KALETRA- lopinavir and ritonavir tablet, film coated
REMEDYREPACK INC.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use KALETRA safely and effectively. See full prescribing information for KALETRA.
KALETRA (lopinavir and ritonavir) tablet, for oral use KALETRA (lopinavir and ritonavir) oral solution Initial U.S. Approval: 2000 RECENT MAJOR CHANGES
INDICATIONS AND USAGEKALETRA is an HIV-1 protease inhibitor indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients (14 days and older). (
1)
DOSAGE AND ADMINISTRATIONTablets: May be taken with or without food, swallowed whole and not chewed, broken, or crushed. ( 2.1) Oral solution: must be taken with food. ( 2.1) Adults ( 2.2):
Pediatric Patients (14 days and older) ( 2.3):
Concomitant Therapy in Adults and Pediatric Patients:
Pregnancy ( 2.4):
DOSAGE FORMS AND STRENGTHSCONTRAINDICATIONS
WARNINGS AND PRECAUTIONSThe following have been observed in patients receiving KALETRA:
ADVERSE REACTIONSCommonly reported adverse reactions to KALETRA included diarrhea, nausea, vomiting, hypertriglyceridemia and hypercholesterolemia. (
6.1)
DRUG INTERACTIONSUSE IN SPECIFIC POPULATIONSLactation: Breastfeeding not recommended. ( 8.2) See 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 7/2016 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
KALETRA is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients (14 days and older).
The following points should be considered when initiating therapy with KALETRA:
- The use of other active agents with KALETRA is associated with a greater likelihood of treatment response [see Microbiology ( 12.4) and Clinical Studies ( 14)] .
- Genotypic or phenotypic testing and/or treatment history should guide the use of KALETRA [see Microbiology ( 12.4)] . The number of baseline lopinavir resistance-associated substitutions affects the virologic response to KALETRA [see Microbiology ( 12.4)] .
2 DOSAGE AND ADMINISTRATION
2.1 General Administration Recommendations
KALETRA tablets may be taken with or without food. The tablets should be swallowed whole and not chewed, broken, or crushed. KALETRA oral solution must be taken with food.
2.2 Dosage Recommendations in Adults
Considerations in Determining KALETRA Once Daily vs. Twice Daily Dosing Regimen:
- KALETRA can be given as once daily or twice daily dosing regimen in patients with less than three lopinavir resistance-associated substitutions.KALETRA can be given as once daily or twice daily dosing regimen in patients with less than three lopinavir resistance-associated substitutions.
- KALETRA must be given as twice daily dosing regimen in patients with three or more resistance-associated substitutions.KALETRA must be given as twice daily dosing regimen in patients with three or more resistance-associated substitutions.
- Table 1 includes the recommended once daily dosing regimen and Tables 2 and 3 include the recommended twice daily dosing regimen.Table 1 includes the recommended once daily dosing regimen and Tables 2 and 3 include the recommended twice daily dosing regimen.
KALETRA once daily dosing regimen is not recommended in:
- Adult patients with three or more of the following lopinavir resistance-associated substitutions: L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T, and I84V [see Microbiology ( 12.4)] . Adult[see Microbiology ( 12.4)]
- In combination with carbamazepine, phenobarbital, or phenytoin [see Drug Interactions ( 7.3)] .
- In combination with efavirenz, nevirapine, or nelfinavir [see Drug Interactions ( 7.3) and Clinical Pharmacology ( 12.3)] . In combination with efavirenz, nevirapine, or nelfinavir [see Drug Interactions ( 7.3) and Clinical Pharmacology ( 12.3)]
- In pregnant women [see Dosage and Administration ( 2.4), Use in Specific Populations ( 8.1) and Clinical Pharmacology ( 12.3)] . In pregnant women [see Dosage and Administration ( 2.4), Use in Specific Populations ( 8.1) and Clinical Pharmacology ( 12.3)] .
administered in combination with efavirenz, nevirapine or nelfinavir. The dose of KALETRA must be increased when administered in combination with efavirenz, nevirapine or nelfinavir.
for twice daily dosing when KALETRA is taken in combination with efavirenz, nevirapine or nelfinavir. Table 3 outlines the dosage recommendations for twice daily dosing when KALETRA is taken in combination with efavirenz, nevirapine or nelfinavir.
| KALETRA Dosage Form | Recommended Dosage |
| 200 mg/50 mg Tablets | 800 mg/200 mg (4 tablets) once daily |
| 80 mg/20 mg per mL Oral Solution | 800 mg/200 mg (10 mL) once daily |
| KALETRA Dosage Form | Recommended Dosage |
| 200 mg/50 mg Tablets | 400 mg/100 mg (2 tablets) twice daily |
| 80 mg/20 mg per mL Oral Solution | 400 mg/100 mg (5 mL) twice daily |
| KALETRA Dosage Form | Recommended Dosage |
| 200 mg/50 mg Tablets and
100 mg/25 mg Tablets | 500 mg/125 mg (2 tablets of 200 mg/50 mg
+ 1 tablet of 100 mg/25 mg) twice daily |
| 80 mg/20 mg per mL Oral Solution | 520 mg/130 mg (6.5 mL) twice daily |
2.3 Dosage Recommendations in Pediatric Patients
KALETRA tablets and oral solution should not be administered once daily in pediatric patients < 18 years of age. KALETRA tablets and oral solution should not be administered once daily in pediatric patients < 18 years of age. The dose of the oral solution should be administered using a calibrated dosing syringe.
Before prescribing KALETRA 100/25 mg tablets, children should be assessed for the ability to swallow intact tablets. If a child is unable to reliably swallow a KALETRA tablet, the KALETRA oral solution formulation should be prescribed.
KALETRA oral solution should not be administered to neonates before a postmenstrual age (first day of the mother’s last menstrual period to birth plus the time elapsed after birth) of 42 weeks and a postnatal age of at least 14 days has been attained . KALETRA oral solution should not be administered to neonates before a postmenstrual age (first day of the mother’s last menstrual period to birth plus the time elapsed after birth) of 42 weeks and a postnatal age of at least 14 days has been attained [see Warnings and Precautions ( 5.2)] .
KALETRA oral solution contains 42.4% (v/v) alcohol and 15.3% (w/v) propylene glycol. Special attention should be given to accurate calculation of the dosage of KALETRA, transcription of the medication order, dispensing information and dosing instructions to minimize the risk for medication errors, and overdose. This is especially important for infants and young children. Total amounts of alcohol and propylene glycol from all medicines that are to be given to pediatric patients 14 days to 6 months of age should be taken into account in order to avoid toxicity from these excipients and KALETRA oral solution contains 42.4% (v/v) alcohol and 15.3% (w/v) propylene glycol. Special attention should be given to accurate calculation of the dosage of KALETRA, transcription of the medication order, dispensing information and dosing instructions to minimize the risk for medication errors, and overdose. This is especially important for infants and young children. Total amounts of alcohol and propylene glycol from all medicines that are to be given to pediatric patients 14 days to 6 months of age should be taken into account in order to avoid toxicity from these excipients [see Warnings and Precautions ( 5.2) and Overdosage ( 10)].
Pediatric Dosage Calculations
Calculate the appropriate dose of KALETRA for each individual based on body weight (kg) or body surface area (BSA) to avoid underdosing or exceeding the recommended adult dose. Calculate the appropriate dose of KALETRA for each individual pediatric patient based on body weight (kg) or body surface area (BSA) to avoid underdosing or exceeding the recommended adult dose.
Body surface area (BSA) can be calculated as follows: Body surface area (BSA) can be calculated as follows:
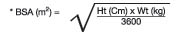
The KALETRA dose can be calculated based on weight or BSA:The KALETRA dose can be calculated based on weight or BSA:
Based on Weight:
Patient Weight (kg) × Prescribed lopinavir dose (mg/kg) = Administered lopinavir dose (mg) Patient Weight (kg) × Prescribed lopinavir dose (mg/kg) = Administered lopinavir dose (mg)
Based on BSA:
Patient BSA (m ) × Prescribed lopinavir dose (mg/m ) = Administered lopinavir dose (mg) Patient BSA (m 2) × Prescribed lopinavir dose (mg/m 2) = Administered lopinavir dose (mg)
If KALETRA oral solution is used, the volume (mL) of KALETRA solution can be determined as follows:If KALETRA oral solution is used, the volume (mL) of KALETRA solution can be determined as follows:
Volume of KALETRA solution (mL) = Administered lopinavir dose (mg) ÷ 80 (mg/mL)Volume of KALETRA solution (mL) = Administered lopinavir dose (mg) ÷ 80 (mg/mL)
Dosage Recommendation in Pediatric Patients 14 Days to 6 Months:
In pediatric patients 14 days to 6 months of age, the recommended dosage of lopinavir/ritonavir using KALETRA oral solution is 16/4 mg/kg or 300/75 mg/m twice daily. Prescribers should calculate the appropriate dose based on body weight or body surface area. Table 4 summarizes the recommended daily dosing regimen for pediatric patients 14 days to 6 months. In pediatric patients 14 days to 6 months of age, the recommended dosage of lopinavir/ritonavir using KALETRA oral solution is 16/4 mg/kg or 300/75 mg/m 2 twice daily. Prescribers should calculate the appropriate dose based on body weight or body surface area. Table 4 summarizes the recommended daily dosing regimen for pediatric patients 14 days to 6 months.
It is recommended that KALETRA not be administered in combination in patients < 6 months of age. It is recommended that KALETRA not be administered in combination with efavirenz, nevirapine, or nelfinavir in patients < 6 months of age.
| Patient Age | Based on Weight (mg/kg) | Based on BSA (mg/m 2) | Frequency |
| 14 days to 6 months | 16/4 | 300/75 | Given twice daily |
Dosage Recommendation in Pediatric Patients 6 Months to 18 Years:
Without Concomitant Efavirenz, Nevirapine, or Nelfinavir
Dosing recommendations using oral solution
In children 6 months to 18 years of age, the recommended dosage of lopinavir/ritonavir using KALETRA oral solution without concomitant efavirenz, nevirapine, or nelfinavir is 230/57.5 mg/m given twice daily, not to exceed the recommended adult dose (400/100 mg [5 mL] twice daily). If weight-based dosing is preferred, the recommended dosage of lopinavir/ritonavir for patients < 15 kg is 12/3 mg/kg given twice daily and the dosage for patients ≥ 15 kg to 40 kg is 10/2.5 mg/kg given twice daily. Table 5 summarizes the recommended daily dosing regimen for pediatric patients 6 months to 18 years. In children 6 months to 18 years of age, the recommended dosage of lopinavir/ritonavir using KALETRA oral solution without concomitant efavirenz, nevirapine, or nelfinavir is 230/57.5 mg/m 2 given twice daily, not to exceed the recommended adult dose (400/100 mg [5 mL] twice daily). If weight-based dosing is preferred, the recommended dosage of lopinavir/ritonavir for patients < 15 kg is 12/3 mg/kg given twice daily and the dosage for patients ≥ 15 kg to 40 kg is 10/2.5 mg/kg given twice daily. Table 5 summarizes the recommended daily dosing regimen for pediatric patients 6 months to 18 years.
| Patient Age | Based on Weight (mg/kg) | Based on BSA (mg/m 2) | Frequency | |
| 6 months to 18 years | <15 kg | 12/3 | 230/57.5 | Given twice daily |
| ≥15 kg to 40 kg | 10/2.5 | |||
Dosing recommendations using tablets
Table 6 provides the dosing recommendations for pediatric patients 6 months to 18 years of age based on body weight or body surface area for KALETRA tablets.Table 6 provides the dosing recommendations for pediatric patients 6 months to 18 years of age based on body weight or body surface area for KALETRA tablets.
| Body Weight (kg) | Body Surface Area (m 2) * | Recommended number of
100/25 mg Tablets Twice Daily |
| 15 to 25 | ≥0.6 to < 0.9 | 2 |
| >25 to 35 | ≥0.9 to < 1.4 | 3 |
| >35 | ≥1.4 | 4 (or two 200/50 mg tablets) |
| * KALETRA oral solution is available for children with a BSA less than 0.6 m 2 or those who are unable to reliably swallow a tablet. | ||
Concomitant Therapy: Efavirenz, Nevirapine, or Nelfinavir
Dosing recommendations using oral solution
A dose increase of KALETRA to 300/75 mg/m using KALETRA oral solution is needed when co-administered with efavirenz, nevirapine, or nelfinavir in children (both treatment-naïve and treatment-experienced) 6 months to 18 years of age, not to exceed the recommended adult dose (533/133 mg [6.5 mL] twice daily). If weight-based dosing is preferred, the recommended dosage for patients <15 kg is 13/3.25 mg/kg given twice daily and the dosage for patients >15 kg to 45 kg is 11/2.75 mg/kg given twice daily. A dose increase of KALETRA to 300/75 mg/m 2 using KALETRA oral solution is needed when co-administered with efavirenz, nevirapine, or nelfinavir in children (both treatment-naïve and treatment-experienced) 6 months to 18 years of age, not to exceed the recommended adult dose (533/133 mg [6.5 mL] twice daily). If weight-based dosing is preferred, the recommended dosage for patients <15 kg is 13/3.25 mg/kg given twice daily and the dosage for patients >15 kg to 45 kg is 11/2.75 mg/kg given twice daily.
Dosing recommendations using tablets
Table 7 provides the dosing recommendations for pediatric patients 6 months to 18 years of age based on body weight or body surface area for KALETRA tablets when given in combination with efavirenz, nevirapine, or nelfinavir.Table 7 provides the dosing recommendations for pediatric patients 6 months to 18 years of age based on body weight or body surface area for KALETRA tablets when given in combination with efavirenz, nevirapine, or nelfinavir.
| Body Weight (kg) | Body Surface Area (m 2) * | Recommended number of
100/25 mg Tablets Twice Daily |
| 15 to 20 | ≥0.6 to < 0.8 | 2 |
| >20 to 30 | ≥0.8 to < 1.2 | 3 |
| >30 to 45 | ≥1.2 to <1.7 | 4 (or two 200/50 mg tablets) |
| >45 | ≥1.7 | 5 [see Dosage and Administration ( 2.2)] |
| * KALETRA oral solution is available for children with a BSA less than 0.6 m
2 or those who are unable to reliably swallow a tablet.
† Please refer to the individual product labels for appropriate dosing in children. |
||
2.4 Dosage Recommendations in Pregnancy
Administer 400/100 mg of KALETRA twice daily in pregnant patients with no documented lopinavir-associated resistance substitutions. Once daily KALETRA dosing is not recommended in pregnancy [see Use in Specific Populations ( 8.1) and Clinical Pharmacology ( 12.3)] .
- There are insufficient data to recommend dosing in pregnant women with any documented lopinavir-associated resistance substitutions. There are insufficient data to recommend dosing in pregnant women with any documented lopinavir-associated resistance substitutions.
- No dosage adjustment of KALETRA is required for patients during the postpartum period.No dosage adjustment of KALETRA is required for patients during the postpartum period.
- Avoid use of KALETRA oral solution in pregnant women [see Use in Specific Populations ( 8.1)] . Avoid use of KALETRA oral solution in pregnant women [see Use in Specific Populations ( 8.1)] .
3 DOSAGE FORMS AND STRENGTHS
-
Tablets, 200 mg lopinavir, 50 mg ritonavir: Yellow, film-coated, ovaloid, debossed with the “a” logo and the code KA providing 200 mg lopinavir and 50 mg ritonavir.
-
Tablets, 100 mg lopinavir, 25 mg ritonavir: Pale yellow, film-coated, ovaloid, debossed with the “a” logo and the code KC providing 100 mg lopinavir and 25 mg ritonavir.
-
Oral Solution: Light yellow to orange colored liquid containing 400 mg lopinavir and 100 mg ritonavir per 5 mL (80 mg lopinavir and 20 mg ritonavir per mL).
4 CONTRAINDICATIONS
- KALETRA is contraindicated in patients with previously demonstrated clinically significant hypersensitivity (e.g., toxic epidermal necrolysis, Stevens-Johnson syndrome, erythema multiforme, urticaria, angioedema) to any of its ingredients, including ritonavir.
- Co-administration of KALETRA is contraindicated with drugs that are highly dependent on CYP3A for clearance and for which elevated plasma concentrations are associated with serious and/or life-threatening reactions.
- Co-administration of KALETRA is contraindicated with potent CYP3A inducers where significantly reduced lopinavir plasma concentrations may be associated with the potential for loss of virologic response and possible resistance and cross-resistance. These drugs are listed in Table 8.
| Drug Class | Drugs Within Class That are Contraindicated with KALETRA | Clinical Comments |
| Alpha 1- Adrenoreceptor Antagonist | Alfuzosin | Potentially increased alfuzosin concentrations can result in hypotension. |
| Antimycobacterial | Rifampin | May lead to loss of virologic response and possible resistance to KALETRA or to the class of protease inhibitors or other co-administered antiretroviral agents [see Drug Interactions ( 7)]. |
| Ergot Derivatives | Dihydroergotamine, ergotamine, methylergonovine | Potential for acute ergot toxicity characterized by peripheral vasospasm and ischemia of the extremities and other tissues. |
| GI Motility Agent | Cisapride | Potential for cardiac arrhythmias. |
| Herbal Products | St. John's Wort (hypericum perforatum) | May lead to loss of virologic response and possible resistance to KALETRA or to the class of protease inhibitors. |
| HMG-CoA Reductase Inhibitors | Lovastatin, simvastatin | Potential for myopathy including rhabdomyolysis. |
| PDE5 Enzyme Inhibitor | Sildenafil a (Revatio ®) when used for the treatment of pulmonary arterial hypertension | A safe and effective dose has not been established when used with KALETRA. There is an increased potential for sildenafil-associated adverse events, including visual abnormalities, hypotension, prolonged erection, and syncope [see Drug Interactions ( 7)] . |
| Neuroleptic | Pimozide | Potential for cardiac arrhythmias. |
| Sedative/Hypnotics | Triazolam;
orally administered midazolam b | Prolonged or increased sedation or respiratory depression. |
| a see Drug Interactions (7),
Table 13 for co-administration of sildenafil in patients with erectile dysfunction.
b see Drug Interactions (7), Table 13 for parenterally administered midazolam. |
||
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Serious Adverse Reactions Due to Drug Interactions
Initiation of KALETRA, a CYP3A inhibitor, in patients receiving medications metabolized by CYP3A or initiation of medications metabolized by CYP3A in patients already receiving KALETRA, may increase plasma concentrations of medications metabolized by CYP3A. Initiation of medications that inhibit or induce CYP3A may increase or decrease concentrations of KALETRA, respectively. These interactions may lead to:
- Clinically significant adverse reactions, potentially leading to severe, life-threatening, or fatal events from greater exposures of concomitant medications.Clinically significant adverse reactions, potentially leading to severe, life-threatening, or fatal events from greater exposures of concomitant medications.
