leak | 1.5 | 1.3 |
| Seroma | 1.3 | 1.9 |
| Wound complication | 2.9 | 2.3 |
| Wound dehiscence | 1.3 | 1.5 |
| Wound secretion | 1.9 | 2.1 |
| Dysuria | 1.1 | 1.3 |
| Atelectasis | 3.4 | 1.9 |
Additional adverse experiences that were reported in this prophylaxis study with INVANZ, regardless of causality, with an incidence <1.0% and >0.5% within each body system are listed below:
Gastrointestinal Disorders: dry mouth, hematochezia;
General Disorders and Administration Site Condition: crepitations;
Infections and Infestations: abdominal abscess, fungal rash, pelvic abscess;
Injury, Poisoning and Procedural Complications: incision site complication, incision site hemorrhage, intestinal stoma complication;
Musculoskeletal and Connective Tissue Disorders: muscle spasms;
Nervous System Disorders: cerebrovascular accident;
Renal and Urinary Disorders: pollakiuria;
Respiratory, Thoracic and Mediastinal Disorders: crackles lung, lung infiltration, pulmonary congestion, pulmonary embolism, wheezing.
Pediatric Patients
Clinical studies enrolled 384 patients treated with ertapenem; in some of the clinical studies, parenteral therapy was followed by a switch to an appropriate oral antimicrobial. (See CLINICAL STUDIES.) The overall adverse experience profile in pediatric patients is comparable to that in adult patients. Table 8 shows the incidence of adverse experiences reported in ≥1.0% of pediatric patients in clinical studies. The most common drug-related adverse experiences in pediatric patients treated with INVANZ, including those who were switched to therapy with an oral antimicrobial, were diarrhea (6.5%), infusion site pain (5.5%), infusion site erythema (2.6%), vomiting (2.1%).
| INVANZ*† | Ceftriaxone* | Ticarcillin/ Clavulanate† | |
| Adverse Events | (N=384) | (N=100) | (N=24) |
|
|||
| Local: | |||
| Infusion Site Erythema | 3.9 | 3.0 | 8.3 |
| Infusion Site Induration | 1.0 | 1.0 | 0.0 |
| Infusion Site Pain | 7.0 | 4.0 | 20.8 |
| Infusion Site Phlebitis | 1.8 | 3.0 | 0.0 |
| Infusion Site Swelling | 1.8 | 1.0 | 4.2 |
| Infusion Site Warmth | 1.3 | 1.0 | 4.2 |
| Systemic: | |||
| Abdominal Pain | 4.7 | 3.0 | 4.2 |
| Upper Abdominal Pain | 1.0 | 2.0 | 0.0 |
| Constipation | 2.3 | 0.0 | 0.0 |
| Diarrhea | 11.7 | 17.0 | 4.2 |
| Loose Stools | 2.1 | 0.0 | 0.0 |
| Nausea | 1.6 | 0.0 | 0.0 |
| Vomiting | 10.2 | 11.0 | 8.3 |
| Pyrexia | 4.9 | 6.0 | 8.3 |
| Abdominal Abscess | 1.0 | 0.0 | 4.2 |
| Herpes Simplex | 1.0 | 1.0 | 4.2 |
| Nasopharyngitis | 1.6 | 6.0 | 0.0 |
| Upper Respiratory Tract Infection | 2.3 | 3.0 | 0.0 |
| Viral Pharyngitis | 1.0 | 0.0 | 0.0 |
| Hypothermia | 1.6 | 1.0 | 0.0 |
| Dizziness | 1.6 | 0.0 | 0.0 |
| Headache | 4.4 | 4.0 | 0.0 |
| Cough | 4.4 | 3.0 | 0.0 |
| Wheezing | 1.0 | 0.0 | 0.0 |
| Dermatitis | 1.0 | 1.0 | 0.0 |
| Pruritus | 1.6 | 0.0 | 0.0 |
| Diaper Dermatitis | 4.7 | 4.0 | 0.0 |
| Rash | 2.9 | 2.0 | 8.3 |
Additional adverse experiences that were reported with INVANZ with an incidence <1.0% and >0.5% within each body system are listed below:
General Disorders and Administration Site Condition: chest pain, infusion site pruritus;
Infections and Infestations: candidiasis, ear infection, oral candidiasis;
Metabolism and Nutrition Disorders: decreased appetite;
Musculoskeletal and Connective Tissue Disorders: arthralgia;
Nervous System Disorders: somnolence;
Psychiatric Disorders: insomnia;
Reproductive System and Breast Disorders: genital rash;
Respiratory, Thoracic and Mediastinal Disorders: pleural effusion, rhinitis, rhinorrhea;
Skin and Subcutaneous Tissue Disorders: dermatitis atopic, rash erythematous, skin lesion;
Vascular Disorders: phlebitis.