- Clinically significant adverse reactions from greater exposures of KALETRA.Clinically significant adverse reactions from greater exposures of KALETRA.
- Loss of therapeutic effect of KALETRA and possible development of resistance.Loss of therapeutic effect of KALETRA and possible development of resistance.
See Table 13 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations [see Drug Interactions ( 7)] . Consider the potential for drug interactions prior to and during KALETRA therapy; review concomitant medications during KALETRA therapy, and monitor for the adverse reactions associated with the concomitant medications [see Contraindications ( 4) and Drug Interactions ( 7)] .
5.2 Toxicity in Preterm Neonates
KALETRA oral solution contains the excipients alcohol (42.4% v/v) and propylene glycol (15.3% w/v). When administered concomitantly with propylene glycol, ethanol competitively inhibits the metabolism of propylene glycol, which may lead to elevated concentrations. Preterm neonates may be at increased risk of propylene glycol-associated adverse events due to diminished ability to metabolize propylene glycol, thereby leading to accumulation and potential adverse events. Postmarketing life-threatening cases of cardiac toxicity (including complete AV block, bradycardia, and cardiomyopathy), lactic acidosis, acute renal failure, CNS depression and respiratory complications leading to death have been reported, predominantly in preterm neonates receiving KALETRA oral solution.
KALETRA oral solution should not be used in preterm neonates in the immediate postnatal period because of possible toxicities. A safe and effective dose of KALETRA oral solution in this patient population has not been established. However, if the benefit of using KALETRA oral solution to treat HIV infection in infants immediately after birth outweighs the potential risks, infants should be monitored closely for increases in serum osmolality and serum creatinine, and for toxicity related to KALETRA oral solution including: hyperosmolality, with or without lactic acidosis, renal toxicity, CNS depression (including stupor, coma, and apnea), seizures, hypotonia, cardiac arrhythmias and ECG changes, and hemolysis. Total amounts of alcohol and propylene glycol from all medicines that are to be given to infants should be taken into account in order to avoid toxicity from these excipients [see Dosage and Administration ( 2.3) and Overdosage ( 10)] .
5.3 Pancreatitis
Pancreatitis has been observed in patients receiving KALETRA therapy, including those who developed marked triglyceride elevations. In some cases, fatalities have been observed. Although a causal relationship to KALETRA has not been established, marked triglyceride elevations are a risk factor for development of pancreatitis [see Warnings and Precautions ( 5.9)] . Patients with advanced HIV-1 disease may be at increased risk of elevated triglycerides and pancreatitis, and patients with a history of pancreatitis may be at increased risk for recurrence during KALETRA therapy.
Pancreatitis should be considered if clinical symptoms (nausea, vomiting, abdominal pain) or abnormalities in laboratory values (such as increased serum lipase or amylase values) suggestive of pancreatitis occur. Patients who exhibit these signs or symptoms should be evaluated and KALETRA and/or other antiretroviral therapy should be suspended as clinically appropriate.
5.4 Hepatotoxicity
Patients with underlying hepatitis B or C or marked elevations in transaminase prior to treatment may be at increased risk for developing or worsening of transaminase elevations or hepatic decompensation with use of KALETRA.
There have been postmarketing reports of hepatic dysfunction, including some fatalities. These have generally occurred in patients with advanced HIV-1 disease taking multiple concomitant medications in the setting of underlying chronic hepatitis or cirrhosis. A causal relationship with KALETRA therapy has not been established.
Elevated transaminases with or without elevated bilirubin levels have been reported in HIV-1 mono-infected and uninfected patients as early as 7 days after the initiation of KALETRA in conjunction with other antiretroviral agents. In some cases, the hepatic dysfunction was serious; however, a definitive causal relationship with KALETRA therapy has not been established.
Appropriate laboratory testing should be conducted prior to initiating therapy with KALETRA and patients should be monitored closely during treatment. Increased AST/ALT monitoring should be considered in the patients with underlying chronic hepatitis or cirrhosis, especially during the first several months of KALETRA treatment [see Use in Specific Populations ( 8.6)].
5.5 QT Interval Prolongation
Postmarketing cases of QT interval prolongation and torsade de pointes have been reported although causality of KALETRA could not be established. Avoid use in patients with congenital long QT syndrome, those with hypokalemia, and with other drugs that prolong the QT interval [see Clinical Pharmacology ( 12.3)] .
5.6 PR Interval Prolongation
Lopinavir/ritonavir prolongs the PR interval in some patients. Cases of second or third degree atrioventricular block have been reported. KALETRA should be used with caution in patients with underlying structural heart disease, pre-existing conduction system abnormalities, ischemic heart disease or cardiomyopathies, as these patients may be at increased risk for developing cardiac conduction abnormalities.
The impact on the PR interval of co-administration of KALETRA with other drugs that prolong the PR interval (including calcium channel blockers, beta-adrenergic blockers, digoxin and atazanavir) has not been evaluated. As a result, co-administration of KALETRA with these drugs should be undertaken with caution, particularly with those drugs metabolized by CYP3A. Clinical monitoring is recommended [see Clinical Pharmacology ( 12.3)] .
5.7 Diabetes Mellitus/Hyperglycemia
New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus, and hyperglycemia have been reported during post-marketing surveillance in HIV-1 infected patients receiving protease inhibitor therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued protease inhibitor therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and a causal relationship between protease inhibitor therapy and these events has not been established.
5.8 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including KALETRA. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis) which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution, however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.9 Lipid Elevations
Treatment with KALETRA has resulted in large increases in the concentration of total cholesterol and triglycerides [see Adverse Reactions ( 6.1)] . Triglyceride and cholesterol testing should be performed prior to initiating KALETRA therapy and at periodic intervals during therapy. Lipid disorders should be managed as clinically appropriate, taking into account any potential drug-drug interactions with KALETRA and HMG-CoA reductase inhibitors [see Contraindications ( 4) and Drug Interactions ( 7.3)].
5.10 Fat Redistribution
Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance" have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
5.11 Patients with Hemophilia
Increased bleeding, including spontaneous skin hematomas and hemarthrosis have been reported in patients with hemophilia type A and B treated with protease inhibitors. In some patients additional factor VIII was given. In more than half of the reported cases, treatment with protease inhibitors was continued or reintroduced. A causal relationship between protease inhibitor therapy and these events has not been established.
5.12 Resistance/Cross-resistance
Because the potential for HIV cross-resistance among protease inhibitors has not been fully explored in KALETRA-treated patients, it is unknown what effect therapy with KALETRA will have on the activity of subsequently administered protease inhibitors [see Microbiology ( 12.4)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling.
- QT Interval Prolongation, PR Interval Prolongation [see Warnings and Precautions ( 5.5, 5.6)]
- Drug Interactions [see Warnings and Precautions ( 5.1)]
- Pancreatitis [see Warnings and Precautions ( 5.3)]
- Hepatotoxicity [see Warnings and Precautions ( 5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of KALETRA has been investigated in about 2,600 patients in Phase II-IV clinical trials, of which about 700 have received a dose of 800/200 mg (6 capsules or 4 tablets) once daily. Along with nucleoside reverse transcriptase inhibitors (NRTIs), in some studies, KALETRA was used in combination with efavirenz or nevirapine.
In clinical studies the incidence of diarrhea in patients treated with either KALETRA capsules or tablets was greater in those patients treated once daily than in those patients treated twice daily. Any grade of diarrhea was reported by at least half of patients taking once daily Kaletra capsules or tablets. At the time of treatment discontinuation, 4.2-6.3% of patients taking once daily Kaletra and 1.8-3.7% of those taking twice daily Kaletra reported ongoing diarrhea.
Commonly reported adverse reactions to KALETRA included diarrhea, nausea, vomiting, hypertriglyceridemia and hypercholesterolemia. Diarrhea, nausea and vomiting may occur at the beginning of the treatment while hypertriglyceridemia and hypercholesterolemia may occur later. The following have been identified as adverse reactions of moderate or severe intensity (Table 9):
| *Represents a medical concept including several similar MedDRA PTs
1. Percentage of male population (N=2,038) 2. Percentage of female population (N=574) |
|||
| System Organ Class (SOC) and Adverse Reaction | n | % | |
| BLOOD AND LYMPHATIC SYSTEM DISORDERS | |||
| anemia* | 54 | 2.1 | |
| leukopenia and neutropenia* | 44 | 1.7 | |
| lymphadenopathy* | 35 | 1.3 | |
| CARDIAC DISORDERS | |||
| atherosclerosis such as myocardial infarction* | 10 | 0.4 | |
| atrioventricular block* | 3 | 0.1 | |
| tricuspid valve incompetence* | 3 | 0.1 | |
| EAR AND LABYRINTH DISORDERS | |||
| vertigo* | 7 | 0.3 | |
| tinnitus | 6 | 0.2 | |
| ENDOCRINE DISORDERS | |||
| hypogonadism* | 16 | 0.8 1 | |
| EYE DISORDERS | |||
| visual impairment* | 8 | 0.3 | |
| GASTROINTESTINAL DISORDERS | |||
| diarrhea* | 510 | 19.5 | |
| nausea | 269 | 10.3 | |
| vomiting* | 177 | 6.8 | |
| abdominal pain (upper and lower)* | 160 | 6.1 | |
| gastroenteritis and colitis* | 66 | 2.5 | |
| dyspepsia | 53 | 2.0 | |
| pancreatitis* | 45 | 1.7 | |
| Gastroesophageal Reflux Disease (GERD)* | 40 | 1.5 | |
| hemorrhoids | 39 | 1.5 | |
| flatulence | 36 | 1.4 | |
| abdominal distension | 34 | 1.3 | |
| constipation* | 26 | 1.0 | |
| stomatitis and oral ulcers* | 24 | 0.9 | |
| duodenitis and gastritis* | 20 | 0.8 | |
| gastrointestinal hemorrhage including rectal hemorrhage* | 13 | 0.5 | |
| dry mouth | 9 | 0.3 | |
| gastrointestinal ulcer* | 6 | 0.2 | |
| fecal incontinence | 5 | 0.2 | |
| GENERAL DISORDERS AND ADMINISTRATION SITE CONDITIONS | |||
| fatigue including asthenia* | 198 | 7.6 | |
| HEPATOBILIARY DISORDERS | |||
| hepatitis including AST, ALT, and GGT increases* | 91 | 3.5 | |
| hepatomegaly | 5 | 0.2 | |
| cholangitis | 3 | 0.1 | |
| hepatic steatosis | 3 | 0.1 | |
| IMMUNE SYSTEM DISORDERS | |||
| hypersensitivity including urticaria and angioedema* | 70 | 2.7 | |
| immune reconstitution syndrome | 3 | 0.1 | |
| INFECTIONS AND INFESTATIONS | |||
| upper respiratory tract infection* | 363 | 13.9 | |
| lower respiratory tract infection* | 202 | 7.7 | |
| skin infections including cellulitis, folliculitis, and furuncle* | 86 | 3.3 | |
| METABOLISM AND NUTRITION DISORDERS | |||
| hypercholesterolemia* | 192 | 7.4 | |
| hypertriglyceridemia* | 161 | 6.2 | |
| weight decreased* | 61 | 2.3 | |
| decreased appetite | 52 | 2.0 | |
| blood glucose disorders including diabetes mellitus* | 30 | 1.1 | |
| weight increased* | 20 | 0.8 | |
| lactic acidosis* | 11 | 0.4 | |
| increased appetite | 5 | 0.2 | |
| MUSCULOSKELETAL AND CONNECTIVE TISSUE DISORDERS | |||
| musculoskeletal pain including arthralgia and back pain* | 166 | 6.4 | |
| myalgia* | 46 | 1.8 | |
| muscle disorders such as weakness and spasms* | 34 | 1.3 | |
| rhabdomyolysis* | 18 | 0.7 | |
| osteonecrosis | 3 | 0.1 | |
| NERVOUS SYSTEM DISORDERS | |||
| headache including migraine* | 165 | 6.3 | |
| insomnia* | 99 | 3.8 | |
| neuropathy and peripheral neuropathy* | 51 | 2.0 | |
| dizziness* | 45 | 1.7 | |
| ageusia* | 19 | 0.7 | |
| convulsion* | 9 | 0.3 | |
| tremor* | 9 | 0.3 | |
| cerebral vascular event* | 6 | 0.2 | |
| PSYCHIATRIC DISORDERS | |||
| anxiety* | 101 | 3.9 | |
| abnormal dreams* | 19 | 0.7 | |
| libido decreased | 19 | 0.7 | |
| RENAL AND URINARY DISORDERS | |||
| renal failure* | 31 | 1.2 | |
| hematuria* | 20 | 0.8 | |
| nephritis* | 3 | 0.1 | |
| REPRODUCTIVE SYSTEM AND BREAST DISORDERS | |||
| erectile dysfunction* | 34 | 1.7 1 | |
| menstrual disorders - amenorrhea, menorrhagia* | 10 | 1.7 2 | |
| SKIN AND SUBCUTANEOUS TISSUE DISORDERS | |||
| rash including maculopapular rash* | 99 | 3.8 | |
| lipodystrophy acquired including facial wasting* | 58 | 2.2 | |
| dermatitis/rash including eczema and seborrheic dermatitis* | 50 | 1.9 | |
| night sweats* | 42 | 1.6 | |
| pruritus* | 29 | 1.1 | |
| alopecia | 10 | 0.4 | |
| capillaritis and vasculitis* | 3 | 0.1 | |
| VASCULAR DISORDERS | |||
| hypertension* | 47 | 1.8 | |
| deep vein thrombosis* | 17 | 0.7 | |
Laboratory Abnormalities in Adults
The percentages of adult patients treated with combination therapy with Grade 3-4 laboratory abnormalities are presented in Table 10 (treatment-naïve patients) and Table 11 (treatment-experienced patients).
| Study 863
(48 Weeks) | Study 720
(360 Weeks) | Study 730
(48 Weeks) |
||||
| Variable | Limit 1 | KALETRA
400/100 mg Twice Daily + d4T +3TC (N = 326) | Nelfinavir
750 mg Three Times Daily + d4T + 3TC (N = 327) | KALETRA
Twice Daily + d4T + 3TC (N = 100) | KALETRA
Once Daily + TDF +FTC (N=333) | KALETRA
Twice Daily + TDF +FTC (N=331) |
| Chemistry | High | |||||
| Glucose | > 250 mg/dL | 2% | 2% | 4% | 0% | <1% |
| Uric Acid | > 12 mg/dL | 2% | 2% | 5% | <1% | 1% |
| SGOT/
AST 2 | > 180 U/L | 2% | 4% | 10% | 1% | 2% |
| SGPT/
ALT 2 | >215 U/L | 4% | 4% | 11% | 1% | 1% |
| GGT | >300 U/L | N/A | N/A | 10% | N/A | N/A |
| Total
Cholesterol | >300 mg/dL | 9% | 5% | 27% | 4% | 3% |
| Triglycerides | >750 mg/dL | 9% | 1% | 29% | 3% | 6% |
| Amylase | >2 x ULN | 3% | 2% | 4% | N/A | N/A |
| Lipase | >2 x ULN | N/A | N/A | N/A | 3% | 5% |
| Chemistry | Low | |||||
| Calculated Creatinine Clearance | <50 mL/min | N/A | N/A | N/A | 2% | 2% |
| Hematology | Low | |||||
| Neutrophils | <0.75 x 10 9/L | 1% | 3% | 5% | 2% | 1% |
| 1 ULN = upper limit of the normal range; N/A = Not Applicable.
2 Criterion for Study 730 was >5x ULN (AST/ALT). |
||||||
| Study 888
(48 Weeks) | Study 957
2 and Study 765
3
(84-144 Weeks) | Study 802
(48 Weeks) |
||||
| Variable | Limit 1 | KALETRA
400/100 mg Twice Daily + NVP + NRTIs (N = 148) | Investigator-Selected Protease Inhibitor(s) + NVP + NRTIs
(N = 140) | KALETRA
Twice Daily + NNRTI + NRTIs (N = 127) | KALETRA
800/200 mg Once Daily +NRTIs (N=300) | KALETRA
400/100 mg Twice Daily +NRTIs (N=299) |
| Chemistry | High | |||||
| Glucose | >250 mg/dL | 1% | 2% | 5% | 2% | 2% |
| Total Bilirubin | >3.48 mg/dL | 1% | 3% | 1% | 1% | 1% |
| SGOT/AST 4 | >180 U/L | 5% | 11% | 8% | 3% | 2% |
| SGPT/ALT 4 | >215 U/L | 6% | 13% | 10% | 2% | 2% |
| GGT | >300 U/L | N/A | N/A | 29% | N/A | N/A |
| Total
Cholesterol | >300 mg/dL | 20% | 21% | 39% | 6% | 7% |
| Triglycerides | >750 mg/dL | 25% | 21% | 36% | 5% | 6% |
| Amylase | >2 x ULN | 4% | 8% | 8% | 4% | 4% |
| Lipase | >2 x ULN | N/A | N/A | N/A | 4% | 1% |
| Creatine
Phosphokinase | >4 x ULN | N/A | N/A | N/A | 4% | 5% |
| Chemistry | Low | |||||
| Calculated
Creatinine Clearance | <50 mL/min | N/A | N/A | N/A | 3% | 3% |
| Inorganic
Phosphorus | <1.5 mg/dL | 1% | 0% | 2% | 1% | <1% |
| Hematology | Low | |||||
| Neutrophils | <0.75 x 10 9/L | 1% | 2% | 4% | 3% | 4% |
| Hemoglobin | <80 g/L | 1% | 1% | 1% | 1% | 2% |
| 1 ULN = upper limit of the normal range; N/A = Not Applicable.
2 Includes clinical laboratory data from patients receiving 400/100 mg twice daily (n = 29) or 533/133 mg twice daily (n = 28) for 84 weeks. Patients received KALETRA in combination with NRTIs and efavirenz. 3 Includes clinical laboratory data from patients receiving 400/100 mg twice daily (n = 36) or 400/200 mg twice daily (n = 34) for 144 weeks. Patients received KALETRA in combination with NRTIs and nevirapine. 4 Criterion for Study 802 was >5x ULN (AST/ALT). |
||||||
Adverse Reactions in Pediatric Patients
KALETRA oral solution dosed up to 300/75 mg/m 2 has been studied in 100 pediatric patients 6 months to 12 years of age. The adverse reaction profile seen during Study 940 was similar to that for adult patients.
Dysgeusia (22%), vomiting (21%), and diarrhea (12%) were the most common adverse reactions of any severity reported in pediatric patients treated with combination therapy for up to 48 weeks in Study 940. A total of 8 patients experienced adverse reactions of moderate to severe intensity. The adverse reactions meeting these criteria and reported for the 8 subjects include: hypersensitivity (characterized by fever, rash and jaundice), pyrexia, viral infection, constipation, hepatomegaly, pancreatitis, vomiting, alanine aminotransferase increased, dry skin, rash, and dysgeusia. Rash was the only event of those listed that occurred in 2 or more subjects (N = 3).