Post-Marketing Experience:
The following post-marketing adverse experiences have been reported:
Immune System: anaphylaxis including anaphylactoid reactions
Psychiatric Disorders: altered mental status (including aggression, delirium)
Nervous System & Psychiatric: dyskinesia, hallucinations, myoclonus, tremor
Adverse Laboratory Changes
Adults
Laboratory adverse experiences that were reported during therapy in ≥1.0% of adult patients treated with INVANZ in clinical studies are presented in Table 9. Drug-related laboratory adverse experiences that were reported during therapy in ≥1.0% of adult patients treated with INVANZ, including those who were switched to therapy with an oral antimicrobial, in clinical studies were ALT increased (6.0%), AST increased (5.2%), serum alkaline phosphatase increased (3.4%), platelet count increased (2.8%), and eosinophils increased (1.1%). Ertapenem was discontinued due to laboratory adverse experiences in 0.3% of patients.
| INVANZ†
1 g daily | Piperacillin/ Tazobactam† 3.375 g q6h | INVANZ‡
1 g daily | Ceftriaxone‡
1 or 2 g daily |
|
| Adverse laboratory experiences | (n§=766) | (n§=755) | (n§=1122) | (n§=920) |
|
||||
| ALT increased | 8.8 | 7.3 | 8.3 | 6.9 |
| AST increased | 8.4 | 8.3 | 7.1 | 6.5 |
| Serum albumin decreased | 1.7 | 1.5 | 0.9 | 1.6 |
| Serum alkaline phosphatase increased | 6.6 | 7.2 | 4.3 | 2.8 |
| Serum creatinine increased | 1.1 | 2.7 | 0.9 | 1.2 |
| Serum glucose increased | 1.2 | 2.3 | 1.7 | 2.0 |
| Serum potassium decreased | 1.7 | 2.8 | 1.8 | 2.4 |
| Serum potassium increased | 1.3 | 0.5 | 0.5 | 0.7 |
| Total serum bilirubin increased | 1.7 | 1.4 | 0.6 | 1.1 |
| Eosinophils increased | 1.1 | 1.1 | 2.1 | 1.8 |
| Hematocrit decreased | 3.0 | 2.9 | 3.4 | 2.4 |
| Hemoglobin decreased | 4.9 | 4.7 | 4.5 | 3.5 |
| Platelet count decreased | 1.1 | 1.2 | 1.1 | 1.0 |
| Platelet count increased | 6.5 | 6.3 | 4.3 | 3.5 |
| Segmented neutrophils decreased | 1.0 | 0.3 | 1.5 | 0.8 |
| Prothrombin time increased | 1.2 | 2.0 | 0.3 | 0.9 |
| WBC decreased | 0.8 | 0.7 | 1.5 | 1.4 |
| Urine RBCs increased | 2.5 | 2.9 | 1.1 | 1.0 |
| Urine WBCs increased | 2.5 | 3.2 | 1.6 | 1.1 |
Additional laboratory adverse experiences that were reported during therapy in >0.1% but <1.0% of patients treated with INVANZ in clinical studies include: increases in BUN, direct and indirect serum bilirubin, serum sodium, monocytes, PTT, urine epithelial cells; decreases in serum bicarbonate.
In a clinical trial for the treatment of diabetic foot infections in which 289 adult diabetic patients were treated with ertapenem, the laboratory adverse experience profile was generally similar to that seen in previous clinical trials.
In a clinical study in adults for the prophylaxis of surgical site infection following elective colorectal surgery in which 476 patients received a 1 g dose of ertapenem 1 hour prior to surgery and were then followed for safety 14 days post surgery, the overall laboratory adverse experience profile was generally comparable to that observed for ertapenem in previous clinical trials. Additional laboratory adverse experiences that were reported during therapy and the 14 days post surgery period in >1.0% of patients, regardless of causality, include: white blood cell count increased and urine protein present.
Pediatric Patients
Laboratory adverse experiences that were reported during therapy in ≥1.0% of pediatric patients treated with INVANZ in clinical studies are presented in Table 10. Drug-related laboratory adverse experiences that were reported during therapy in ≥2.0% of pediatric patients treated with INVANZ, including those who were switched to therapy with an oral antimicrobial, in clinical studies were neutrophil count decreased (3.0%), ALT increased (2.2%), and AST increased (2.1%).
| INVANZ | Ceftriaxone | Ticarcillin/ Clavulanate |
|
| Adverse laboratory experiences | (n†=379) | (n†=97) | (n†=24) |
| ALT Increased | 3.8 | 1.1 | 4.3 |
| Alkaline Phosphatase Increased | 1.1 | 0.0 | 0.0 |
| AST Increased | 3.8 | 1.1 | 4.3 |
| Eosinophil Count Increased | 1.1 | 2.1 | 0.0 |
| Neutrophil Count Decreased | 5.8 | 3.1 | 0.0 |
| Platelet Count Increased | 1.3 | 0.0 | 8.7 |
Additional laboratory adverse experiences that were reported during therapy in >0.5% but <1.0% of patients treated with INVANZ in clinical studies include: white blood cell count decreased and urine protein present.
OVERDOSAGE
No specific information is available on the treatment of overdosage with INVANZ. Intentional overdosing of INVANZ is unlikely. Intravenous administration of INVANZ at a dose of 2 g over 30 min or 3 g over 1-2h in healthy adult volunteers resulted in an increased incidence of nausea. In clinical studies in adults, inadvertent administration of three 1 g doses of INVANZ in a 24 hour period resulted in diarrhea and transient dizziness in one patient. In pediatric clinical studies, a single IV dose of 40 mg/kg up to a maximum of 2 g did not result in toxicity.
In the event of an overdose, INVANZ should be discontinued and general supportive treatment given until renal elimination takes place.
INVANZ can be removed by hemodialysis; the plasma clearance of the total fraction of ertapenem was increased 30% in subjects with end-stage renal insufficiency when hemodialysis (4 hour session) was performed immediately following administration. However, no information is available on the use of hemodialysis to treat overdosage.
DOSAGE AND ADMINISTRATION
The dose of INVANZ in patients 13 years of age and older is 1 gram (g) given once a day. The dose of INVANZ in patients 3 months to 12 years of age is 15 mg/kg twice daily (not to exceed 1 g/day). INVANZ may be administered by intravenous infusion for up to 14 days or intramuscular injection for up to 7 days. When administered intravenously, INVANZ should be infused over a period of 30 minutes.
Intramuscular administration of INVANZ may be used as an alternative to intravenous administration in the treatment of those infections for which intramuscular therapy is appropriate.
DO NOT MIX OR CO-INFUSE INVANZ WITH OTHER MEDICATIONS. DO NOT USE DILUENTS CONTAINING DEXTROSE (α-D-GLUCOSE).
Table 11 presents treatment guidelines for INVANZ.
| Infection† | Daily Dose (IV or IM) Adults and Pediatric Patients 13 years of age and older | Daily Dose (IV or IM) Pediatric Patients 3 months to 12 years of age | Recommended Duration of Total Antimicrobial Treatment |
|
|||
| Complicated intra-abdominal infections | 1 g | 15 mg/kg twice daily‡ | 5 to 14 days |
| Complicated skin and skin structure infections, including diabetic foot infections§ | 1 g | 15 mg/kg twice daily‡ | 7 to 14 days¶ |
| Community acquired pneumonia | 1 g | 15 mg/kg twice daily‡ | 10 to 14 days# |
| Complicated urinary tract infections, including pyelonephritis | 1 g | 15 mg/kg twice daily‡ | 10 to 14 days# |
| Acute pelvic infections including postpartum endomyometritis, septic abortion and post surgical gynecologic infections | 1 g | 15 mg/kg twice daily‡ | 3 to 10 days |
Table 12 presents prophylaxis guidelines for INVANZ.
| Indication |
Daily Dose (IV) Adults |
Recommended Duration of Total Antimicrobial Treatment |
| Prophylaxis of surgical site infection following elective colorectal surgery | 1 g |
Single intravenous dose given 1 hour prior to surgical incision |
Patients with Renal Insufficiency: INVANZ may be used for the treatment of infections in adult patients with renal insufficiency. In patients whose creatinine clearance is >30 mL/min/1.73 m2, no dosage adjustment is necessary. Adult patients with advanced renal insufficiency (creatinine clearance ≤30 mL/min/1.73 m2) and end-stage renal insufficiency (creatinine clearance ≤10 mL/min/1.73 m2) should receive 500 mg daily. There are no data in pediatric patients with renal insufficiency.
Patients on Hemodialysis: When adult patients on hemodialysis are given the recommended daily dose of 500 mg of INVANZ within 6 hours prior to hemodialysis, a supplementary dose of 150 mg is recommended following the hemodialysis session. If INVANZ is given at least 6 hours prior to hemodialysis, no supplementary dose is needed. There are no data in patients undergoing peritoneal dialysis or hemofiltration. There are no data in pediatric patients on hemodialysis.
When only the serum creatinine is available, the following formula2 may be used to estimate creatinine clearance. The serum creatinine should represent a steady state of renal function.