KALETRA oral solution dosed at 300/75 mg/m 2 has been studied in 31 pediatric patients 14 days to 6 months of age. The adverse reaction profile in Study 1030 was similar to that observed in older children and adults. No adverse reaction was reported in greater than 10% of subjects. Adverse drug reactions of moderate to severe intensity occurring in 2 or more subjects included decreased neutrophil count (N=3), anemia (N=2), high potassium (N=2), and low sodium (N=2).
KALETRA oral solution and soft gelatin capsules dosed at higher than recommended doses including 400/100 mg/m 2 (without concomitant NNRTI) and 480/120 mg/m 2 (with concomitant NNRTI) have been studied in 26 pediatric patients 7 to 18 years of age in Study 1038. Patients also had saquinavir mesylate added to their regimen at Week 4. Rash (12%), blood cholesterol abnormal (12%) and blood triglycerides abnormal (12%) were the only adverse reactions reported in greater than 10% of subjects. Adverse drug reactions of moderate to severe intensity occurring in 2 or more subjects included rash (N=3), blood triglycerides abnormal (N=3), and electrocardiogram QT prolonged (N=2). Both subjects with QT prolongation had additional predisposing conditions such as electrolyte abnormalities, concomitant medications, or pre-existing cardiac abnormalities.
Laboratory Abnormalities in Pediatric Patients
The percentages of pediatric patients treated with combination therapy including KALETRA with Grade 3-4 laboratory abnormalities are presented in Table 12.
| Variable | Limit 1 | KALETRA Twice Daily + RTIs
(N = 100) |
| Chemistry | High | |
| Sodium | > 149 mEq/L | 3% |
| Total Bilirubin | ≥ 3.0 x ULN | 3% |
| SGOT/AST | > 180 U/L | 8% |
| SGPT/ALT | > 215 U/L | 7% |
| Total Cholesterol | > 300 mg/dL | 3% |
| Amylase | > 2.5 x ULN | 7% 2 |
| Chemistry | Low | |
| Sodium | < 130 mEq/L | 3% |
| Hematology | Low | |
| Platelet Count | < 50 x 10 9/L | 4% |
| Neutrophils | < 0.40 x 10 9/L | 2% |
| 1 ULN = upper limit of the normal range.
2 Subjects with Grade 3-4 amylase confirmed by elevations in pancreatic amylase. |
||
6.2 Postmarketing Experience
The following adverse reactions have been reported during postmarketing use of KALETRA. Because these reactions are reported voluntarily from a population of unknown size, it is not possible to reliably estimate their frequency or establish a causal relationship to KALETRA exposure.
Body as a Whole
Redistribution/accumulation of body fat has been reported
[see Warnings and Precautions (
5.10)]
.
Cardiovascular
Bradyarrhythmias. First-degree AV block, second-degree AV block, third-degree AV block, QTc interval prolongation, torsades (torsade) de pointes
[see Warnings and Precautions (
5.5,
5.6)]
.
Skin and Appendages
Toxic epidermal necrolysis (TEN), Stevens-Johnson syndrome and erythema multiforme.
7 DRUG INTERACTIONS
See also Contraindications ( 4), Warnings and Precautions ( 5.1), Clinical Pharmacology ( 12.3)
7.1 Potential for KALETRA to Affect Other Drugs
Lopinavir/ritonavir is an inhibitor of CYP3A and may increase plasma concentrations of agents that are primarily metabolized by CYP3A. Agents that are extensively metabolized by CYP3A and have high first pass metabolism appear to be the most susceptible to large increases in AUC (> 3-fold) when co-administered with KALETRA. Thus, co-administration of KALETRA with drugs highly dependent on CYP3A for clearance and for which elevated plasma concentrations are associated with serious and/or life-threatening events is contraindicated. Co-administration with other CYP3A substrates may require a dose adjustment or additional monitoring as shown in Table 13.
7.2 Potential for Other Drugs to Affect Lopinavir
Lopinavir/ritonavir is a CYP3A substrate; therefore, drugs that induce CYP3A may decrease lopinavir plasma concentrations and reduce KALETRA’s therapeutic effect. Although not observed in the KALETRA/ketoconazole drug interaction study, co-administration of KALETRA and other drugs that inhibit CYP3A may increase lopinavir plasma concentrations.
7.3 Established and Other Potentially Significant Drug Interactions
Table 13 provides a listing of established or potentially clinically significant drug interactions. Alteration in dose or regimen may be recommended based on drug interaction studies or predicted interaction [see Clinical Pharmacology ( 12.3) for magnitude of interaction] .
| Concomitant Drug Class:
Drug Name | Effect on Concentration of Lopinavir or Concomitant Drug | Clinical Comments |
| HIV-1 Antiviral Agents | ||
| HIV-1 Protease Inhibitor:
fosamprenavir/ritonavir | ↓ amprenavir
↓ lopinavir | An increased rate of adverse reactions has been observed with co-administration of these medications. Appropriate doses of the combinations with respect to safety and efficacy have not been established. |
| HIV-1 Protease Inhibitor:
indinavir* | ↑ indinavir | Decrease indinavir dose to 600 mg twice daily, when co-administered with KALETRA 400/100 mg twice daily [see Clinical Pharmacology ( 12.3)] . KALETRA once daily has not been studied in combination with indinavir. |
| HIV-1 Protease Inhibitor:
nelfinavir* | ↑ nelfinavir
↑ M8 metabolite of nelfinavir ↓ lopinavir | KALETRA should not be administered once daily in combination with nelfinavir
[see Dosage and Administration ( 2) and Clinical Pharmacology ( 12.3)] . |
| HIV-1 Protease Inhibitor:
ritonavir* | ↑ lopinavir | Appropriate doses of additional ritonavir in combination with KALETRA with respect to safety and efficacy have not been established. |
| HIV-1 Protease Inhibitor:
saquinavir* | ↑ saquinavir | The saquinavir dose is 1000 mg twice daily, when co-administered with KALETRA 400/100 mg twice daily.
KALETRA once daily has not been studied in combination with saquinavir. |
| HIV-1 Protease Inhibitor:
tipranavir | ↓ lopinavir AUC and C min | KALETRA should not be administered with tipranavir (500 mg twice daily) co-administered with ritonavir (200 mg twice daily). |
| HIV CCR5 – Antagonist:
maraviroc | ↑ maraviroc | Concurrent administration of maraviroc with KALETRA will increase plasma levels of maraviroc. When co-administered, patients should receive 150 mg twice daily of maraviroc. For further details see complete prescribing information for Selzentry ® (maraviroc). |
| Non-nucleoside Reverse Transcriptase Inhibitor:
etravirine | ↓ etravirine | Because the reduction in the mean systemic exposures of etravirine in the presence of lopinavir/ritonavir is similar to the reduction in mean systemic exposures of etravirine in the presence of darunavir/ritonavir, no dose adjustment is required. |
| Non-nucleoside Reverse Transcriptase Inhibitors:
efavirenz*, nevirapine* | ↓ lopinavir | KALETRA dose increase is recommended in all patients
[see Dosage and Administration (
2)
and
Clinical Pharmacology (
12.3)]
.
Increasing the dose of KALETRA tablets to 500/125 mg (given as two 200/50 mg tablets and one 100/25 mg tablet) twice daily co-administered with efavirenz resulted in similar lopinavir concentrations compared to KALETRA tablets 400/100 mg (given as two 200/50 mg tablets) twice daily without efavirenz. Increasing the dose of KALETRA tablets to 600/150 mg (given as three 200/50 mg tablets) twice daily co-administered with efavirenz resulted in significantly higher lopinavir plasma concentrations compared to KALETRA tablets 400/100 mg twice daily without efavirenz. KALETRA should not be administered once daily in combination with efavirenz or nevirapine [see Dosage and Administration ( 2) and Clinical Pharmacology ( 12.3)] . |
| Non-nucleoside Reverse Transcriptase Inhibitor:
delavirdine | ↑ lopinavir | Appropriate doses of the combination with respect to safety and efficacy have not been established. |
| Non-nucleoside Reverse Transcriptase Inhibitor:
rilpivirine | ↑ rilpivirine | No dose adjustment is required. |
| Nucleoside Reverse Transcriptase Inhibitor:
didanosine | KALETRA tablets can be administered simultaneously with didanosine without food.
For KALETRA oral solution, it is recommended that didanosine be administered on an empty stomach; therefore, didanosine should be given one hour before or two hours after KALETRA oral solution (given with food). |
|
| Nucleoside Reverse Transcriptase Inhibitor:
tenofovir | ↑ tenofovir | KALETRA increases tenofovir concentrations. The mechanism of this interaction is unknown. Patients receiving KALETRA and tenofovir should be monitored for adverse reactions associated with tenofovir. |
| Nucleoside Reverse Transcriptase Inhibitors:
abacavir zidovudine | ↓ abacavir
↓ zidovudine | KALETRA induces glucuronidation; therefore, KALETRA has the potential to reduce zidovudine and abacavir plasma concentrations. The clinical significance of this potential interaction is unknown. |
| Other Agents | ||
| Antiarrhythmics e.g.:
amiodarone, bepridil, lidocaine (systemic), quinidine | ↑ antiarrhythmics | Caution is warranted and therapeutic concentration monitoring (if available) is recommended for antiarrhythmics when co-administered with KALETRA. |
| Anticancer Agents:
vincristine, vinblastine, dasatinib, nilotinib | ↑ anticancer agents | Concentrations of these drugs may be increased when co-administered with KALETRA resulting in the potential for increased adverse events usually associated with these anticancer agents.
For vincristine and vinblastine, consideration should be given to temporarily withholding the ritonavir-containing antiretroviral regimen in patients who develop significant hematologic or gastrointestinal side effects when KALETRA is administered concurrently with vincristine or vinblastine. If the antiretroviral regimen must be withheld for a prolonged period, consideration should be given to initiating a revised regimen that does not include a CYP3A or P-gp inhibitor. A decrease in the dosage or an adjustment of the dosing interval of nilotinib and dasatinib may be necessary for patients requiring co-administration with strong CYP3A inhibitors such as KALETRA. Please refer to the nilotinib and dasatinib prescribing information for dosing instructions. |
| Anticoagulants:
warfarin, rivaroxaban | ↑ rivaroxaban | Concentrations of warfarin may be affected. It is recommended that INR (international normalized ratio) be monitored.
Avoid concomitant use of rivaroxaban and KALETRA. Co-administration of KALETRA and rivaroxaban is expected to result in increased exposure of rivaroxaban which may lead to risk of increased bleeding. |
| Anticonvulsants:
carbamazepine, phenobarbital, phenytoin | ↓ lopinavir
↓ phenytoin | KALETRA may be less effective due to decreased lopinavir plasma concentrations in patients taking these agents concomitantly and should be used with caution.
KALETRA should not be administered once daily in combination with carbamazepine, phenobarbital, or phenytoin. In addition, co-administration of phenytoin and KALETRA may cause decreases in steady-state phenytoin concentrations. Phenytoin levels should be monitored when co-administering with KALETRA. |
| Anticonvulsants:
lamotrigine, valproate | ↓ lamotrigine
↓ or ↔ valproate | Co-administration of KALETRA and lamotrigine or valproate may decrease the exposure of lamotrigine or valproate. A dose increase of lamotrigine or valproate may be needed when co-administered with KALETRA and therapeutic concentration monitoring for lamotrigine may be indicated; particularly during dosage adjustments. |
| Antidepressant:
bupropion | ↓ bupropion
↓ active metabolite, hydroxybupropion | Concurrent administration of bupropion with KALETRA may decrease plasma levels of both bupropion and its active metabolite (hydroxybupropion). Patients receiving KALETRA and bupropion concurrently should be monitored for an adequate clinical response to bupropion. |
| Antidepressant:
trazodone | ↑ trazodone | Concomitant use of trazodone and KALETRA may increase concentrations of trazodone. Adverse reactions of nausea, dizziness, hypotension and syncope have been observed following co-administration of trazodone and ritonavir. If trazodone is used with a CYP3A4 inhibitor such as ritonavir, the combination should be used with caution and a lower dose of trazodone should be considered. |
| Anti-infective:
clarithromycin | ↑ clarithromycin | For patients with renal impairment, the following dosage adjustments should be considered:
|
| Antifungals:
ketoconazole*, itraconazole, voriconazole | ↑ ketoconazole
↑ itraconazole ↓ voriconazole | High doses of ketoconazole (>200 mg/day) or itraconazole (> 200 mg/day) are not recommended.
Co-administration of voriconazole with KALETRA has not been studied. However, a study has been shown that administration of voriconazole with ritonavir 100 mg every 12 hours decreased voriconazole steady-state AUC by an average of 39%; therefore, co-administration of KALETRA and voriconazole may result in decreased voriconazole concentrations and the potential for decreased voriconazole effectiveness and should be avoided, unless an assessment of the benefit/risk to the patient justifies the use of voriconazole. Otherwise, alternative antifungal therapies should be considered in these patients. |
| Anti-gout:
colchicine | ↑ colchicine | Patients with renal or hepatic impairment should not be given colchicine with KALETRA.
Treatment of gout flares-co-administration of colchicine in patients on KALETRA: 0.6 mg (1 tablet) x 1 dose, followed by 0.3 mg (half tablet) 1 hour later. Dose to be repeated no earlier than 3 days. Prophylaxis of gout flares-co-administration of colchicine in patients on KALETRA: If the original colchicine regimen was 0.6 mg twice a day, the regimen should be adjusted to 0.3 mg once a day. If the original colchicine regimen was 0.6 mg once a day, the regimen should be adjusted to 0.3 mg once every other day. Treatment of familial Mediterranean fever (FMF)-co-administration of colchicine in patients on KALETRA: Maximum daily dose of 0.6 mg (may be given as 0.3 mg twice a day). |
| Antimycobacterial:
bedaquiline | ↑ bedaquiline | Bedaquiline should only be used with KALETRA if the benefit of co-administration outweighs the risk [see Pharmacokinetics ( 12.3)] . |
| Antimycobacterial:
rifabutin* | ↑ rifabutin and rifabutin metabolite | Dosage reduction of rifabutin by at least 75% of the usual dose of 300 mg/day is recommended (i.e., a maximum dose of 150 mg every other day or three times per week). Increased monitoring for adverse reactions is warranted in patients receiving the combination. Further dosage reduction of rifabutin may be necessary. |
| Antimycobacterial:
rifampin | ↓ lopinavir | May lead to loss of virologic response and possible resistance to KALETRA or to the class of protease inhibitors or other co-administered antiretroviral agents. A study evaluated combination of rifampin 600 mg once daily, with KALETRA 800/200 mg twice daily or KALETRA 400/100 mg + ritonavir 300 mg twice daily. Pharmacokinetic and safety results from this study do not allow for a dose recommendation. Nine subjects (28%) experienced a ≥ grade 2 increase in ALT/AST, of which seven (21%) prematurely discontinued study per protocol. Based on the study design, it is not possible to determine whether the frequency or magnitude of the ALT/AST elevations observed is higher than what would be seen with rifampin alone [see Clinical Pharmacology ( 12.3) for magnitude of interaction ]. |
| Antiparasitic:
atovaquone | ↓ atovaquone | Clinical significance is unknown; however, increase in atovaquone doses may be needed. |
| Antipsychotics: quetiapine | ↑ quetiapine |
Initiation of KALETRA in patients taking quetiapine:
Initiation of quetiapine in patients taking KALETRA:
|
| Benzodiazepines: parenterally administered midazolam | ↑ midazolam | Midazolam is extensively metabolized by CYP3A4. Increases in the concentration of midazolam are expected to be significantly higher with oral than parenteral administration. Therefore, KALETRA should not be given with orally administered midazolam [see Contraindications ( 4)] . If KALETRA is co-administered with parenteral midazolam, close clinical monitoring for respiratory depression and/or prolonged sedation should be exercised and dosage adjustment should be considered. |
| Contraceptive:
ethinyl estradiol* | ↓ ethinyl estradiol | Because contraceptive steroid concentrations may be altered when KALETRA is co-administered with oral contraceptives or with the contraceptive patch, alternative methods of nonhormonal contraception are recommended. |
| Corticosteroids (systemic): e.g.
budesonide, dexamethasone, prednisone | ↓ lopinavir
↑ glucocorticoids | Use with caution. KALETRA may be less effective due to decreased lopinavir plasma concentrations in patients taking these agents concomitantly.
Concomitant use may result in increased steroid concentrations and reduced serum cortisol concentrations. Concomitant use of glucocorticoids that are metabolized by CYP3A, particularly for long-term use, should consider the potential benefit of treatment versus the risk of systemic corticosteroid effects. Concomitant use may increase the risk for development of systemic corticosteroid effects including Cushing’s syndrome and adrenal suppression. |
| Dihydropyridine Calcium Channel Blockers: e.g.
felodipine, nifedipine, nicardipine | ↑ dihydropyridine calcium channel blockers | Caution is warranted and clinical monitoring of patients is recommended. |
| Disulfiram/metronidazole | KALETRA oral solution contains alcohol, which can produce disulfiram-like reactions when co-administered with disulfiram or other drugs that produce this reaction (e.g., metronidazole). | |
| Endothelin Receptor Antagonists:
bosentan | ↑ bosentan | Co-administration of bosentan in patients on KALETRA:
In patients who have been receiving KALETRA for at least 10 days, start bosentan at 62.5 mg once daily or every other day based upon individual tolerability. Co-administration of KALETRA in patients on bosentan: Discontinue use of bosentan at least 36 hours prior to initiation of KALETRA. After at least 10 days following the initiation of KALETRA, resume bosentan at 62.5 mg once daily or every other day based upon individual tolerability. |
| HCV-Protease Inhibitor:
boceprevir | ↓ lopinavir
↓ boceprevir ↓ ritonavir | It is not recommended to co-administer KALETRA and boceprevir. Concomitant administration of KALETRA and boceprevir reduced boceprevir, lopinavir and ritonavir steady-state exposures [see Clinical Pharmacology ( 12.3)] . |
| HCV-Protease Inhibitor:
simeprevir | ↑ simeprevir | It is not recommended to co-administer KALETRA and simeprevir. |
| HMG-CoA Reductase Inhibitors:
atorvastatin rosuvastatin | ↑ atorvastatin
↑ rosuvastatin | Use atorvastatin with caution and at the lowest necessary dose. Titrate rosuvastatin dose carefully and use the lowest necessary dose; do not exceed rosuvastatin 10 mg/day. See Drugs with No Observed or Predicted Interactions with KALETRA ( 7.4) and Clinical Pharmacology ( 12.3) for drug interaction data with other HMG-CoA reductase inhibitors. |
| Immunosuppressants: e.g.
cyclosporine, tacrolimus, sirolimus | ↑ immunosuppressants | Therapeutic concentration monitoring is recommended for immunosuppressant agents when co-administered with KALETRA. |
| Inhaled or Intranasal Steroids e.g.:
fluticasone, budesonide | ↑ glucocorticoids | Concomitant use of KALETRA and fluticasone or other glucocorticoids that are metabolized by CYP3A is not recommended unless the potential benefit of treatment outweighs the risk of systemic corticosteroid effects. Concomitant use may result in increased steroid concentrations and reduce serum cortisol concentrations.
Systemic corticosteroid effects including Cushing's syndrome and adrenal suppression have been reported during postmarketing use in patients when certain ritonavir-containing products have been co-administered with fluticasone propionate or budesonide. |
| Long-acting beta-adrenoceptor Agonist:
salmeterol | ↑ salmeterol | Concurrent administration of salmeterol and KALETRA is not recommended. The combination may result in increased risk of cardiovascular adverse events associated with salmeterol, including QT prolongation, palpitations and sinus tachycardia. |
| Narcotic Analgesics:
methadone,* fentanyl | ↓ methadone
↑ fentanyl | Dosage of methadone may need to be increased when co-administered with KALETRA.
Concentrations of fentanyl are expected to increase. Careful monitoring of therapeutic and adverse effects (including potentially fatal respiratory depression) is recommended when fentanyl is concomitantly administered with KALETRA. |
| PDE5 inhibitors:
avanafil, sildenafil, tadalafil, vardenafil | ↑ avanafil
↑ sildenafil ↑ tadalafil ↑ vardenafil | Do not use KALETRA with avanafil because a safe and effective avanafil dosage regimen has not been established.
Particular caution should be used when prescribing sildenafil, tadalafil, or vardenafil in patients receiving KALETRA. Co-administration of KALETRA with these drugs is expected to substantially increase their concentrations and may result in an increase in PDE5 inhibitor associated adverse reactions including hypotension, syncope, visual changes and prolonged erection. Use of PDE5 inhibitors for pulmonary arterial hypertension (PAH): Sildenafil (Revatio ®) is contraindicated when used for the treatment of pulmonary arterial hypertension (PAH) because a safe and effective dose has not been established when used with KALETRA [see Contraindications ( 4)] . The following dose adjustments are recommended for use of tadalafil (Adcirca ®) with KALETRA: Co-administration of ADCIRCA in patients on KALETRA: In patients receiving KALETRA for at least one week, start ADCIRCA at 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability. Co-administration of KALETRA in patients on ADCIRCA: Avoid use of ADCIRCA during the initiation of KALETRA. Stop ADCIRCA at least 24 hours prior to starting KALETRA. After at least one week following the initiation of KALETRA, resume ADCIRCA at 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability. Use of PDE5 inhibitors for erectile dysfunction: It is recommended not to exceed the following doses: • Sildenafil: 25 mg every 48 hours • Tadalafil: 10 mg every 72 hours • Vardenafil: 2.5 mg every 72 hours Use with increased monitoring for adverse events. |
| * see Clinical Pharmacology ( 12.3) for magnitude of interaction. | ||
7.4 Drugs with No Observed or Predicted Interactions with KALETRA
Drug interaction or clinical studies reveal no clinically significant interaction between KALETRA and desipramine (CYP2D6 probe), pitavastatin, pravastatin, stavudine, lamivudine, omeprazole, raltegravir, or ranitidine.
Based on known metabolic profiles, clinically significant drug interactions are not expected between KALETRA and dapsone, trimethoprim/sulfamethoxazole, azithromycin, erythromycin, or fluconazole.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to KALETRA during pregnancy. Physicians are encouraged to register patients by calling the Antiretroviral Pregnancy Registry at 1-800-258-4263.
Available data from the Antiretroviral Pregnancy Registry show no difference in the risk of overall major birth defects compared to the background rate for major birth defects of 2.7% in the U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP). No treatment-related malformations were observed when lopinavir in combination with ritonavir was administered to pregnant rats or rabbits; however embryonic and fetal developmental toxicities occurred in rats administered maternally toxic doses.
Dose Adjustments During Pregnancy and the Postpartum Period
Administer 400/100 mg of KALETRA twice daily in pregnant patients with no documented lopinavir-associated resistance substitutions [see Dosage and Administration ( 2.4) and Clinical Pharmacology ( 12.3)] . There are insufficient data to recommend KALETRA dosing for pregnant patients with any documented lopinavir-associated resistance substitutions. No dose adjustment of KALETRA is required for patients during the postpartum period.
Once daily KALETRA dosing is not recommended in pregnancy.
Avoid use of KALETRA oral solution during pregnancy due to the alcohol content. KALETRA oral solution contains the excipients alcohol (42.4% v/v) and propylene glycol (15.3% w/v).
KALETRA was evaluated in 12 HIV-infected pregnant women in an open-label pharmacokinetic trial [see Clinical Pharmacology ( 12.3)] . No new trends in the safety profile were identified in pregnant women dosed with KALETRA compared to the safety described in non-pregnant adults, based on the review of these limited data.
Antiretroviral Pregnancy Registry Data: Based on prospective reports from the Antiretroviral Pregnancy Registry (APR) of over 3,000 exposures to lopinavir containing regimens (including over 1,000 exposed in the first trimester), there was no difference between lopinavir and overall birth defects compared with the background birth defect rate of 2.7% in the U.S. reference population of the Metropolitan Atlanta Congenital Defects Program. Based on prospective reports from the APR of over 5,000 exposures to ritonavir containing regimens (including over 2,000 exposures in the first trimester) there was no difference between ritonavir and overall birth defects compared with the U.S. background rate (MACDP). For both lopinavir and ritonavir, sufficient numbers of first trimester exposures have been monitored to detect at least a 1.5 fold increase in risk of overall birth defects and a 2 fold increase in risk of birth defects in the cardiovascular and genitourinary systems.
Embryonic and fetal developmental toxicities (early resorption, decreased fetal viability, decreased fetal body weight, increased incidence of skeletal variations and skeletal ossification delays) occurred in rats at a maternally toxic dosage. Based on AUC measurements, the drug exposures in rats at the toxic doses were approximately 0.7-fold for lopinavir and 1.8-fold for ritonavir for males and females that of the exposures in humans at the recommended therapeutic dose (400/100 mg twice daily). In a peri- and postnatal study in rats, a developmental toxicity (a decrease in survival in pups between birth and postnatal Day 21) occurred.
No embryonic and fetal developmental toxicities were observed in rabbits at a maternally toxic dosage. Based on AUC measurements, the drug exposures in rabbits at the toxic doses were approximately 0.6-fold for lopinavir and 1.0-fold for ritonavir that of the exposures in humans at the recommended therapeutic dose (400/100 mg twice daily).
8.2 Lactation
The Centers for Disease Control and Prevention recommend that HIV-1 infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV-1. Because of the potential for HIV-1 transmission in breastfed infants, advise women not to breastfeed.
8.4 Pediatric Use
The safety, efficacy, and pharmacokinetic profiles of KALETRA in pediatric patients below the age of 14 days have not been established. KALETRA should not be administered once daily in pediatric patients.
An open-label, multi-center, dose-finding trial was performed to evaluate the pharmacokinetic profile, tolerability, safety and efficacy of KALETRA oral solution containing lopinavir 80 mg/mL and ritonavir 20 mg/mL at a dose of 300/75 mg/m 2 twice daily plus two NRTIs in HIV-infected infants ≥14 days and < 6 months of age. Results revealed that infants younger than 6 months of age generally had lower lopinavir AUC 12 than older children (6 months to 12 years of age), however, despite the lower lopinavir drug exposure observed, antiviral activity was demonstrated as reflected in the proportion of subjects who achieved HIV-1 RNA <400 copies/mL at Week 24 [see Adverse Reactions ( 6.2), Clinical Pharmacology ( 12.3), Clinical Studies ( 14.4)] .
Safety and efficacy in pediatric patients > 6 months of age was demonstrated in a clinical trial in 100 patients. The clinical trial was an open-label, multicenter trial evaluating the pharmacokinetic profile, tolerability, safety, and efficacy of KALETRA oral solution containing lopinavir 80 mg/mL and ritonavir 20 mg/mL in 100 antiretroviral naïve and experienced pediatric patients ages 6 months to 12 years. Dose selection for patients 6 months to 12 years of age was based on the following results. The 230/57.5 mg/m 2 oral solution twice daily regimen without nevirapine and the 300/75 mg/m 2 oral solution twice daily regimen with nevirapine provided lopinavir plasma concentrations similar to those obtained in adult patients receiving the 400/100 mg twice daily regimen (without nevirapine) [see Adverse Reactions ( 6.2), Clinical Pharmacology ( 12.3), Clinical Studies ( 14.4)] .
A prospective multicenter, open-label trial evaluated the pharmacokinetic profile, tolerability, safety and efficacy of high-dose KALETRA with or without concurrent NNRTI therapy (Group 1: 400/100 mg/m 2 twice daily + ≥ 2 NRTIs; Group 2: 480/120 mg/m 2 twice daily + ≥ 1 NRTI + 1 NNRTI) in 26 children and adolescents ≥ 2 years to < 18 years of age who had failed prior therapy. Patients also had saquinavir mesylate added to their regimen. This strategy was intended to assess whether higher than approved doses of KALETRA could overcome protease inhibitor cross-resistance. High doses of KALETRA exhibited a safety profile similar to those observed in previous trials; changes in HIV-1 RNA were less than anticipated; three patients had HIV-1 RNA <400 copies/mL at Week 48. CD4+ cell count increases were noted in the eight patients who remained on treatment for 48 weeks [see Adverse Reactions ( 6.2), Clinical Pharmacology ( 12.3)] .
A prospective multicenter, randomized, open-label study evaluated the efficacy and safety of twice-daily versus once-daily dosing of KALETRA tablets dosed by weight as part of combination antiretroviral therapy (cART) in virologically suppressed HIV-1 infected children (n=173). Children were eligible when they were aged < 18 years, ≥ 15 kg in weight, receiving cART that included KALETRA, HIV-1 ribonucleic acid (RNA) < 50 copies/mL for at least 24 weeks and able to swallow tablets. At week 24, efficacy (defined as the proportion of subjects with plasma HIV-1 RNA less than 50 copies per mL) was significantly higher in subjects receiving twice daily dosing compared to subjects receiving once daily dosing. The safety profile was similar between the two treatment arms although there was a greater incidence of diarrhea in the once daily treated subjects.
8.5 Geriatric Use
Clinical studies of KALETRA did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, appropriate caution should be exercised in the administration and monitoring of KALETRA in elderly patients reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Hepatic Impairment
KALETRA is principally metabolized by the liver; therefore, caution should be exercised when administering this drug to patients with hepatic impairment, because lopinavir concentrations may be increased [see Warnings and Precautions ( 5.4) and Clinical Pharmacology ( 12.3)] .
10 OVERDOSAGE
Overdoses with KALETRA oral solution have been reported. One of these reports described fatal cardiogenic shock in a 2.1 kg infant who received a single dose of 6.5 mL of KALETRA oral solution (520 mg lopinavir, approximately 10-fold above the recommended lopinavir dose) nine days prior. The following events have been reported in association with unintended overdoses in preterm neonates: complete AV block, cardiomyopathy, lactic acidosis, and acute renal failure [see Warnings and Precautions ( 5.2)] . Healthcare professionals should be aware that KALETRA oral solution is highly concentrated and therefore, should pay special attention to accurate calculation of the dose of KALETRA, transcription of the medication order, dispensing information and dosing instructions to minimize the risk for medication errors and overdose. This is especially important for infants and young children.
KALETRA oral solution contains 42.4% alcohol (v/v) and 15.3% propylene glycol (w/v). Ingestion of the product over the recommended dose by an infant or a young child could result in significant toxicity and could potentially be lethal.
Human experience of acute overdosage with KALETRA is limited. Treatment of overdose with KALETRA should consist of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. There is no specific antidote for overdose with KALETRA. If indicated, elimination of unabsorbed drug should be achieved by gastric lavage. Administration of activated charcoal may also be used to aid in removal of unabsorbed drug. Since lopinavir is highly protein bound, dialysis is unlikely to be beneficial in significant removal of the drug. However, dialysis can remove both alcohol and propylene glycol in the case of overdose with KALETRA oral solution.
11 DESCRIPTION
KALETRA is a co-formulation of lopinavir and ritonavir. Lopinavir is an inhibitor of the HIV-1 protease. As co-formulated in KALETRA, ritonavir inhibits the CYP3A-mediated metabolism of lopinavir, thereby providing increased plasma levels of lopinavir.
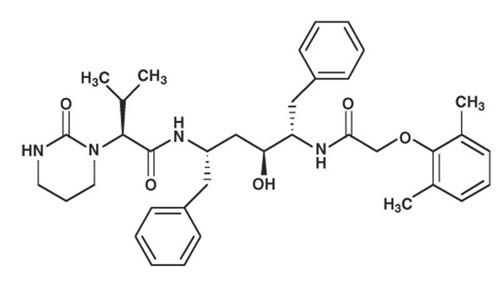
Lopinavir is chemically designated as [1 S-[1 R*,( R*), 3 R*, 4 R*]]- N-[4-[[(2,6-dimethylphenoxy)acetyl]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]tetrahydro-alpha-(1-methylethyl)-2-oxo-1(2 H)-pyrimidineacetamide. Its molecular formula is C 37H 48N 4O 5, and its molecular weight is 628.80. Lopinavir is a white to light tan powder. It is freely soluble in methanol and ethanol, soluble in isopropanol and practically insoluble in water. Lopinavir has the following structural formula:
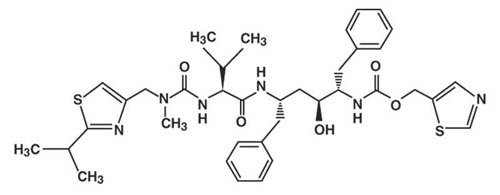
Ritonavir is chemically designated as 10-hydroxy-2-methyl-5-(1-methylethyl)-1- [2-(1-methylethyl)-4-thiazolyl]-3,6-dioxo-8,11-bis(phenylmethyl)-2,4,7,12-tetraazatridecan-13-oic acid, 5-thiazolylmethyl ester, [5 S-(5 R*,8 R*,10 R*,11 R*)]. Its molecular formula is C 37H 48N 6O 5S 2, and its molecular weight is 720.95. Ritonavir is a white to light tan powder. It is freely soluble in methanol and ethanol, soluble in isopropanol and practically insoluble in water. Ritonavir has the following structural formula:
KALETRA tablets are available for oral administration in two strengths:
- Yellow tablets containing 200 mg of lopinavir and 50 mg of ritonavir
- Pale yellow tablets containing 100 mg of lopinavir and 25 mg of ritonavir.
The yellow, 200 mg lopinavir and 50 mg ritonavir, tablets contain the following inactive ingredients: copovidone, sorbitan monolaurate, colloidal silicon dioxide, and sodium stearyl fumarate. The following are the ingredients in the film coating: hypromellose, titanium dioxide, polyethylene glycol 400, hydroxypropyl cellulose, talc, colloidal silicon dioxide, polyethylene glycol 3350, yellow ferric oxide E172, and polysorbate 80.
The pale yellow, 100 mg lopinavir and 25 mg ritonavir, tablets contain the following inactive ingredients: copovidone, sorbitan monolaurate, colloidal silicon dioxide, and sodium stearyl fumarate. The following are the ingredients in the film coating: polyvinyl alcohol, titanium dioxide, talc, polyethylene glycol 3350, and yellow ferric oxide E172.
KALETRA oral solution is available for oral administration as 80 mg lopinavir and 20 mg ritonavir per milliliter with the following inactive ingredients: acesulfame potassium, alcohol, artificial cotton candy flavor, citric acid, glycerin, high fructose corn syrup, Magnasweet-110 flavor, menthol, natural & artificial vanilla flavor, peppermint oil, polyoxyl 40 hydrogenated castor oil, povidone, propylene glycol, saccharin sodium, sodium chloride, sodium citrate, and water.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lopinavir is an antiviral drug [see Microbiology ( 12.4)] . As co-formulated in KALETRA, ritonavir inhibits the CYP3A-mediated metabolism of lopinavir, thereby providing increased plasma levels of lopinavir.
12.3 Pharmacokinetics
The pharmacokinetic properties of lopinavir co-administered with ritonavir have been evaluated in healthy adult volunteers and in HIV-1 infected patients; no substantial differences were observed between the two groups. Lopinavir is essentially completely metabolized by CYP3A. Ritonavir inhibits the metabolism of lopinavir, thereby increasing the plasma levels of lopinavir. Across studies, administration of KALETRA 400/100 mg twice daily yields mean steady-state lopinavir plasma concentrations 15- to 20-fold higher than those of ritonavir in HIV-1 infected patients. The plasma levels of ritonavir are less than 7% of those obtained after the ritonavir dose of 600 mg twice daily. The in vitro antiviral EC 50 of lopinavir is approximately 10-fold lower than that of ritonavir. Therefore, the antiviral activity of KALETRA is due to lopinavir.
Figure 1 displays the mean steady-state plasma concentrations of lopinavir and ritonavir after KALETRA 400/100 mg twice daily with food for 3 weeks from a pharmacokinetic study in HIV-1 infected adult subjects (n = 19).
Figure 1. Mean Steady-State Plasma Concentrations with 95% Confidence Intervals (CI) for HIV-1 Infected Adult Subjects (N = 19)
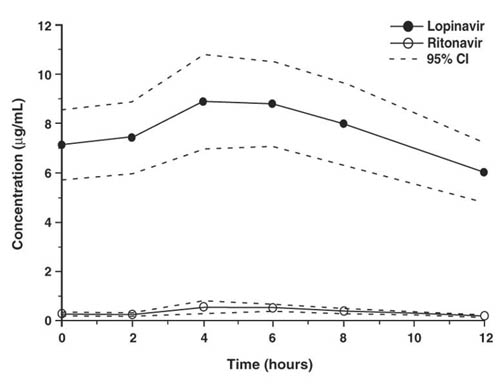
In a pharmacokinetic study in HIV-1 positive subjects (n = 19), multiple dosing with 400/100 mg KALETRA twice daily with food for 3 weeks produced a mean ± SD lopinavir peak plasma concentration (C max) of 9.8 ± 3.7 µg/mL, occurring approximately 4 hours after administration. The mean steady-state trough concentration prior to the morning dose was 7.1 ± 2.9 µg/mL and minimum concentration within a dosing interval was 5.5 ± 2.7 µg/mL. Lopinavir AUC over a 12 hour dosing interval averaged 92.6 ± 36.7 µg •h/mL. The absolute bioavailability of lopinavir co-formulated with ritonavir in humans has not been established. Under nonfasting conditions (500 kcal, 25% from fat), lopinavir concentrations were similar following administration of KALETRA co-formulated capsules and oral solution. When administered under fasting conditions, both the mean AUC and C max of lopinavir were 22% lower for the KALETRA oral solution relative to the capsule formulation.