Males: (weight in kg) x (140-age in years)
(72) x serum creatinine (mg/100 mL)
Females: (0.85) x (value calculated for males)
Patients with Hepatic Insufficiency: No dose adjustment recommendations can be made in patients with impaired hepatic function. (See CLINICAL PHARMACOLOGY, Special Populations, Hepatic Insufficiency and PRECAUTIONS.)
No dosage adjustment is recommended based on age (13 years of age and older) or gender. (SeeCLINICAL PHARMACOLOGY, Special Populations.)
- 2
-
Cockcroft and Gault equation: Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976.
PREPARATION OF SOLUTION
Vials
Adults and pediatric patients 13 years of age and older
Preparation for intravenous administration:
DO NOT MIX OR CO-INFUSE INVANZ WITH OTHER MEDICATIONS. DO NOT USE DILUENTS CONTAINING DEXTROSE (α-D-GLUCOSE).
INVANZ MUST BE RECONSTITUTED AND THEN DILUTED PRIOR TO ADMINISTRATION.
- Reconstitute the contents of a 1 g vial of INVANZ with 10 mL of one of the following: Water for Injection, 0.9% Sodium Chloride Injection or Bacteriostatic Water for Injection.
- Shake well to dissolve and immediately transfer contents of the reconstituted vial to 50 mL of 0.9% Sodium Chloride Injection.
- Complete the infusion within 6 hours of reconstitution.
Preparation for intramuscular administration:
INVANZ MUST BE RECONSTITUTED PRIOR TO ADMINISTRATION.
- Reconstitute the contents of a 1 g vial of INVANZ with 3.2 mL of 1.0% lidocaine HCl injection3 (without epinephrine). Shake vial thoroughly to form solution.
- Immediately withdraw the contents of the vial and administer by deep intramuscular injection into a large muscle mass (such as the gluteal muscles or lateral part of the thigh).
- The reconstituted IM solution should be used within 1 hour after preparation. NOTE: THE RECONSTITUTED SOLUTION SHOULD NOT BE ADMINISTERED INTRAVENOUSLY.
- 3
-
Refer to the prescribing information for lidocaine HCl.
Pediatric patients 3 months to 12 years of age:
Preparation for intravenous administration:
DO NOT MIX OR CO-INFUSE INVANZ WITH OTHER MEDICATIONS. DO NOT USE DILUENTS CONTAINING DEXTROSE (α-D-GLUCOSE).
INVANZ MUST BE RECONSTITUTED AND THEN DILUTED PRIOR TO ADMINISTRATION.
- Reconstitute the contents of a 1 g vial of INVANZ with 10 mL of one of the following: Water for Injection, 0.9% Sodium Chloride Injection or Bacteriostatic Water for Injection.
- Shake well to dissolve and immediately withdraw a volume equal to 15 mg/kg of body weight (not to exceed 1 g/day) and dilute in 0.9% Sodium Chloride Injection to a final concentration of 20 mg/mL or less.
- Complete the infusion within 6 hours of reconstitution.
Preparation for intramuscular administration:
INVANZ MUST BE RECONSTITUTED PRIOR TO ADMINISTRATION.
- Reconstitute the contents of a 1 g vial of INVANZ with 3.2 mL of 1.0% lidocaine HCl injection3 (without epinephrine). Shake vial thoroughly to form solution.
- Immediately withdraw a volume equal to 15 mg/kg of body weight (not to exceed 1 g/day) and administer by deep intramuscular injection into a large muscle mass (such as the gluteal muscles or lateral part of the thigh).
- The reconstituted IM solution should be used within 1 hour after preparation. NOTE: THE RECONSTITUTED SOLUTION SHOULD NOT BE ADMINISTERED INTRAVENOUSLY.
ADD-Vantage®4 Vials
See separate INSTRUCTIONS FOR USE OF INVANZ (Ertapenem for Injection) IN ADD-Vantage® VIALS. INVANZ in ADD-Vantage® vials should be reconstituted with ADD-Vantage® diluent containers containing 50 mL or 100 mL of 0.9% Sodium Chloride Injection.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to use, whenever solution and container permit. Solutions of INVANZ range from colorless to pale yellow. Variations of color within this range do not affect the potency of the product.
- 4
-
Registered trademark of Hospira Laboratories, Inc
STORAGE AND STABILITY
Before reconstitution
Do not store lyophilized powder above 25°C (77°F).
Reconstituted and infusion solutions
The reconstituted solution, immediately diluted in 0.9% Sodium Chloride Injection (see DOSAGE AND ADMINISTRATION, PREPARATION OF SOLUTION) may be stored at room temperature (25°C) and used within 6 hours or stored for 24 hours under refrigeration (5°C) and used within 4 hours after removal from refrigeration. Solutions of INVANZ should not be frozen.
HOW SUPPLIED
INVANZ is supplied as a sterile lyophilized powder in single dose vials containing ertapenem for intravenous infusion or for intramuscular injection as follows:
No. 3843—1 g ertapenem equivalent
NDC 0006-3843-71 in trays of 10 vials
INVANZ is supplied as a sterile lyophilized powder in single dose ADD-Vantage® vials containing ertapenem for intravenous infusion as follows:
No. 3845—1 g ertapenem equivalent
NDC 0006-3845-71 in trays of 10 ADD-Vantage® vials.
CLINICAL STUDIES
Adults
Complicated Intra-Abdominal Infections
Ertapenem was evaluated in adults for the treatment of complicated intra-abdominal infections in a clinical trial. This study compared ertapenem (1 g intravenously once a day) with piperacillin/tazobactam (3.375 g intravenously every 6 hours) for 5 to 14 days and enrolled 665 patients with localized complicated appendicitis, and any other complicated intra-abdominal infection including colonic, small intestinal, and biliary infections and generalized peritonitis. The combined clinical and microbiologic success rates in the microbiologically evaluable population at 4 to 6 weeks posttherapy (test-of-cure) were 83.6% (163/195) for ertapenem and 80.4% (152/189) for piperacillin/tazobactam.
Complicated Skin and Skin Structure Infections
Ertapenem was evaluated in adults for the treatment of complicated skin and skin structure infections in a clinical trial. This study compared ertapenem (1 g intravenously once a day) with piperacillin/tazobactam (3.375 g intravenously every 6 hours) for 7 to 14 days and enrolled 540 patients including patients with deep soft tissue abscess, posttraumatic wound infection and cellulitis with purulent drainage. The clinical success rates at 10 to 21 days posttherapy (test-of-cure) were 83.9% (141/168) for ertapenem and 85.3% (145/170) for piperacillin/tazobactam.
Diabetic Foot Infections
Ertapenem was evaluated in adults for the treatment of diabetic foot infections without concomitant osteomyelitis in a multicenter, randomized, double-blind clinical trial. This study compared ertapenem (1 g intravenously once a day) with piperacillin/tazobactam (3.375 g intravenously every 6 hours). Test-of-cure was defined as clinical response between treatment groups in the clinically evaluable population at the 10-day posttherapy follow-up visit. The study included 295 patients randomized to ertapenem and 291 patients to piperacillin/tazobactam. Both regimens allowed the option to switch to oral amoxicillin/clavulanate for a total of 5 to 28 days of treatment (parenteral and oral). All patients were eligible to receive appropriate adjunctive treatment methods, such as debridement, as is typically required in the treatment of diabetic foot infections, and most patients received these treatments. Patients with suspected osteomyelitis could be enrolled if all the infected bone was removed within 2 days of initiation of study therapy, and preferably within the prestudy period. Investigators had the option to add open-label vancomycin if enterococci or methicillin-resistant Staphylococcus aureus (MRSA) were among the pathogens isolated or if patients had a history of MRSA infection and additional therapy was indicated in the opinion of the investigator. Two hundred and four (204) patients randomized to ertapenem and 202 patients randomized to piperacillin/tazobactam were clinically evaluable. The clinical success rates at 10 days posttherapy were 75.0% (153/204) for ertapenem and 70.8% (143/202) for piperacillin/tazobactam.
Community Acquired Pneumonia
Ertapenem was evaluated in adults for the treatment of community acquired pneumonia in two clinical trials. Both studies compared ertapenem (1 g parenterally once a day) with ceftriaxone (1 g parenterally once a day) and enrolled a total of 866 patients. Both regimens allowed the option to switch to oral amoxicillin/clavulanate for a total of 10 to 14 days of treatment (parenteral and oral). In the first study the primary efficacy parameter was the clinical success rate in the clinically evaluable population and success rates were 92.3% (168/182) for ertapenem and 91.0% (183/201) for ceftriaxone at 7 to 14 days posttherapy (test-of-cure). In the second study the primary efficacy parameter was the clinical success rate in the microbiologically evaluable population and success rates were 91% (91/100) for ertapenem and 91.8% (45/49) for ceftriaxone at 7 to 14 days posttherapy (test-of-cure).
Complicated Urinary Tract Infections Including Pyelonephritis
Ertapenem was evaluated in adults for the treatment of complicated urinary tract infections including pyelonephritis in two clinical trials. Both studies compared ertapenem (1 g parenterally once a day) with ceftriaxone (1 g parenterally once a day) and enrolled a total of 850 patients. Both regimens allowed the option to switch to oral ciprofloxacin (500 mg twice daily) for a total of 10 to 14 days of treatment (parenteral and oral). The microbiological success rates (combined studies) at 5 to 9 days posttherapy (test-of-cure) were 89.5% (229/256) for ertapenem and 91.1% (204/224) for ceftriaxone.