Plasma concentrations of lopinavir and ritonavir after administration of two 200/50 mg KALETRA tablets are similar to three 133.3/33.3 mg KALETRA capsules under fed conditions with less pharmacokinetic variability.
Effects of Food on Oral Absorption
No clinically significant changes in C max and AUC were observed following administration of KALETRA tablets under fed conditions compared to fasted conditions. Relative to fasting, administration of KALETRA tablets with a moderate fat meal (500 - 682 Kcal, 23 to 25% calories from fat) increased lopinavir AUC and C max by 26.9% and 17.6%, respectively. Relative to fasting, administration of KALETRA tablets with a high fat meal (872 Kcal, 56% from fat) increased lopinavir AUC by 18.9% but not C max. Therefore, KALETRA tablets may be taken with or without food.
Relative to fasting, administration of KALETRA oral solution with a moderate fat meal (500 - 682 Kcal, 23 to 25% calories from fat) increased lopinavir AUC and C max by 80 and 54%, respectively. Relative to fasting, administration of KALETRA oral solution with a high fat meal (872 Kcal, 56% from fat) increased lopinavir AUC and C max by 130% and 56%, respectively. To enhance bioavailability and minimize pharmacokinetic variability KALETRA oral solution should be taken with food.
At steady state, lopinavir is approximately 98-99% bound to plasma proteins. Lopinavir binds to both alpha-1-acid glycoprotein (AAG) and albumin; however, it has a higher affinity for AAG. At steady state, lopinavir protein binding remains constant over the range of observed concentrations after 400/100 mg KALETRA twice daily, and is similar between healthy volunteers and HIV-1 positive patients.
In vitro experiments with human hepatic microsomes indicate that lopinavir primarily undergoes oxidative metabolism. Lopinavir is extensively metabolized by the hepatic cytochrome P450 system, almost exclusively by the CYP3A isozyme. Ritonavir is a potent CYP3A inhibitor which inhibits the metabolism of lopinavir, and therefore increases plasma levels of lopinavir. A 14C-lopinavir study in humans showed that 89% of the plasma radioactivity after a single 400/100 mg KALETRA dose was due to parent drug. At least 13 lopinavir oxidative metabolites have been identified in man. Ritonavir has been shown to induce metabolic enzymes, resulting in the induction of its own metabolism. Pre-dose lopinavir concentrations decline with time during multiple dosing, stabilizing after approximately 10 to 16 days.
Following a 400/100 mg 14C-lopinavir/ritonavir dose, approximately 10.4 ± 2.3% and 82.6 ± 2.5% of an administered dose of 14C-lopinavir can be accounted for in urine and feces, respectively, after 8 days. Unchanged lopinavir accounted for approximately 2.2 and 19.8% of the administered dose in urine and feces, respectively. After multiple dosing, less than 3% of the lopinavir dose is excreted unchanged in the urine. The apparent oral clearance (CL/F) of lopinavir is 5.98 ± 5.75 L/hr (mean ± SD, n = 19).
The pharmacokinetics of once daily KALETRA have been evaluated in HIV-1 infected subjects naïve to antiretroviral treatment. KALETRA 800/200 mg was administered in combination with emtricitabine 200 mg and tenofovir DF 300 mg as part of a once daily regimen. Multiple dosing of 800/200 mg KALETRA once daily for 4 weeks with food (n = 24) produced a mean ± SD lopinavir peak plasma concentration (C max) of 11.8 ± 3.7 µg/mL, occurring approximately 6 hours after administration. The mean steady-state lopinavir trough concentration prior to the morning dose was 3.2 ± 2.1 µg/mL and minimum concentration within a dosing interval was 1.7 ± 1.6 µg/mL. Lopinavir AUC over a 24 hour dosing interval averaged 154.1 ± 61.4 µg• h/mL.
The pharmacokinetics of once daily KALETRA has also been evaluated in treatment experienced HIV-1 infected subjects. Lopinavir exposure (C max, AUC [0-24h], C trough) with once daily KALETRA administration in treatment experienced subjects is comparable to the once daily lopinavir exposure in treatment naïve subjects.
QTcF interval was evaluated in a randomized, placebo and active (moxifloxacin 400 mg once daily) controlled crossover study in 39 healthy adults, with 10 measurements over 12 hours on Day 3. The maximum mean time-matched (95% upper confidence bound) differences in QTcF interval from placebo after baseline-correction were 5.3 (8.1) and 15.2 (18.0) mseconds (msec) for 400/100 mg twice daily and supratherapeutic 800/200 mg twice daily KALETRA, respectively. KALETRA 800/200 mg twice daily resulted in a Day 3 mean C max approximately 2-fold higher than the mean C max observed with the approved once daily and twice daily KALETRA doses at steady state.
PR interval prolongation was also noted in subjects receiving KALETRA in the same study on Day 3. The maximum mean (95% upper confidence bound) difference from placebo in the PR interval after baseline-correction were 24.9 (21.5, 28.3) and 31.9 (28.5, 35.3) msec for 400/100 mg twice daily and supratherapeutic 800/200 mg twice daily KALETRA, respectively [see Warnings and Precautions ( 5.5, 5.6)] .
No gender related pharmacokinetic differences have been observed in adult patients. No clinically important pharmacokinetic differences due to race have been identified. Lopinavir pharmacokinetics have not been studied in elderly patients.
The pharmacokinetics of KALETRA oral solution 300/75 mg/m 2 twice daily and 230/57.5 mg/m 2 twice daily have been studied in a total of 53 pediatric patients in Study 940, ranging in age from 6 months to 12 years [see Clinical Studies ( 14.4)] . The 230/57.5 mg/m 2 twice daily regimen without nevirapine and the 300/75 mg/m 2 twice daily regimen with nevirapine provided lopinavir plasma concentrations similar to those obtained in adult patients receiving the 400/100 mg twice daily regimen (without nevirapine).
The mean steady-state lopinavir AUC, C max, and C min were 72.6 ± 31.1 µg •h/mL, 8.2 ± 2.9 and 3.4 ± 2.1 µg/mL, respectively after KALETRA oral solution 230/57.5 mg/m 2 twice daily without nevirapine (n = 12), and were 85.8 ± 36.9 µg • h/mL, 10.0 ± 3.3 and 3.6 ± 3.5 µg/mL, respectively, after 300/75 mg/m 2 twice daily with nevirapine (n = 12). The nevirapine regimen was 7 mg/kg twice daily (6 months to 8 years) or 4 mg/kg twice daily (> 8 years).
The pharmacokinetics of KALETRA oral solution at approximately 300/75 mg/m 2 twice daily have also been evaluated in infants at approximately 6 weeks of age (n = 9) and between 6 weeks and 6 months of age (n = 18) in Study 1030. The mean steady-state lopinavir AUC 12, C max, and C 12 were 43.4 ± 14.8 µg• h/mL, 5.2 ± 1.8 µg/mL and 1.9 ± 1.1 µg/mL, respectively, in infants at approximately 6 weeks of age, and 74.5 ± 37.9 µg• h/mL, 9.4 ± 4.9 and 3.1 ± 1.8 µg/mL, respectively, in infants between 6 weeks and 6 months of age after KALETRA oral solution was administered at approximately 300/75 mg/m 2 twice daily without concomitant NNRTI therapy.
The pharmacokinetics of KALETRA soft gelatin capsule and oral solution (Group 1: 400/100 mg/m 2 twice daily + 2 NRTIs; Group 2: 480/120 mg/m 2 twice daily + ≥ 1 NRTI + 1 NNRTI) have been evaluated in children and adolescents age ≥ 2 years to < 18 years of age who had failed prior therapy (n=26) in Study 1038. KALETRA doses of 400/100 and 480/120 mg/m 2 resulted in high lopinavir exposure, as almost all subjects had lopinavir AUC 12 above 100 µg•h/mL. Both groups of subjects also achieved relatively high average minimum lopinavir concentrations.
In an open-label pharmacokinetic study, 12 HIV-infected pregnant women received KALETRA 400 mg/100 mg (two 200/50 mg tablets) twice daily as part of an antiretroviral regimen. Plasma concentrations of lopinavir were measured over 12-hour periods during the second trimester (20-24 weeks gestation), the third trimester (30 weeks gestation) and at 8 weeks post-partum. The C 12h values of lopinavir were lower during the second and third trimester by approximately 40% as compared to post-partum, but this decrease is not considered clinically relevant in patients with no documented KALETRA-associated resistance substitutions receiving 400 mg/100 mg twice daily.
Lopinavir pharmacokinetics have not been studied in patients with renal impairment; however, since the renal clearance of lopinavir is negligible, a decrease in total body clearance is not expected in patients with renal impairment.
Lopinavir is principally metabolized and eliminated by the liver. Multiple dosing of KALETRA 400/100 mg twice daily to HIV-1 and HCV co-infected patients with mild to moderate hepatic impairment (n = 12) resulted in a 30% increase in lopinavir AUC and 20% increase in C max compared to HIV-1 infected subjects with normal hepatic function (n = 12). Additionally, the plasma protein binding of lopinavir was statistically significantly lower in both mild and moderate hepatic impairment compared to controls (99.09 vs. 99.31%, respectively). Caution should be exercised when administering KALETRA to subjects with hepatic impairment. KALETRA has not been studied in patients with severe hepatic impairment [see Warnings and Precautions ( 5.4) and Use in Specific Populations ( 8.6)] .
KALETRA is an inhibitor of the P450 isoform CYP3A in vitro. Co-administration of KALETRA and drugs primarily metabolized by CYP3A may result in increased plasma concentrations of the other drug, which could increase or prolong its therapeutic and adverse effects [see Contraindications ( 4) and Drug Interactions ( 7)] .
KALETRA does not inhibit CYP2D6, CYP2C9, CYP2C19, CYP2E1, CYP2B6 or CYP1A2 at clinically relevant concentrations.
KALETRA has been shown in vivo to induce its own metabolism and to increase the biotransformation of some drugs metabolized by cytochrome P450 enzymes and by glucuronidation.
KALETRA is metabolized by CYP3A. Drugs that induce CYP3A activity would be expected to increase the clearance of lopinavir, resulting in lowered plasma concentrations of lopinavir. Although not noted with concurrent ketoconazole, co-administration of KALETRA and other drugs that inhibit CYP3A may increase lopinavir plasma concentrations.
Drug interaction studies were performed with KALETRA and other drugs likely to be co-administered and some drugs commonly used as probes for pharmacokinetic interactions. The effects of co-administration of KALETRA on the AUC, C max and C min are summarized in Table 14 (effect of other drugs on lopinavir) and Table 15 (effect of KALETRA on other drugs). The effects of other drugs on ritonavir are not shown since they generally correlate with those observed with lopinavir (if lopinavir concentrations are decreased, ritonavir concentrations are decreased) unless otherwise indicated in the table footnotes. For information regarding clinical recommendations, see Table 13 in Drug Interactions ( 7) .
| Co-administered Drug | Dose of Co-administered Drug
(mg) | Dose of KALETRA
(mg) | n | Ratio (in combination with Co-administered drug/alone) of Lopinavir Pharmacokinetic Parameters (90% CI); No Effect = 1.00 | ||
| C max | AUC | C min | ||||
| Boceprevir | 800 q8h,
6 d | 400/100 tablet twice daily,
22 d | 13 | 0.70
(0.65, 0.77) | 0.66
12
(0.60, 0.72) | 0.57
(0.49, 0.65) |
| Efavirenz 1,2 | 600 at bedtime,
9 d | 400/100 capsule twice daily,
9 d | 11, 7* | 0.97
(0.78, 1.22) | 0.81
(0.64, 1.03) | 0.61
(0.38, 0.97) |
| 600 at bedtime,
9 d | 500/125 tablet
twice daily, 10 d | 19 | 1.12
(1.02, 1.23) | 1.06
(0.96, 1.17) | 0.90
(0.78, 1.04) |
|
| 600 at bedtime,
9 d | 600/150 tablet
twice daily, 10 d | 23 | 1.36
(1.28, 1.44) | 1.36
(1.28, 1.44) | 1.32
(1.21, 1.44) |
|
| Etravirine | 200 twice daily | 400/100 mg twice day (tablets) | 16 | 0.89
(0.82-0.96) | 0.87
(0.83-0.92) | 0.80
(0.73-0.88) |
| Fosamprenavir 3 | 700 twice daily plus ritonavir 100 twice daily,
14 d | 400/100 capsule twice daily,
14 d | 18 | 1.30
(0.85, 1.47) | 1.37
(0.80, 1.55) | 1.52
(0.72, 1.82) |
| Ketoconazole | 200 single dose | 400/100 capsule twice daily,
16 d | 12 | 0.89
(0.80, 0.99) | 0.87
(0.75, 1.00) | 0.75
(0.55, 1.00) |
| Nelfinavir | 1000 twice daily,
10 d | 400/100 capsule twice daily,
21 d | 13 | 0.79
(0.70, 0.89) | 0.73
(0.63, 0.85) | 0.62
(0.49, 0.78) |
| Nevirapine | 200 twice daily, steady-state
(> 1 yr) 4# | 400/100 capsule twice daily,
steady-state | 22, 19* | 0.81
(0.62, 1.05) | 0.73
(0.53, 0.98) | 0.49
(0.28, 0.74) |
| 7 mg/kg or 4 mg/kg once daily, 2 wk; twice daily
1 wk 5 | (> 1 yr) 300/75 mg/m
2
oral solution twice daily, 3 wk | 12, 15* | 0.86
(0.64, 1.16) | 0.78
(0.56, 1.09) | 0.45
(0.25, 0.81) |
|
| Omeprazole | 40 once daily,
5 d | 400/100 tablet twice daily,
10 d | 12 | 1.08
(0.99, 1.17) | 1.07
(0.99, 1.15) | 1.03
(0.90, 1.18) |
| 40 once daily,
5 d | 800/200 tablet once daily,
10 d | 12 | 0.94
(0.88, 1.00) | 0.92
(0.86, 0.99) | 0.71
(0.57, 0.89) |
|
| Pitavastatin 6 | 4 once daily,
5 d | 400/100 tablet twice daily,
16 d | 23 | 0.93
(0.88-0.98) | 0.91
(0.86-0.97) | N/A |
| Pravastatin | 20 once daily,
4 d | 400/100 capsule twice daily,
14 d | 12 | 0.98
(0.89, 1.08) | 0.95
(0.85, 1.05) | 0.88
(0.77, 1.02) |
| Rifabutin | 150 once daily,
10 d | 400/100 capsule twice daily,
20 d | 14 | 1.08
(0.97, 1.19) | 1.17
(1.04, 1.31) | 1.20
(0.96, 1.65) |
| Ranitidine | 150 single dose | 400/100 tablet twice daily,
10 d | 12 | 0.99
(0.95, 1.03) | 0.97
(0.93, 1.01) | 0.90
(0.85, 0.95) |
| 150 single dose | 800/200 tablet once daily,
10 d | 10 | 0.97
(0.95, 1.00) | 0.95
(0.91, 0.99) | 0.82
(0.74, 0.91) |
|
| Rifampin | 600 once daily,
10 d | 400/100 capsule twice daily,
20 d | 22 | 0.45
(0.40, 0.51) | 0.25
(0.21, 0.29) | 0.01
(0.01, 0.02) |
| 600 once daily,
14 d | 800/200 capsule twice daily,
9 d 7 | 10 | 1.02
(0.85, 1.23) | 0.84
(0.64, 1.10) | 0.43
(0.19, 0.96) |
|
| 600 once daily,
14 d | 400/400 capsule twice daily,
9 d 8 | 9 | 0.93
(0.81, 1.07) | 0.98
(0.81, 1.17) | 1.03
(0.68, 1.56) |
|
| Rilpivirine | 150 once daily 13 | 400/100 twice daily (capsules) | 15 | 0.96
(0.88-1.05) | 0.99
(0.89-1.10) | 0.89
(0.73-1.08) |
| Ritonavir 4 | 100 twice daily,
3-4 wk # | 400/100 capsule twice daily,
3-4 wk | 8, 21* | 1.28
(0.94, 1.76) | 1.46
(1.04, 2.06) | 2.16
(1.29, 3.62) |
| Tenofovir 9 | 300 once daily,
14 d | 400/100 capsule twice daily,
14 d | 24 | NC † | NC † | NC † |
| Tipranavir/ ritonavir 4 | 500/200 twice daily
(28 doses) # | 400/100 capsule twice daily
(27 doses) | 21
69 | 0.53
(0.40, 0.69) 10 | 0.45
(0.32, 0.63) 10 | 0.30 (0.17, 0.51)
10
0.48 (0.40, 0.58) 11 |
| All interaction studies conducted in healthy, HIV-1 negative subjects unless otherwise indicated.