Acute Pelvic Infections Including Endomyometritis, Septic Abortion and Post-Surgical Gynecological Infections
Ertapenem was evaluated in adults for the treatment of acute pelvic infections in a clinical trial. This study compared ertapenem (1 g intravenously once a day) with piperacillin/tazobactam (3.375 g intravenously every 6 hours) for 3 to 10 days and enrolled 412 patients including 350 patients with obstetric/postpartum infections and 45 patients with septic abortion. The clinical success rates in the clinically evaluable population at 2 to 4 weeks posttherapy (test-of-cure) were 93.9% (153/163) for ertapenem and 91.5% (140/153) for piperacillin/tazobactam.
Prophylaxis of Surgical Site Infections Following Elective Colorectal Surgery
Ertapenem was evaluated in adults for prophylaxis of surgical site infection following elective colorectal surgery in a multicenter, randomized, double-blind clinical trial. This study compared a single intravenous dose of ertapenem (1 g) versus cefotetan (2 g) administered over 30 minutes, 1 hour before elective colorectal surgery. Test-of-prophylaxis was defined as no evidence of surgical site infection, post-operative anastomotic leak, or unexplained antibiotic use in the clinically evaluable population up to and including at the 4-week posttreatment follow-up visit. The study included 500 patients randomized to ertapenem and 502 patients randomized to cefotetan. The modified intent-to-treat (MITT) population consisted of 451 ertapenem patients and 450 cefotetan patients and included all patients who were randomized, treated, and underwent elective colorectal surgery with adequate bowel preparation. The clinically evaluable population was a subset of the MITT population and consisted of patients who received a complete dose of study therapy no more than two hours prior to surgical incision and no more than six hours before surgical closure. Clinically evaluable patients had sufficient information to determine outcome at the 4-week follow-up assessment and had no confounding factors that interfered with the assessment of that outcome. Examples of confounding factors included prior or concomitant antibiotic violations, the need for a second surgical procedure during the study period, and identification of a distant site infection with concomitant antibiotic administration and no evidence of subsequent wound infection. Three-hundred forty-six (346) patients randomized to ertapenem and 339 patients randomized to cefotetan were clinically evaluable. The prophylactic success rates at 4 weeks posttreatment in the clinically evaluable population were 70.5% (244/346) for ertapenem and 57.2% (194/339) for cefotetan (difference 13.3%, [95% C.I.: 6.1, 20.4], p<0.001). Prophylaxis failure due to surgical site infections occurred in 18.2% (63/346) ertapenem patients and 31.0% (105/339) cefotetan patients. Post-operative anastomotic leak occurred in 2.9% (10/346) ertapenem patients and 4.1% (14/339) cefotetan patients. Unexplained antibiotic use occurred in 8.4% (29/346) ertapenem patients and 7.7% (26/339) cefotetan patients. Though patient numbers were small in some subgroups, in general, clinical response rates by age, gender, and race were consistent with the results found in the clinically evaluable population. In the MITT analysis, the prophylactic success rates at 4 weeks posttreatment were 58.3% (263/451) for ertapenem and 48.9% (220/450) for cefotetan (difference 9.4%, [95% C.I.: 2.9, 15.9], p=0.002). A statistically significant difference favoring ertapenem over cefotetan with respect to the primary endpoint has been observed at a significance level of 5% in this study. A second adequate and well-controlled study to confirm these findings has not been conducted; therefore, the clinical superiority of ertapenem over cefotetan has not been demonstrated.
Pediatric Patients
Ertapenem was evaluated in pediatric patients 3 months to 17 years of age in two randomized, multicenter clinical trials. The first study enrolled 404 patients and compared ertapenem (15 mg/kg IV every 12 hours in patients 3 months to 12 years of age, and 1 g IV once a day in patients 13 to 17 years of age) to ceftriaxone (50 mg/kg/day IV in two divided doses in patients 3 months to 12 years of age and 50 mg/kg/day IV as a single daily dose in patients 13 to 17 years of age) for the treatment of complicated urinary tract infection (UTI), skin and soft tissue infection (SSTI), or community-acquired pneumonia (CAP). Both regimens allowed the option to switch to oral amoxicillin/clavulanate for a total of up to 14 days of treatment (parenteral and oral). The microbiological success rates in the evaluable per protocol (EPP) analysis in patients treated for UTI were 87.0% (40/46) for ertapenem and 90.0% (18/20) for ceftriaxone. The clinical success rates in the EPP analysis in patients treated for SSTI were 95.5% (64/67) for ertapenem and 100% (26/26) for ceftriaxone, and in patients treated for CAP were 96.1% (74/77) for ertapenem and 96.4% (27/28) for ceftriaxone.
The second study enrolled 112 patients and compared ertapenem (15 mg/kg IV every 12 hours in patients 3 months to 12 years of age, and 1 g IV once a day in patients 13 to 17 years of age) to ticarcillin/clavulanate (50 mg/kg for patients <60 kg or 3.0 g for patients >60 kg, 4 or 6 times a day) up to 14 days for the treatment of complicated intra-abdominal infections (IAI) and acute pelvic infections (API). In patients treated for IAI (primarily patients with perforated or complicated appendicitis), the clinical success rates were 83.7% (36/43) for ertapenem and 63.6% (7/11) for ticarcillin/clavulanate in the EPP analysis. In patients treated for API (post-operative or spontaneous obstetrical endomyometritis, or septic abortion), the clinical success rates were 100% (23/23) for ertapenem and 100% (4/4) for ticarcillin/clavulanate in the EPP analysis.
REFERENCES
- Clinical and Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically. Seventh Edition; Approved Standard, CLSI Document M7-A7. Clinical and Laboratory Standards Institute, Wayne, PA, January 2006.
- Clinical And Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing – Sixteenth Informational Supplement. Approved Standard, CLSI Document M100-S16. Clinical and Laboratory Standards Institute, Wayne, PA, January 2006.
- Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Disk Susceptibility Tests. Ninth Edition; Approved Standard, CLSI Document M2-A9. Clinical and Laboratory Standards Institute, Wayne, PA, January 2006.
- Clinical and Laboratory Standards Institute (CLSI). Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria – Sixth Edition; Approved Standard, CLSI Document M11-A6. Clinical and Laboratory Standards Institute, Wayne, PA, January 2004.
Manuf for:
Merck & Co., Inc., Whitehouse Station, NJ 08889, USA
By: Laboratories Merck Sharp & Dohme-Chibret
63963 Clermont-Ferrand Cedex 9, France
US Patent Nos.: 5,478,820; 5,952,323; 5,652,233
Issued May 2009
9709707
047A-03/09 515045Z
INSTRUCTIONS FOR USE OF INVANZ®5
(Ertapenem for Injection)
IN ADD-Vantage®4 VIALS
For I.V. Use Only.
INSTRUCTIONS FOR USE
To Open Diluent Container:
Peel overwrap from the corner and remove container. Some opacity of the plastic due to moisture absorption during the sterilization process may be observed. This is normal and does not affect the solution quality or safety. The opacity will diminish gradually.
To Assemble Vial and Flexible Diluent Container:
(Use Aseptic Technique)
- Remove the protective covers from the top of the vial and the vial port on the diluent container as follows:
- To remove the breakaway vial cap, swing the pull ring over the top of the vial and pull down far enough to start the opening. (SEE FIGURE 1.) Pull the ring approximately half way around the cap and then pull straight up to remove the cap. (SEE FIGURE 2.) NOTE: DO NOT ACCESS VIAL WITH SYRINGE.
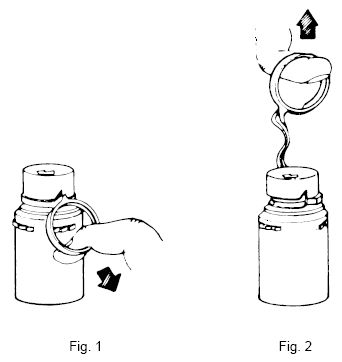
- To remove the vial port cover, grasp the tab on the pull ring, pull up to break the three tie strings, then pull back to remove the cover. (SEE FIGURE 3.)
- To remove the breakaway vial cap, swing the pull ring over the top of the vial and pull down far enough to start the opening. (SEE FIGURE 1.) Pull the ring approximately half way around the cap and then pull straight up to remove the cap. (SEE FIGURE 2.) NOTE: DO NOT ACCESS VIAL WITH SYRINGE.
- Screw the vial into the vial port until it will go no further. THE VIAL MUST BE SCREWED IN TIGHTLY TO ASSURE A SEAL. This occurs approximately ½ turn (180°) after the first audible click. (SEE FIGURE 4.) The clicking sound does not assure a seal; the vial must be turned as far as it will go. NOTE: Once vial is seated, do not attempt to remove. (SEE FIGURE 4.)
- Recheck the vial to assure that it is tight by trying to turn it further in the direction of assembly.
- Label appropriately.
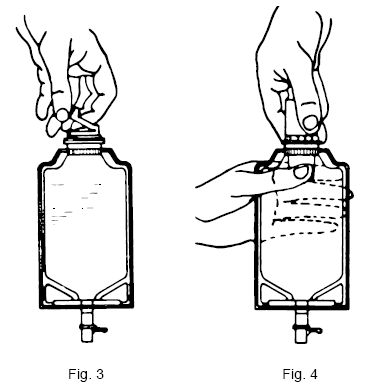
To Prepare Admixture:
- Squeeze the bottom of the diluent container gently to inflate the portion of the container surrounding the end of the drug vial.