1 The pharmacokinetics of ritonavir are unaffected by concurrent efavirenz. 2 Reference for comparison is lopinavir/ritonavir 400/100 mg twice daily without efavirenz. 3 Data extracted from the fosamprenavir package insert. 4 Study conducted in HIV-1 positive adult subjects. 5 Study conducted in HIV-1 positive pediatric subjects ranging in age from 6 months to 12 years. 6 Data extracted from the pitavastatin package insert and results presented at the 2011 International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention (Morgan , et al, poster #MOPE170). 7 Titrated to 800/200 twice daily as 533/133 twice daily x 1 d, 667/167 twice daily x 1 d, then 800/200 twice daily x 7 d, compared to 400/100 twice daily x 10 days alone. 8 Titrated to 400/400 twice daily as 400/200 twice daily x 1 d, 400/300 twice daily x 1 d, then 400/400 twice daily x 7 d, compared to 400/100 twice daily x 10 days alone. 9 Data extracted from the tenofovir package insert. 10 Intensive PK analysis. 11 Drug levels obtained at 8-16 hrs post-dose. 12 AUC parameter is AUC (0-last) 13 This interaction study has been performed with a dose higher than the recommended dose for rilpivirine (25 mg once daily) assessing the maximal effect on the co-administered drug. * Parallel group design; n for KALETRA + co-administered drug, n for KALETRA alone. N/A = Not available. † NC = No change. # For the nevirapine 200 mg twice daily study, ritonavir, and tipranavir/ritonavir studies, KALETRA was administered with or without food. For all other studies, KALETRA was administered with food. |
||||||
| Co-administered Drug | Dose of Co-administered Drug
(mg) | Dose of KALETRA
(mg) | n | Ratio (in combination with KALETRA/alone) of Co-administered Drug Pharmacokinetic Parameters (90% CI); No Effect = 1.00 | ||
| C max | AUC | C min | ||||
| Bedaquiline 1 | 400 single dose | 400/100 twice daily, 24 d | N/A | N/A | 1.22
(1.11, 1.34) | N/A |
| Boceprevir | 800 q8h, 6 d | 400/100 tablet twice daily, 22 d | 13 9 | 0.50
(0.45, 0.55) | 0.55
(0.49, 0.61) | 0.43
(0.36, 0.53) |
| Desipramine 3 | 100 single dose | 400/100
capsule twice daily, 10 d | 15 | 0.91
(0.84, 0.97) | 1.05
(0.96, 1.16) | N/A |
| Efavirenz | 600 at bedtime, 9 d | 400/100
capsule twice daily, 9 d | 11, 12* | 0.91
(0.72, 1.15) | 0.84
(0.62, 1.15) | 0.84
(0.58, 1.20) |
| Ethinyl Estradiol | 35 µg once daily, 21 d
(Ortho Novum ®) | 400/100
capsule twice daily, 14 d | 12 | 0.59
(0.52, 0.66) | 0.58
(0.54, 0.62) | 0.42
(0.36, 0.49) |
| Etravirine | 200 twice daily | 400/100
twice day (tablets) | 16 | 0.70
(0.64-0.78) | 0.65
(0.59-0.71) | 0.55
(0.49-0.62) |
| Fosamprenavir 4 | 700 twice daily plus ritonavir 100 twice daily, 14 d | 400/100
capsule twice daily, 14 d | 18 | 0.42
(0.30, 0.58) | 0.37
(0.28, 0.49) | 0.35
(0.27, 0.46) |
| Indinavir 2 | 600 twice daily, 10 d combo nonfasting vs. 800 three times daily, 5 d alone fasting | 400/100
capsule twice daily, 15 d | 13 | 0.71
(0.63, 0.81) | 0.91
(0.75, 1.10) | 3.47
(2.60, 4.64) |
| Ketoconazole | 200 single dose | 400/100
capsule twice daily, 16 d | 12 | 1.13
(0.91, 1.40) | 3.04
(2.44, 3.79) | N/A |
| Methadone | 5 single dose | 400/100
capsule twice daily, 10 d | 11 | 0.55
(0.48, 0.64) | 0.47
(0.42, 0.53) | N/A |
| Nelfinavir 2 | 1000 twice daily, 10 d combo vs. 1250 twice daily 14 d alone | 400/100
capsule twice daily, 21 d | 13 | 0.93
(0.82, 1.05) | 1.07
(0.95, 1.19) | 1.86
(1.57, 2.22) |
| M8 metabolite | 2.36
(1.91, 2.91) | 3.46
(2.78, 4.31) | 7.49
(5.85, 9.58) |
|||
| Nevirapine | 200 once daily, 14 d; twice daily, 6 d | 400/100
capsule twice daily, 20 d | 5, 6* | 1.05
(0.72, 1.52) | 1.08
(0.72, 1.64) | 1.15
(0.71, 1.86) |
| Norethindrone | 1 once daily, 21 d
(Ortho Novum ®) | 400/100
capsule twice daily, 14 d | 12 | 0.84
(0.75, 0.94) | 0.83
(0.73, 0.94) | 0.68
(0.54, 0.85) |
| Pitavastatin 5 | 4 once daily, 5 d | 400/100 tablet twice daily, 16 d | 23 | 0.96
(0.84-1.10) | 0.80
(0.73-0.87) | N/A |
| Pravastatin | 20 once daily, 4 d | 400/100 capsule twice daily, 14 d | 12 | 1.26
(0.87, 1.83) | 1.33
(0.91, 1.94) | N/A |
| Rifabutin | 150 once daily, 10 d; combo vs. 300 once daily, 10 d; alone | 400/100 capsule twice daily, 10 d | 12 | 2.12
(1.89, 2.38) | 3.03
(2.79, 3.30) | 4.90
(3.18, 5.76) |
| 25- O-desacetyl rifabutin | 23.6
(13.7, 25.3) | 47.5
(29.3, 51.8) | 94.9
(74.0, 122) |
|||
| Rifabutin + 25- O-desacetyl rifabutin 6 | 3.46
(3.07, 3.91) | 5.73
(5.08, 6.46) | 9.53
(7.56, 12.01) |
|||
| Rilpivirine | 150 once daily 10 | 400/100
twice daily (capsules) | 15 | 1.29
(1.18-1.40) | 1.52
(1.36-1.70) | 1.74
(1.46-2.08) |
| Rosuvastatin 7 | 20 once daily, 7 d | 400/100 tablet twice daily, 7 d | 15 | 4.66
(3.4, 6.4) | 2.08
(1.66, 2.6) | 1.04
(0.9, 1.2) |
| Tenofovir 8 | 300 once daily, 14 d | 400/100 capsule twice daily, 14 d | 24 | NC † | 1.32
(1.26, 1.38) | 1.51
(1.32, 1.66) |
| All interaction studies conducted in healthy, HIV-1 negative subjects unless otherwise indicated.
1 Data extracted from the bedaquiline package insert. 2 Ratio of parameters for indinavir, and nelfinavir, are not normalized for dose. 3 Desipramine is a probe substrate for assessing effects on CYP2D6-mediated metabolism. 4 Data extracted from the fosamprenavir package insert. 5 Data extracted from the pitavastatin package insert and results presented at the 2011 International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention (Morgan , et al, poster #MOPE170). 6 Effect on the dose-normalized sum of rifabutin parent and 25- O-desacetyl rifabutin active metabolite. 7 Kiser, et al. J Acquir Immune Defic Syndr. 2008 Apr 15;47(5):570-8. 8 Data extracted from the tenofovir package insert. 9 N=12 for C min (test arm) 10 This interaction study has been performed with a dose higher than the recommended dose for rilpivirine (25 mg once daily) assessing the maximal effect on the co-administered drug. * Parallel group design; n for KALETRA + co-administered drug, n for co-administered drug alone. N/A = Not available. † NC = No change. |
||||||
12.4 Microbiology
Lopinavir, an inhibitor of the HIV-1 protease, prevents cleavage of the Gag-Pol polyprotein, resulting in the production of immature, non-infectious viral particles.
The antiviral activity of lopinavir against laboratory HIV strains and clinical HIV-1 isolates was evaluated in acutely infected lymphoblastic cell lines and peripheral blood lymphocytes, respectively. In the absence of human serum, the mean 50% effective concentration (EC 50) values of lopinavir against five different HIV-1 subtype B laboratory strains ranged from 10-27 nM (0.006-0.017 µg/mL, 1 µg/mL = 1.6 µM) and ranged from 4-11 nM (0.003-0.007 µg/mL) against several HIV-1 subtype B clinical isolates (n = 6). In the presence of 50% human serum, the mean EC 50 values of lopinavir against these five HIV-1 laboratory strains ranged from 65-289 nM (0.04-0.18 µg/mL), representing a 7 to 11-fold attenuation. Combination antiviral drug activity studies with lopinavir in cell cultures demonstrated additive to antagonistic activity with nelfinavir and additive to synergistic activity with amprenavir, atazanavir, indinavir, saquinavir and tipranavir. The EC 50 values of lopinavir against three different HIV-2 strains ranged from 12-180 nM (0.008-113 μg/mL).
HIV-1 isolates with reduced susceptibility to lopinavir have been selected in cell culture. The presence of ritonavir does not appear to influence the selection of lopinavir-resistant viruses in cell culture.
The selection of resistance to KALETRA in antiretroviral treatment naïve patients has not yet been characterized. In a study of 653 antiretroviral treatment naïve patients (Study 863), plasma viral isolates from each patient on treatment with plasma HIV-1 RNA > 400 copies/mL at Week 24, 32, 40 and/or 48 were analyzed. No evidence of resistance to KALETRA was observed in 37 evaluable KALETRA-treated patients (0%). Evidence of genotypic resistance to nelfinavir, defined as the presence of the D30N and/or L90M substitution in HIV-1 protease, was observed in 25/76 (33%) of evaluable nelfinavir-treated patients. The selection of resistance to KALETRA in antiretroviral treatment naïve pediatric patients (Study 940) appears to be consistent with that seen in adult patients (Study 863).
Resistance to KALETRA has been noted to emerge in patients treated with other protease inhibitors prior to KALETRA therapy. In studies of 227 antiretroviral treatment naïve and protease inhibitor experienced patients, isolates from 4 of 23 patients with quantifiable (> 400 copies/mL) viral RNA following treatment with KALETRA for 12 to 100 weeks displayed significantly reduced susceptibility to lopinavir compared to the corresponding baseline viral isolates. Three of these patients had previously received treatment with a single protease inhibitor (indinavir, nelfinavir, or saquinavir) and one patient had received treatment with multiple protease inhibitors (indinavir, ritonavir, and saquinavir). All four of these patients had at least 4 substitutions associated with protease inhibitor resistance immediately prior to KALETRA therapy. Following viral rebound, isolates from these patients all contained additional substitutions, some of which are recognized to be associated with protease inhibitor resistance. However, there are insufficient data at this time to identify patterns of lopinavir resistance-associated substitutions in isolates from patients on KALETRA therapy. The assessment of these patterns is under study.
Cross-resistance - Preclinical Studies
Varying degrees of cross-resistance have been observed among HIV-1 protease inhibitors. Little information is available on the cross-resistance of viruses that developed decreased susceptibility to lopinavir during KALETRA therapy.
The antiviral activity in cell culture of lopinavir against clinical isolates from patients previously treated with a single protease inhibitor was determined. Isolates that displayed > 4-fold reduced susceptibility to nelfinavir (n = 13) and saquinavir (n = 4), displayed < 4-fold reduced susceptibility to lopinavir. Isolates with > 4-fold reduced susceptibility to indinavir (n = 16) and ritonavir (n = 3) displayed a mean of 5.7- and 8.3-fold reduced susceptibility to lopinavir, respectively. Isolates from patients previously treated with two or more protease inhibitors showed greater reductions in susceptibility to lopinavir, as described in the following paragraph.
Clinical Studies - Antiviral Activity of KALETRA in Patients with Previous Protease Inhibitor Therapies
The clinical relevance of reduced susceptibility in cell culture to lopinavir has been examined by assessing the virologic response to KALETRA therapy in treatment-experienced patients, with respect to baseline viral genotype in three studies and baseline viral phenotype in one study.
Virologic response to KALETRA has been shown to be affected by the presence of three or more of the following amino acid substitutions in protease at baseline: L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T, and I84V. Table 16 shows the 48-week virologic response (HIV-1 RNA <400 copies/mL) according to the number of the above protease inhibitor resistance-associated substitutions at baseline in studies 888 and 765 [see Clinical Studies ( 14.2) and ( 14.3)] and study 957 (see below). Once daily administration of KALETRA for adult patients with three or more of the above substitutions is not recommended.
| Number of protease inhibitor substitutions at baseline 1 | Study 888 (Single protease inhibitor-experienced 2, NNRTI-naïve) n=130 | Study 765 (Single protease inhibitor-experienced 3, NNRTI-naïve) n=56 | Study 957 (Multiple protease inhibitor-experienced 4, NNRTI-naïve) n=50 |
| 0-2 | 76/103 (74%) | 34/45 (76%) | 19/20 (95%) |
| 3-5 | 13/26 (50%) | 8/11 (73%) | 18/26 (69%) |
| 6 or more | 0/1 (0%) | N/A | 1/4 (25%) |
| 1 Substitutions considered in the analysis included L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T, and I84V.
2 43% indinavir, 42% nelfinavir, 10% ritonavir, 15% saquinavir. 3 41% indinavir, 38% nelfinavir, 4% ritonavir, 16% saquinavir. 4 86% indinavir, 54% nelfinavir, 80% ritonavir, 70% saquinavir. |
|||
Virologic response to KALETRA therapy with respect to phenotypic susceptibility to lopinavir at baseline was examined in Study 957. In this study 56 NNRTI-naïve patients with HIV-1 RNA >1,000 copies/mL despite previous therapy with at least two protease inhibitors selected from indinavir, nelfinavir, ritonavir, and saquinavir were randomized to receive one of two doses of KALETRA in combination with efavirenz and nucleoside reverse transcriptase inhibitors (NRTIs). The EC 50 values of lopinavir against the 56 baseline viral isolates ranged from 0.5- to 96-fold the wild-type EC 50 value. Fifty-five percent (31/56) of these baseline isolates displayed >4-fold reduced susceptibility to lopinavir. These 31 isolates had a median reduction in lopinavir susceptibility of 18-fold. Response to therapy by baseline lopinavir susceptibility is shown in Table 17.
| Lopinavir susceptibility 2 at baseline | HIV-1 RNA <400 copies/mL (%) | HIV-1 RNA <50 copies/mL (%) |
| < 10 fold | 25/27 (93%) | 22/27 (81%) |
| > 10 and < 40 fold | 11/15 (73%) | 9/15 (60%) |
| ≥ 40 fold | 2/8 (25%) | 2/8 (25%) |
| 1 Lopinavir susceptibility was determined by recombinant phenotypic technology performed by Virologic.
2 Fold change in susceptibility from wild type. |
||
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Lopinavir/ritonavir combination was evaluated for carcinogenic potential by oral gavage administration to mice and rats for up to 104 weeks. Results showed an increase in the incidence of benign hepatocellular adenomas and an increase in the combined incidence of hepatocellular adenomas plus carcinoma in both males and females in mice and males in rats at doses that produced approximately 1.6-2.2 times (mice) and 0.5 times (rats) the human exposure (based on AUC 0-24hr measurement) at the recommended dose of 400/100 mg KALETRA twice daily. Administration of lopinavir/ritonavir did not cause a statistically significant increase in the incidence of any other benign or malignant neoplasm in mice or rats.
Carcinogenicity studies in mice and rats have been carried out on ritonavir. In male mice, there was a dose dependent increase in the incidence of both adenomas and combined adenomas and carcinomas in the liver. Based on AUC measurements, the exposure at the high dose was approximately 4-fold for males that of the exposure in humans with the recommended therapeutic dose (400/100 mg KALETRA twice daily). There were no carcinogenic effects seen in females at the dosages tested. The exposure at the high dose was approximately 9-fold for the females that of the exposure in humans. There were no carcinogenic effects in rats. In this study, the exposure at the high dose was approximately 0.7-fold that of the exposure in humans with the 400/100 mg KALETRA twice daily regimen. Based on the exposures achieved in the animal studies, the significance of the observed effects is not known.
Neither lopinavir nor ritonavir was found to be mutagenic or clastogenic in a battery of in vitro and in vivo assays including the Ames bacterial reverse mutation assay using S. typhimurium and E. coli, the mouse lymphoma assay, the mouse micronucleus test and chromosomal aberration assays in human lymphocytes.
Lopinavir in combination with ritonavir at a 2:1 ratio produced no effects on fertility in male and female rats at levels of 10/5, 30/15 or 100/50 mg/kg/day. Based on AUC measurements, the exposures in rats at the high doses were approximately 0.7-fold for lopinavir and 1.8-fold for ritonavir of the exposures in humans at the recommended therapeutic dose (400/100 mg twice daily).
14 CLINICAL STUDIES
14.1 Adult Patients without Prior Antiretroviral Therapy
Study 863: KALETRA Capsules twice daily + stavudine + lamivudine compared to nelfinavir three times daily + stavudine + lamivudine
Study 863 was a randomized, double-blind, multicenter trial comparing treatment with KALETRA capsules (400/100 mg twice daily) plus stavudine and lamivudine versus nelfinavir (750 mg three times daily) plus stavudine and lamivudine in 653 antiretroviral treatment naïve patients. Patients had a mean age of 38 years (range: 19 to 84), 57% were Caucasian, and 80% were male. Mean baseline CD4+ cell count was 259 cells/mm 3 (range: 2 to 949 cells/mm 3) and mean baseline plasma HIV-1 RNA was 4.9 log 10 copies/mL (range: 2.6 to 6.8 log 10 copies/mL).
Treatment response and outcomes of randomized treatment are presented in Table 18.
| Outcome | KALETRA+d4T+3TC
(N = 326) | Nelfinavir+d4T+3TC
(N = 327) |
| Responder 1 | 75% | 62% |
| Virologic failure
2
Rebound Never suppressed through Week 48 | 9%
7% 2% | 25%
15% 9% |
| Death | 2% | 1% |
| Discontinued due to adverse events | 4% | 4% |
| Discontinued for other reasons 3 | 10% | 8% |
| 1 Patients achieved and maintained confirmed HIV-1 RNA < 400 copies/mL through Week 48.
2 Includes confirmed viral rebound and failure to achieve confirmed < 400 copies/mL through Week 48. 3 Includes lost to follow-up, patient's withdrawal, non-compliance, protocol violation and other reasons. Overall discontinuation through Week 48, including patients who discontinued subsequent to virologic failure, was 17% in the KALETRA arm and 24% in the nelfinavir arm. |
||
Through 48 weeks of therapy, there was a statistically significantly higher proportion of patients in the KALETRA arm compared to the nelfinavir arm with HIV-1 RNA < 400 copies/mL (75% vs. 62%, respectively) and HIV-1 RNA < 50 copies/mL (67% vs. 52%, respectively). Treatment response by baseline HIV-1 RNA level subgroups is presented in Table 19.
| Baseline Viral Load (HIV-1 RNA copies/mL) | KALETRA +d4T+3TC | Nelfinavir +d4T+3TC | ||||
| <400 copies/mL 1 | <50 copies/mL 2 | n | <400 copies/mL 1 | <50 copies/mL 2 | n | |
| < 30,000 | 74% | 71% | 82 | 79% | 72% | 87 |
| ≥ 30,000 to < 100,000 | 81% | 73% | 79 | 67% | 54% | 79 |
| ≥ 100,000 to < 250,000 | 75% | 64% | 83 | 60% | 47% | 72 |
| ≥ 250,000 | 72% | 60% | 82 | 44% | 33% | 89 |
| 1 Patients achieved and maintained confirmed HIV-1 RNA < 400 copies/mL through Week 48.
2 Patients achieved HIV-1 RNA < 50 copies/mL at Week 48. |
||||||
Through 48 weeks of therapy, the mean increase from baseline in CD4+ cell count was 207 cells/mm 3 for the KALETRA arm and 195 cells/mm 3 for the nelfinavir arm.
Study 730: KALETRA Tablets once daily + tenofovir DF + emtricitabine compared to KALETRA Tablets twice daily + tenofovir DF + emtricitabine
Study 730 was a randomized, open-label, multicenter trial comparing treatment with KALETRA 800/200 mg once daily plus tenofovir DF and emtricitabine versus KALETRA 400/100 mg twice daily plus tenofovir DF and emtricitabine in 664 antiretroviral treatment-naïve patients. Patients were randomized in a 1:1 ratio to receive either KALETRA 800/200 mg once daily (n = 333) or KALETRA 400/100 mg twice daily (n = 331). Further stratification within each group was 1:1 (tablet vs. capsule). Patients administered the capsule were switched to the tablet formulation at Week 8 and maintained on their randomized dosing schedule. Patients were administered emtricitabine 200 mg once daily and tenofovir DF 300 mg once daily. Mean age of patients enrolled was 39 years (range: 19 to 71); 75% were Caucasian, and 78% were male. Mean baseline CD4+ cell count was 216 cells/mm 3 (range: 20 to 775 cells/mm 3) and mean baseline plasma HIV-1 RNA was 5.0 log 10 copies/mL (range: 1.7 to 7.0 log 10 copies/mL).
Treatment response and outcomes of randomized treatment through Week 48 are presented in Table 20.
| Outcome | KALETRA Once Daily + TDF + FTC
(n = 333) | KALETRA Twice Daily + TDF + FTC
(n = 331) |
| Responder 1 | 78% | 77% |
| Virologic failure
2
Rebound Never suppressed through Week 48 | 10%
5% 5% | 8%
5% 3% |
| Death | 1% | <1% |
| Discontinued due to adverse events | 4% | 3% |
| Discontinued for other reasons 3 | 8% | 11% |
| 1 Patients achieved and maintained confirmed HIV-1 RNA < 50 copies/mL through Week 48.
2 Includes confirmed viral rebound and failure to achieve confirmed < 50 copies/mL through Week 48. 3 Includes lost to follow-up, patient's withdrawal, non-compliance, protocol violation and other reasons. |
||
Through 48 weeks of therapy, 78% in the KALETRA once daily arm and 77% in the KALETRA twice daily arm achieved and maintained HIV-1 RNA < 50 copies/mL (95% confidence interval for the difference, -5.9% to 6.8%). Mean CD4+ cell count increases at Week 48 were 186 cells/mm 3 for the KALETRA once daily arm and 198 cells/mm 3 for the KALETRA twice daily arm.
14.2 Adult Patients with Prior Antiretroviral Therapy
Study 888: KALETRA Capsules twice daily + nevirapine + NRTIs compared to investigator-selected protease inhibitor(s) + nevirapine + NRTIs
Study 888 was a randomized, open-label, multicenter trial comparing treatment with KALETRA capsules (400/100 mg twice daily) plus nevirapine and nucleoside reverse transcriptase inhibitors versus investigator-selected protease inhibitor(s) plus nevirapine and nucleoside reverse transcriptase inhibitors in 288 single protease inhibitor-experienced, non-nucleoside reverse transcriptase inhibitor (NNRTI)-naïve patients. Patients had a mean age of 40 years (range: 18 to 74), 68% were Caucasian, and 86% were male. Mean baseline CD4+ cell count was 322 cells/mm 3 (range: 10 to 1059 cells/mm 3) and mean baseline plasma HIV-1 RNA was 4.1 log 10 copies/mL (range: 2.6 to 6.0 log 10 copies/mL).
Treatment response and outcomes of randomized treatment through Week 48 are presented in Table 21.
| Outcome | KALETRA + nevirapine + NRTIs
(n = 148) | Investigator-Selected Protease Inhibitor(s) + nevirapine + NRTIs
(n = 140) |
| Responder 1 | 57% | 33% |
| Virologic failure
2
Rebound Never suppressed through Week 48 | 24%
11% 13% | 41%
19% 23% |
| Death | 1% | 2% |
| Discontinued due to adverse events | 5% | 11% |
| Discontinued for other reasons 3 | 14% | 13% |
| 1 Patients achieved and maintained confirmed HIV-1 RNA < 400 copies/mL through Week 48.
2 Includes confirmed viral rebound and failure to achieve confirmed < 400 copies/mL through Week 48. 3 Includes lost to follow-up, patient's withdrawal, non-compliance, protocol violation and other reasons. |
||
Through 48 weeks of therapy, there was a statistically significantly higher proportion of patients in the KALETRA arm compared to the investigator-selected protease inhibitor(s) arm with HIV-1 RNA < 400 copies/mL (57% vs. 33%, respectively).
Through 48 weeks of therapy, the mean increase from baseline in CD4+ cell count was 111 cells/mm 3 for the KALETRA arm and 112 cells/mm 3 for the investigator-selected protease inhibitor(s) arm.
Study 802: KALETRA Tablets 800/200 mg Once Daily Versus 400/100 mg Twice Daily when Co-administered with Nucleoside/Nucleotide Reverse Transcriptase Inhibitors in Antiretroviral-Experienced, HIV-1 Infected Subjects
M06-802 was a randomized open-label study comparing the safety, tolerability, and antiviral activity of once daily and twice daily dosing of KALETRA tablets in 599 subjects with detectable viral loads while receiving their current antiviral therapy. Of the enrolled subjects, 55% on both treatment arms had not been previously treated with a protease inhibitor and 81 – 88% had received prior NNRTIs as part of their anti-HIV treatment regimen. Patients were randomized in a 1:1 ratio to receive either KALETRA 800/200 mg once daily (n = 300) or KALETRA 400/100 mg twice daily (n = 299). Patients were administered at least two nucleoside/nucleotide reverse transcriptase inhibitors selected by the investigator. Mean age of patients enrolled was 41 years (range: 21 to 73); 51% were Caucasian, and 66% were male. Mean baseline CD4+ cell count was 254 cells/mm 3 (range: 4 to 952 cells/mm 3) and mean baseline plasma HIV-1 RNA was 4.3 log 10 copies/mL (range: 1.7 to 6.6 log 10 copies/mL).
Treatment response and outcomes of randomized treatment through Week 48 are presented in Table 22.
| Outcome | KALETRA Once Daily + NRTIs
(n = 300) | KALETRA Twice Daily + NRTIs
(n = 299) |
| Virologic Success (HIV-1 RNA <50 copies/mL) | 57% | 54% |
| Virologic failure 1 | 22% | 24% |
| No virologic data in Week 48 window | ||
| Discontinued study due to adverse event or death 2 | 5% | 7% |
| Discontinued study for other reasons 3 | 13% | 12% |
| Missing data during window but on study | 3% | 3% |
| 1 Includes patients who discontinued prior to Week 48 for lack or loss of efficacy and patients with HIV-1 RNA ≥ 50 copies/mL at Week 48.
2 Includes patients who discontinued due to adverse events or death at any time from Day 1 through Week 48 if this resulted in no virologic data on treatment at Week 48. 3 Includes withdrawal of consent, loss to follow-up, non-compliance, protocol violation and other reasons. |
||
Through 48 weeks of treatment, the mean change from baseline for CD4 + cell count was 135 cells/mm 3 for the once daily group and 122 cells/mm 3 for the twice daily group.
14.3 Other Studies Supporting Approval in Adult Patients
Study 720: KALETRA twice daily + stavudine + lamivudine
Study 765: KALETRA twice daily + nevirapine + NRTIs
Study 720 (patients without prior antiretroviral therapy) and study 765 (patients with prior protease inhibitor therapy) were randomized, blinded, multi-center trials evaluating treatment with KALETRA at up to three dose levels (200/100 mg twice daily [720 only], 400/100 mg twice daily, and 400/200 mg twice daily). In Study 720, all patients switched to 400/100 mg twice daily between Weeks 48-72. Patients in study 720 had a mean age of 35 years, 70% were Caucasian, and 96% were male, while patients in study 765 had a mean age of 40 years, 73% were Caucasian, and 90% were male. Mean (range) baseline CD4+ cell counts for patients in study 720 and study 765 were 338 (3-918) and 372 (72-807) cells/mm 3, respectively. Mean (range) baseline plasma HIV-1 RNA levels for patients in study 720 and study 765 were 4.9 (3.3 to 6.3) and 4.0 (2.9 to 5.8) log 10 copies/mL, respectively.
Through 360 weeks of treatment in study 720, the proportion of patients with HIV-1 RNA < 400 (< 50) copies/mL was 61% (59%) [n = 100]. Among patients completing 360 weeks of treatment with CD4+ cell count measurements [n=60], the mean (median) increase in CD4+ cell count was 501 (457) cells/mm 3. Thirty-nine patients (39%) discontinued the study, including 13 (13%) discontinuations due to adverse reactions and 1 (1%) death.
Through 144 weeks of treatment in study 765, the proportion of patients with HIV-1 RNA < 400 (< 50) copies/mL was 54% (50%) [n = 70], and the corresponding mean increase in CD4+ cell count was 212 cells/mm 3. Twenty-seven patients (39%) discontinued the study, including 5 (7%) discontinuations secondary to adverse reactions and 2 (3%) deaths.
14.4 Pediatric Studies
Study 1030 was an open-label, multicenter, dose-finding trial evaluating the pharmacokinetic profile, tolerability, safety and efficacy of KALETRA oral solution containing lopinavir 80 mg/mL and ritonavir 20 mg/mL at a dose of 300/75 mg/m 2 twice daily plus 2 NRTIs in HIV-1 infected infants ≥14 days and <6 months of age.
Ten infants, ≥14 days and <6 wks of age, were enrolled at a median (range) age of 5.7 (3.6-6.0) weeks and all completed 24 weeks. At entry, median (range) HIV-1 RNA was 6.0 (4.7-7.2) log 10 copies/mL. Seven of 10 infants had HIV-1 RNA <400 copies/mL at Week 24. At entry, median (range) CD4+ percentage was 41 (16-59) with a median decrease of 1% (95% CI: -10, 18) from baseline to week 24 in 6 infants with available data.
Twenty-one infants, between 6 weeks and 6 months of age, were enrolled at a median (range) age of 14.7 (6.9-25.7) weeks and 19 of 21 infants completed 24 weeks. At entry, median (range) HIV RNA level was 5.8 (3.7-6.9) log 10 copies/mL. Ten of 21 infants had HIV RNA <400 copies/mL at Week 24. At entry, the median (range) CD4+ percentage was 32 (11-54) with a median increase of 4% (95% CI: -1, 9) from baseline to week 24 in 19 infants with available data.
See Clinical Pharmacology ( 12.3) for pharmacokinetic results .
Study 940 was an open-label, multicenter trial evaluating the pharmacokinetic profile, tolerability, safety and efficacy of KALETRA oral solution containing lopinavir 80 mg/mL and ritonavir 20 mg/mL in 100 antiretroviral naïve (44%) and experienced (56%) pediatric patients. All patients were non-nucleoside reverse transcriptase inhibitor naïve. Patients were randomized to either 230 mg lopinavir/57.5 mg ritonavir per m 2 or 300 mg lopinavir/75 mg ritonavir per m 2. Naïve patients also received lamivudine and stavudine. Experienced patients received nevirapine plus up to two nucleoside reverse transcriptase inhibitors.
Safety, efficacy and pharmacokinetic profiles of the two dose regimens were assessed after three weeks of therapy in each patient. After analysis of these data, all patients were continued on the 300 mg lopinavir/75 mg ritonavir per m 2 dose. Patients had a mean age of 5 years (range 6 months to 12 years) with 14% less than 2 years. Mean baseline CD4+ cell count was 838 cells/mm 3 and mean baseline plasma HIV-1 RNA was 4.7 log 10 copies/mL.
Through 48 weeks of therapy, the proportion of patients who achieved and sustained an HIV-1 RNA < 400 copies/mL was 80% for antiretroviral naïve patients and 71% for antiretroviral experienced patients. The mean increase from baseline in CD4+ cell count was 404 cells/mm 3 for antiretroviral naïve and 284 cells/mm 3 for antiretroviral experienced patients treated through 48 weeks. At 48 weeks, two patients (2%) had prematurely discontinued the study. One antiretroviral naïve patient prematurely discontinued secondary to an adverse reaction, while one antiretroviral experienced patient prematurely discontinued secondary to an HIV-1 related event.
Dose selection in pediatric patients was based on the following:
- Among patients 14 days to 6 months of age receiving 300/75 mg/m 2 twice daily without nevirapine, plasma concentrations were lower than those observed in adults or in older children. This dose resulted in HIV-1 RNA < 400 copies/mL in 55% of patients (70% in those initiating treatment at <6 weeks of age).
- Among patients 6 months to 12 years of age, the 230/57.5 mg/m 2 oral solution twice daily regimen without nevirapine and the 300/75 mg/m 2 oral solution twice daily regimen with nevirapine provided lopinavir plasma concentrations similar to those obtained in adult patients receiving the 400/100 mg twice daily regimen (without nevirapine). These doses resulted in treatment benefit (proportion of patients with HIV-1 RNA < 400 copies/mL) similar to that seen in the adult clinical trials.
- Among patients 12 to 18 years of age receiving 400/100 mg/m 2 or 480/120 mg/m 2 (with efavirenz) twice daily, plasma concentrations were 60-100% higher than among 6 to 12 year old patients receiving 230/57.5 mg/m 2. Mean apparent clearance was similar to that observed in adult patients receiving standard dose and in patients 6 to 12 years of age. Although changes in HIV-1 RNA in patients with prior treatment failure were less than anticipated, the pharmacokinetic data supports use of similar dosing as in patients 6 to 12 years of age, not to exceed the recommended adult dose.
- For all age groups, the body surface area dosing was converted to body weight dosing using the patient’s prescribed lopinavir dose.
16 HOW SUPPLIED/STORAGE AND HANDLING
KALETRA ® (lopinavir and ritonavir) tablets and oral solution are available in the following strengths and package sizes:
16.1 KALETRA Tablets, 200 mg lopinavir and 50 mg ritonavir
Yellow film-coated ovaloid tablets debossed with the “a” logo and the code KA:
Bottles of 120 tablets ….…………… (NDC 0074-6799-22)
Store KALETRA tablets at 20°-25°C (68°-77°F); excursions permitted to 15°-30°C (59° to 86°F) [see USP controlled room temperature]. Dispense in original container or USP equivalent tight container (250 mL or less). For patient use: exposure of this product to high humidity outside the original container or USP equivalent tight container (250 mL or less) for longer than 2 weeks is not recommended.
16.2 KALETRA Tablets, 100 mg lopinavir and 25 mg ritonavir
Pale yellow film-coated ovaloid tablets debossed with the “a” logo and the code KC:
Bottles of 60 tablets ….…………… (NDC 0074-0522-60)
Store KALETRA tablets at 20°-25°C (68°-77°F); excursions permitted to 15°-30°C (59° to 86°F)[see USP controlled room temperature]. Dispense in original container or USP equivalent tight container (100 mL or less). For patient use: exposure of this product to high humidity outside the original container or USP equivalent tight container (100 mL or less) for longer than 2 weeks is not recommended.
16.3 KALETRA Oral Solution
KALETRA (lopinavir and ritonavir) oral solution is a light yellow to orange colored liquid supplied in amber-colored multiple-dose bottles containing 400 mg lopinavir and 100 mg ritonavir per 5 mL (80 mg lopinavir and 20 mg ritonavir per mL) packaged with a marked dosing cup in the following size:
160 mL bottle……………………………….(NDC 0074-3956-46)
Store KALETRA oral solution at 2°-8°C (36°-46°F) until dispensed. Avoid exposure to excessive heat. For patient use, refrigerated KALETRA oral solution remains stable until the expiration date printed on the label. If stored at room temperature up to 25°C (77°F), oral solution should be used within 2 months.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide)
Patients or parents of patients should be informed that:
They should pay special attention to accurate administration of their dose to minimize the risk of accidental overdose or underdose of KALETRA.
They should inform their healthcare provider if their children’s weight changes in order to make sure that the child’s KALETRA dose is the correct one.
They should take the prescribed dose of KALETRA as directed and to set up a daily routine in order to do so.
KALETRA tablets may be taken with or without food. KALETRA oral solution should be taken with food to enhance absorption.
Sustained decreases in plasma HIV-1 RNA have been associated with a reduced risk of progression to AIDS and death. Patients should remain under the care of a physician while using KALETRA. Patients should be advised to take KALETRA and other concomitant antiretroviral therapy every day as prescribed. KALETRA must always be used in combination with other antiretroviral drugs. Patients should not alter the dose or discontinue therapy without consulting with their doctor. If a dose of KALETRA is missed patients should take the dose as soon as possible and then return to their normal schedule. However, if a dose is skipped the patient should not double the next dose. The amount of HIV-1 virus in their blood may increase if the medicine is stopped for even a short time. The virus may become resistant to KALETRA and become harder to treat.
KALETRA is not a cure for HIV-1 infection and patients may continue to experience illnesses associated with HIV-1 infection, including opportunistic infections. Patients should remain under the care of a physician when using KALETRA.
Patients should be advised to avoid doing things that can spread HIV-1 infection to others.
- Do not share needles or other injection equipment.
- Do not share personal items that can have blood or body fluids on them, like toothbrushes and razor blades.
- Do not have any kind of sex without protection. Always practice safe sex by using a latex or polyurethane condom to lower the chance of sexual contact with semen, vaginal secretions, or blood.
- Do not breastfeed. Mothers with HIV-1 should not breastfeed because HIV-1 can be passed to the baby in the breast milk.
KALETRA may interact with some drugs; therefore, patients should be advised to report to their doctor the use of any other prescription, non-prescription medication or herbal products, particularly St. John's Wort.
KALETRA tablets can be taken at the same time as didanosine without food. Patients taking didanosine should take didanosine one hour before or two hours after KALETRA oral solution.
If they are receiving avanafil, sildenafil, tadalafil, or vardenafil for the treatment of erectile dysfunction, there may be an increased risk of associated adverse reactions including hypotension, visual changes, and sustained erection, and should promptly report any symptoms to their doctor. If they are currently using or planning to use avanafil or tadalafil (for the treatment of pulmonary arterial hypertension) they should ask their doctor about potential adverse reactions these medications may cause when taken with KALETRA. The doctor may choose not to keep them on avanafil, or may adjust the dose of tadalafil while initiating treatment with KALETRA.
If they are receiving estrogen-based hormonal contraceptives, additional or alternate contraceptive measures should be used during therapy with KALETRA.
If they are taking or before they begin using Serevent ® (salmeterol) and KALETRA, they should talk to their doctor about problems these two medications may cause when taken together. The doctor may choose not to keep someone on Serevent ® (salmeterol).
If they are taking or before they begin taking Advair ® (salmeterol in combination with fluticasone propionate) and KALETRA, they should talk to their doctor about problems these two medications may cause when taken together. The doctor may choose not to keep someone on Advair® (salmeterol in combination with fluticasone propionate).
Skin rashes ranging in severity from mild to toxic epidermal necrolysis (TEN), Stevens-Johnson syndrome, erythema multiforme, urticaria, and angioedema have been reported in patients receiving KALETRA or its components lopinavir and/or ritonavir. Patients should be advised to contact their healthcare provider if they develop a rash while taking KALETRA. The healthcare provider will determine if treatment should be continued or an alternative antiretroviral regimen used.