- With the other hand, push the drug vial down into the container telescoping the walls of the container. Grasp the inner cap of the vial through the walls of the container. (SEE FIGURE 5.)
- Pull the inner cap from the drug vial. (SEE FIGURE 6.) Verify that the rubber stopper has been pulled out, allowing the drug and diluent to mix.
- Mix container contents thoroughly and use within the specified time.
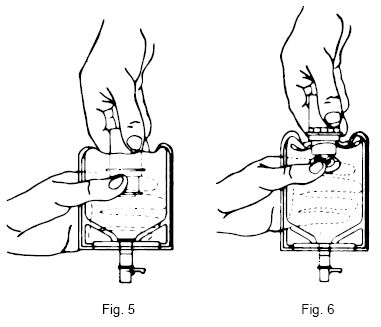
Preparation for Administration:
(Use Aseptic Technique)
- Confirm the activation and admixture of vial contents.
- Check for leaks by squeezing container firmly. If leaks are found, discard unit as sterility may be impaired.
- Close flow control clamp of administration set.
- Remove cover from outlet port at bottom of container.
- Insert piercing pin of administration set into port with a twisting motion until the pin is firmly seated. NOTE: See full directions on administration set carton.
- Lift the free end of the hanger loop on the bottom of the vial, breaking the two tie strings. Bend the loop outward to lock it in the upright position, then suspend container from hanger.
- Squeeze and release drip chamber to establish proper fluid level in chamber.
- Open flow control clamp and clear air from set. Close clamp.
- Attach set to venipuncture device. If device is not indwelling, prime and make venipuncture.
- Regulate rate of administration with flow control clamp.
WARNING: Do not use flexible container in series connections.
Storage
INVANZ (Ertapenem for Injection) 1 g single dose ADD-Vantage® vials should be prepared with ADD-Vantage® diluent containers containing 50 mL or 100 mL of 0.9% Sodium Chloride Injection. When prepared with this diluent, INVANZ (Ertapenem for Injection) maintains satisfactory potency for 6 hours at room temperature (25°C) or for 24 hours under refrigeration (5°C) and used within 4 hours after removal from refrigeration. Solutions of INVANZ should not be frozen.
Before administering, see accompanying package circular for INVANZ (Ertapenem for Injection).
Manuf for:
Merck & Co., Inc., Whitehouse Station, NJ 08889, USA
By: Laboratories Merck Sharp & Dohme-Chibret
63963 Clermont-Ferrand Cedex 9, France
US Patent Nos.: 5,478,820; 5,952,323; 5,652,233
Issued May 2009
9709707
047A-03/09 515045Z
- 5
-
Registered trademark of MERCK & CO., Inc. COPYRIGHT © 2006 MERCK & CO., Inc. All rights reserved
PRINCIPAL DISPLAY PANEL - Single Dose Vial Label 1g
1 Single dose vial
1 g
Rx only
INVANZ®
(ertapenem sodium) IV/IM
No. 3843
Inactive ingredients: 175 mg sodium bicarbonate and sodium hydroxide to adjust pH to 7.5. Solutions range from colorless to pale yellow. Variations of color within this range do not affect the potency of the product.
FOR INTRAVENOUS OR INTRAMUSCULAR USE
Manuf. for:
MERCK & CO., INC., Whitehouse Station, NJ 08889, USA
By: Laboratories Merck Sharp & Dohme-Chibret
63963 Clermont-Ferrand Cedex 9, France
9712301

PRINCIPAL DISPLAY PANEL - Single Dose ADD-Vantage Vial Label 1g
Single dose ADD-Vantage®* vial
1 g
Rx only
INVANZ®
(ertapenem sodium) IV/IM
Each vial contains 1.046 grams ertapenem sodium, equiv. to 1 gram ertapenem.
FOR I.V. USE ONLY
Manuf. for:
MERCK & CO., INC., Whitehouse Station, NJ 08889, USA
By: Laboratories Merck Sharp & Dohme-Chibret
63963 Clermont-Ferrand Cedex 9, France
Formulated in France.
9785802

| INVANZ
ertapenem sodium injection, powder, lyophilized, for solution |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA021337 | 11/21/2001 | |
| INVANZ
ertapenem sodium injection, powder, lyophilized, for solution |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA021337 | 11/21/2001 | |
| Labeler - Merck & Co., Inc. (001317064) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Laboratories Merck, Sharp & Dohme - Chibret | 493743686 | MANUFACTURE | |