Patients should be advised that appropriate liver function testing will be conducted prior to initiating and during therapy with KALETRA. Pre-existing liver disease including Hepatitis B or C can worsen with use of KALETRA. This can be seen as worsening of transaminase elevations or hepatic decompensation. Patients should be advised that their liver function tests will need to be monitored closely especially during the first several months of KALETRA treatment and that they should notify their healthcare provider if they develop the signs and symptoms of worsening liver disease including loss of appetite, abdominal pain, jaundice, and itchy skin.
New onset of diabetes or exacerbation of pre-existing diabetes mellitus, and hyperglycemia have been reported during KALETRA use. Patients should be advised to notify their healthcare provider if they develop the signs and symptoms of diabetes mellitus including frequent urination, excessive thirst, extreme hunger or unusual weight loss and/or an increased blood sugar while on KALETRA as they may require a change in their diabetes treatment or new treatment.
KALETRA might produce changes in the electrocardiogram (e.g., PR and/or QT prolongation). Patients should consult their physician if they experience symptoms such as dizziness, lightheadedness, abnormal heart rhythm or loss of consciousness.
They should seek medical assistance immediately if they develop a sustained penile erection lasting more than 4 hours while taking KALETRA and a PDE 5 Inhibitor such as Viagra, Cialis or Levitra.
Redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy and that the cause and long term health effects of these conditions are not known at this time.
Patients should be informed that there may be a greater chance of developing diarrhea with the once daily regimen as compared with the twice daily regimen.
KALETRA Tablets, 200 mg lopinavir and 50 mg ritonavir
Manufactured by AbbVie LTD, Barceloneta, PR 00617
for AbbVie Inc., North Chicago, IL 60064 USA
KALETRA Tablets, 100 mg lopinavir and 25 mg ritonavir and KALETRA Oral Solution
AbbVie Inc., North Chicago, IL 60064 USA
The brands listed are trademarks of their respective owners and are not trademarks of AbbVie Inc. The makers of these brands are not affiliated with and do not endorse AbbVie Inc. or its products.
© 2015 AbbVie Inc. All rights reserved.
Read this Medication Guide before you start taking KALETRA and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or treatment. You and your doctor should talk about your treatment with KALETRA before you start taking it and at regular check-ups. You should stay under your doctor’s care when taking KALETRA.
What is the most important information I should know about KALETRA?
KALETRA may cause serious side effects, including:
- Interactions with other medicines. It is important to know the medicines that should not be taken with KALETRA. For more information, see "Who should not take KALETRA?”
-
Changes in your heart rhythm and the electrical activity of your heart. These changes may be seen on an EKG (electrocardiogram) and can lead to serious heart problems. Your risk for these problems may be higher if you:
- already have a history of abnormal heart rhythm or other types of heart disease.
- take other medicines that can affect your heart rhythm while you take KALETRA.
Tell your doctor right away if you have any of these symptoms while taking KALETRA:
- dizziness
- lightheadedness
- fainting
- sensation of abnormal heartbeats
See “What are the possible side effects of KALETRA?” for more information about serious side effects.
KALETRA is a prescription HIV-1 medicine that is used with other HIV medicines to treat HIV-1 (Human Immunodeficiency Virus) infection in adults and children 14 days of age and older. HIV is the virus that causes AIDS (Acquired Immune Deficiency Syndrome). KALETRA is a type of HIV medicine called a protease inhibitor. KALETRA contains two medicines: lopinavir and ritonavir.
When used with other HIV medicines, KALETRA may help to reduce the amount of HIV in your blood (called “viral load”). KALETRA may also help to increase the number of white blood cells called CD4 (T) cell which help fight off other infections. Reducing the amount of HIV and increasing the CD4 (T) cell count may improve your immune system. This may reduce your risk of death or infections that can happen when your immune system is weak (opportunistic infections).
It is not known if KALETRA is safe and effective in children under 14 days old.
KALETRA does not cure HIV infection or AIDS. People taking KALETRA may develop infections or other conditions associated with HIV infection, including opportunistic infections (for example, pneumonia and herpes virus infections).
Avoid doing things that can spread HIV-1 infection to others:
- Do not share needles or other injection equipment.
- Do not share personal items that can have blood or body fluids on them, like toothbrushes and razor blades.
- Do not have any kind of sex without protection. Always practice safer sex by using a latex or polyurethane condom to lower the chance of sexual contact with semen, vaginal secretions, or blood.
Ask your doctor if you have any questions on how to prevent passing HIV to other people.
Do not take KALETRA if you take any of the following medicines:
- alfuzosin (Uroxatral ®)
- cisapride (Propulsid ®, Quicksolv ®)
- ergot containing medicines including
- ergotamine tartrate (Cafergot ®, Migergot ®, Ergomar ®, Ergostat ®, Medihaler ®, Ergotamine, Wigraine ®, Wigrettes ®)
- dihydroergotamine mesylate (D.H.E. 45 ®, Migranal ®)
- methylergonovine (Methergine ®)
- lovastatin (Advicor ®, Altoprev ®, Mevacor ®)
- midazolam oral syrup
- pimozide (Orap ®)
- rifampin (Rifadin ®, Rifamate ®, Rifater ®, Rimactane ®)
- sildenafil (Revatio ®), when used for the treatment of pulmonary arterial hypertension
- simvastatin (Zocor ®, Vytorin ®, Simcor ®)
- St. John’s Wort (Hypericum perforatum)
- triazolam (Halcion ®)
Serious problems can happen if you or your child take any of the medicines listed above with KALETRA.
- Do not take KALETRA if you are allergic to lopinavir, ritonavir or any of the ingredients in KALETRA. See the end of this Medication Guide for a complete list of ingredients in KALETRA.
What should I tell my doctor before taking KALETRA?
KALETRA may not be right for you. Tell your doctor about all your medical conditions, including if you:
- have any heart problems, including if you have a condition called Congenital Long QT Syndrome.
- have or had pancreas problems.
- have liver problems, including Hepatitis B or Hepatitis C.
- have diabetes.
- have hemophilia. People who take KALETRA may have increased bleeding.
- have low potassium in your blood.
- are pregnant or plan to become pregnant. Taking KALETRA during pregnancy has not been associated with an increased risk of birth defects. You and your doctor should decide if KALETRA is right for you.
Pregnancy Registry. There is a pregnancy registry for women who take antiretroviral medicines during pregnancy. The purpose of the pregnancy registry is to collect information about the health of you and your baby. Talk to your doctor about how you can take part in this registry. - are breastfeeding or plan to breastfeed.
Do not breastfeed if you take KALETRA.
- You should not breastfeed if you have HIV-1 because of the risk of passing HIV-1 to your baby.
- Talk to your doctor about the best way to feed your baby.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Many medicines interact with KALETRA. Do not start taking a new medicine without telling your doctor or pharmacist. Your doctor can tell you if it is safe to take KALETRA with other medicines. Your doctor may need to change the dose of other medicines while you take KALETRA.
Especially tell your doctor if you take:
- medicine to treat HIV
- estrogen-based contraceptives (birth control pills and patches). KALETRA may reduce the effectiveness of estrogen-based contraceptives. During treatment with KALETRA, you should use a different type or an extra form of birth control. Talk to your doctor about what types of birth control you can use to prevent pregnancy while taking KALETRA.
- medicines to prevent organ transplant rejection
- medicines to treat cancer
- amiodarone (Cordarone ®, Pacerone ®)
- atorvastatin (Lipitor ®)
- atovaquone (Marlarone ®, Mepron ®)
- avanafil (Stendra ®), sildenafil (Viagra ®), tadalafil (Cialis ®), or vardenafil (Levitra ®) for the treatment of erectile dysfunction (ED). If you get dizzy or faint (low blood pressure), have vision changes or have an erection that last longer than 4 hours, call your doctor or get medical help right away.
- bedaquiline (Sirturo ®)
- bepridil (Bepadin ®, Vascor ®)
- boceprevir (Victrelis ®)
- bosentan (Tracleer ®)
- budesonide (Rhinocort ®, Symbicort ®, Pulmicort ®, Entocort EC ®)
- bupropion (Aplenzin ®, Forfivo XL ®, Wellbutrin ®, Zyban ®)
- carbamazepine (Carbatrol ®, Epitol ®, Equetro ®, Tegretol ®)
- clarithromycin (Biaxin ®, Prevpac ®)
- colchicine (Colcrys ®)
- dexamethasone (Maxidex ®, Ozurdex ®)
- disulfiram
- felodipine
- fentanyl (Abstral ®, Actiq ®, Duragesic ®, Fentora ®, Lazanda ®, Onsolis ®, Subsys ®)
- fluticasone (Cutivate ®, Flonase ®, Flovent ®, Flovent Diskus ®, Flovent HFA ®, Veramyst ®)
- itraconazole (Onmel ®, Sporanox ®)
- ketoconazole (Extina ®, Ketozole ®, Nizoral ®, Xolegel ®)
- lamotrigine (Lamictal ®)
- lidocaine
- methadone hydrochloride (Dolphine hydrochloride, Methadose ®)
- metronidazole
- nicardipine (Cardene ®)
- nifedipine (Adalat CC ®, Afeditab CR ®, Procardia ®)
- phenobarbital
- phenytoin (Dilantin ®, Phenytek ®)
- prednisone
- quinidine (Quinidex ®)
- quetiapine (Seroquel®)
- rifabutin (Mycobutin ®)
- rivaroxaban (Xarelto ®)
- rosuvastatin (Crestor ®)
- salmeterol (Serevent ®) or salmeterol when taken in combination with fluticasone (Advair Diskus ®, Advair HFA ®)
- simeprevir (Olysio ®)
- tadalafil (Adcirca ®) for the treatment of pulmonary arterial hypertension
- trazodone (Oleptro ®)
- valproate (Depakote ®, Depakene ®, Depacon ®)
- voriconazole (Vfend ®)
- warfarin (Coumadin ®, Jantoven ®)
KALETRA should not be administered once daily in combination with carbamazepine (Carbatrol ®, Epitol ®, Equetro ®, Tegretol ®), phenobarbital, or phenytoin (Dilantin ®, Phenytek ®)
Ask your doctor or pharmacist if you are not sure if your medicine is one that is listed above.
Know all the medicines that you take. Keep a list of them with you to show doctors and pharmacists when you get a new medicine.
If you are not sure if you are taking a medicine above, ask your doctor.
- Take KALETRA every day exactly as prescribed by your doctor.
- It is very important to set up a dosing schedule and follow it every day.
- Do not change your treatment or stop treatment without first talking with your doctor.
- KALETRA tablets should not be taken 1 time each day if you are pregnant. You should not take KALETRA oral solution if you are pregnant.
- Swallow KALETRA tablets whole. Do not chew, break, or crush KALETRA tablets.
- KALETRA tablets can be taken with or without food.
- If you are taking both didanosine (Videx
®) and KALETRA:
- didanosine can be taken at the same time as KALETRA tablets, without food.
- take didanosine either one hour before or two hours after taking KALETRA oral solution.
- Do not miss a dose of KALETRA. This could make the virus harder to treat. If you forget to take KALETRA, take the missed dose right away. If it is almost time for your next dose, do not take the missed dose. Instead, follow your regular dosing schedule by taking your next dose at its regular time. Do not take more than one dose of KALETRA at one time.
- If you take more than the prescribed dose of KALETRA, call your doctor or go to the nearest emergency room right away.
- Take KALETRA oral solution with food to help it work better.
- If your child is prescribed KALETRA, tell your doctor if your child’s weight changes.
- KALETRA should not be given one time each day in children. When giving KALETRA to your child, give KALETRA exactly as prescribed.
- KALETRA oral solution contains propylene glycol and a large amount of alcohol. KALETRA oral solution
should not be given to babies younger than 14 days of age unless your doctor thinks it is right for your baby.
- If a young child drinks more than the recommended dose, it could make them sick. Contact your local poison control center or emergency room right away.
- Talk with your doctor if you take or plan to take metronidazole or disulfiram. You can have severe nausea and vomiting if you take these medicines with KALETRA.
- When your KALETRA supply starts to run low, get more from your doctor or pharmacy. It is important not to run out of KALETRA. The amount of HIV-1 virus in your blood may increase if the medicine is stopped for even a short time. The virus may become resistant to KALETRA and become harder to treat.
What are the possible side effects of KALETRA?
KALETRA can cause serious side effects, including:
- See “What is the most important information I should know about KALETRA?”
- Inflammation of the pancreas (pancreatitis). Some people who take KALETRA get inflammation of the pancreas which may be serious and cause death. You have a higher chance of getting pancreatitis if you have had it before. Tell your doctor if you have nausea, vomiting, or abdominal pain while taking KALETRA. These may be signs of pancreatitis.
-
Liver problems. Liver problems, including death, can happen in people who take KALETRA. Your doctor should do blood tests before and during your treatment with KALETRA to check your liver function. Some people with liver disease such as Hepatitis B and Hepatitis C who take KALETRA may have worsening liver disease. Tell your doctor right away if you have any of these signs and symptoms of liver problems:
- loss of appetite
- yellow skin and whites of eyes (jaundice)
- dark-colored urine
- pale colored stools
- itchy skin
- stomach area (abdominal) pain.
- Diabetes and high blood sugar (hyperglycemia). Some people who take protease inhibitors including KALETRA get new or more serious diabetes, or high blood sugar. Tell your doctor if you notice an increase in thirst or urinate often while taking KALETRA.
- Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Call your doctor right away if you start having new symptoms after starting your HIV medicine.
- Increases in certain fat (triglycerides and cholesterol) levels in your blood. Large increases of triglycerides and cholesterol can be seen in blood test results of some people who take KALETRA. Your doctor should do blood tests to check your cholesterol and triglyceride levels before you start taking KALETRA and during your treatment.
- Changes in body fat. Changes in body fat in some people who take antiretroviral therapy. These changes may include increased amount of fat in the upper back and neck ("buffalo hump"), breast, and around the trunk. Loss of fat from the legs, arms and face may also happen. The cause and long-term health effects of these conditions are not known at this time.
- Increased bleeding for hemophiliacs. Some people with hemophilia have increased bleeding with protease inhibitors including KALETRA.
- Allergic reactions. Skin rashes, some of them severe, can occur in people who take KALETRA. Tell your doctor if you had a rash when you took another medicine for your HIV-1 infection or if you notice any skin rash when you take KALETRA.
- Babies taking KALETRA oral solution may have side effects. KALETRA oral solution contains alcohol and propylene glycol. Call your doctor right away if your baby appears too sleepy or their breathing has changed.
Common side effects of KALETRA include:
- diarrhea
- nausea
- increased fats in blood (triglycerides or cholesterol)
- vomiting
Tell your doctor about any side effect that bothers you or that does not go away.
These are not all of the possible side effects of KALETRA. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
- Store KALETRA tablets at room temperature, between 59°F to 86°F (15°C to 30°C).
- Do not keep KALETRA tablets out of the container it comes in for longer than 2 weeks, especially in areas where there is a lot of humidity. Keep the container closed tightly.
- Store KALETRA oral solution in a refrigerator, between 36°F to 46°F (2°C to 8°C). KALETRA oral solution that is kept refrigerated may be used until the expiration date printed on the label.
- KALETRA oral solution that is stored at room temperature (less than 77°F or 25°C) should be used within 2 months.
- Keep KALETRA away from high heat.
Throw away any medicine that is out of date or that you no longer need.
Keep KALETRA and all medicines out of the reach of children.
General information about KALETRA
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use KALETRA for a condition for which it was not prescribed. Do not give KALETRA to other people, even if they have the same condition you have. It may harm them.
This Medication Guide summarizes the most important information about KALETRA. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about KALETRA that is written for health professionals.
For more information about KALETRA call 1-800-633-9110 or go to www.KALETRA.com.
What are the ingredients in KALETRA?
Active ingredients: lopinavir and ritonavir
KALETRA 200 mg lopinavir and 50 mg ritonavir tablets: copovidone, sorbitan monolaurate, colloidal silicon dioxide, and sodium stearyl fumarate. The film coating contains: hypromellose, titanium dioxide, polyethylene glycol 400, hydroxypropyl cellulose, talc, colloidal silicon dioxide, polyethylene glycol 3350, yellow ferric oxide 172, and polysorbate 80.
KALETRA 100 mg lopinavir and 25 mg ritonavir tablets: copovidone, sorbitan monolaurate, colloidal silicon dioxide, and sodium stearyl fumarate. The film coating contains: polyvinyl alcohol, titanium dioxide, talc, polytheylene glycol 3350, and yellow ferric oxide E172.
KALETRA oral solution: acesulfame potassium, alcohol, artificial cotton candy flavor, citric acid, glycerin, high fructose corn syrup, Magnasweet-110 flavor, menthol, natural and artificial vanilla flavor, peppermint oil, polyoxyl 40 hydrogenated castor oil, povidone, propylene glycol, saccharin sodium, sodium chloride, sodium citrate, and water.
KALETRA oral solution contains 42.4% alcohol (v/v). “See How should I take KALETRA?”.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
KALETRA Tablets, 200 mg lopinavir and 50 mg ritonavir
Manufactured by AbbVie LTD, Barceloneta, PR 00617
for AbbVie Inc., North Chicago, IL 60064 USA
KALETRA Tablets, 100 mg lopinavir and 25 mg ritonavir and KALETRA Oral Solution
AbbVie Inc., North Chicago, IL 60064 USA
The brands listed are trademarks of their respective owners and are not trademarks of AbbVie Inc. The makers of these brands are not affiliated with and do not endorse AbbVie Inc. or its products.
© 2015 AbbVie Inc. All rights reserved.
DRUG: Kaletra
GENERIC: Lopinavir and Ritonavir
DOSAGE: TABLET, FILM COATED
ADMINSTRATION: ORAL
NDC: 24236-676-06
COLOR: yellow
SHAPE: OVAL
SCORE: No score
SIZE: 19 mm
IMPRINT: a;KA
PACKAGING: 28 in 1 BLISTER PACK
ACTIVE INGREDIENT(S):
- LOPINAVIR 200mg in 1
- RITONAVIR 50mg in 1
INACTIVE INGREDIENT(S):
- COPOVIDONE K25-31
- FERRIC OXIDE YELLOW
- POLYSORBATE 80
- HYDROXYPROPYL CELLULOSE (TYPE H)
- HYPROMELLOSES
- POLYETHYLENE GLYCOL 3350
- SILICON DIOXIDE
- SODIUM STEARYL FUMARATE
- SORBITAN MONOLAURATE
- POLYETHYLENE GLYCOL 400
- TALC
- TITANIUM DIOXIDE

| KALETRA
lopinavir and ritonavir tablet, film coated |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| Labeler - REMEDYREPACK INC. (829572556) |