FARXIGA- dapagliflozin tablet, film coated
E.R. Squibb & Sons, L.L.C.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use FARXIGA safely and effectively. See full prescribing information for FARXIGA.
FARXIGA (dapagliflozin) tablets, for oral use Initial U.S. Approval: 2014 INDICATIONS AND USAGEDOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb at 1-800-721-5072 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 8/2014 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
FARXIGA (dapagliflozin) is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus [see Clinical Studies (14)].
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
The recommended starting dose of FARXIGA is 5 mg once daily, taken in the morning, with or without food. In patients tolerating FARXIGA 5 mg once daily who require additional glycemic control, the dose can be increased to 10 mg once daily.
In patients with volume depletion, correcting this condition prior to initiation of FARXIGA is recommended [see Warnings and Precautions (5.1), Use in Specific Populations (8.5, 8.6), and Patient Counseling Information (17)].
2.2 Patients with Renal Impairment
Assessment of renal function is recommended prior to initiation of FARXIGA therapy and periodically thereafter.
FARXIGA should not be initiated in patients with an eGFR less than 60 mL/min/1.73 m2.
No dose adjustment is needed in patients with mild renal impairment (eGFR of 60 mL/min/1.73 m2 or greater).
FARXIGA should be discontinued when eGFR is persistently less than 60 mL/min/1.73 m2 [see Warnings and Precautions (5.2) and Use in Specific Populations (8.6)].
3 DOSAGE FORMS AND STRENGTHS
- •
- FARXIGA 5 mg tablets are yellow, biconvex, round, film-coated tablets with “5” engraved on one side and “1427” engraved on the other side.
- •
- FARXIGA 10 mg tablets are yellow, biconvex, diamond-shaped, film-coated tablets with “10” engraved on one side and “1428” engraved on the other side.
4 CONTRAINDICATIONS
- •
- History of a serious hypersensitivity reaction to FARXIGA [see Adverse Reactions (6.1)].
- •
- Severe renal impairment, end-stage renal disease (ESRD), or patients on dialysis [see Use in Specific Populations (8.6)].
5 WARNINGS AND PRECAUTIONS
5.1 Hypotension
FARXIGA causes intravascular volume contraction. Symptomatic hypotension can occur after initiating FARXIGA [see Adverse Reactions (6.1)] particularly in patients with impaired renal function (eGFR less than 60 mL/min/1.73 m2), elderly patients, or patients on loop diuretics. Before initiating FARXIGA in patients with one or more of these characteristics, volume status should be assessed and corrected. Monitor for signs and symptoms of hypotension after initiating therapy.
5.2 Impairment in Renal Function
FARXIGA increases serum creatinine and decreases eGFR. Elderly patients and patients with impaired renal function may be more susceptible to these changes. Adverse reactions related to renal function can occur after initiating FARXIGA [see Adverse Reactions (6.1)]. Renal function should be evaluated prior to initiation of FARXIGA and monitored periodically thereafter.
5.3 Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues
Insulin and insulin secretagogues are known to cause hypoglycemia. FARXIGA can increase the risk of hypoglycemia when combined with insulin or an insulin secretagogue [see Adverse Reactions (6.1)]. Therefore, a lower dose of insulin or insulin secretagogue may be required to minimize the risk of hypoglycemia when these agents are used in combination with FARXIGA.
5.4 Genital Mycotic Infections
FARXIGA increases the risk of genital mycotic infections. Patients with a history of genital mycotic infections were more likely to develop genital mycotic infections [see Adverse Reactions (6.1)]. Monitor and treat appropriately.
5.5 Increases in Low-Density Lipoprotein Cholesterol (LDL-C)
Increases in LDL‑C occur with FARXIGA [see Adverse Reactions (6.1)]. Monitor LDL‑C and treat per standard of care after initiating FARXIGA.
5.6 Bladder Cancer
Across 22 clinical studies, newly diagnosed cases of bladder cancer were reported in 10/6045 patients (0.17%) treated with FARXIGA and 1/3512 patient (0.03%) treated with placebo/comparator. After excluding patients in whom exposure to study drug was less than one year at the time of diagnosis of bladder cancer, there were 4 cases with FARXIGA and no cases with placebo/comparator. Bladder cancer risk factors and hematuria (a potential indicator of pre-existing tumors) were balanced between treatment arms at baseline. There were too few cases to determine whether the emergence of these events is related to FARXIGA.
There are insufficient data to determine whether FARXIGA has an effect on pre-existing bladder tumors. Consequently, FARXIGA should not be used in patients with active bladder cancer. In patients with prior history of bladder cancer, the benefits of glycemic control versus unknown risks for cancer recurrence with FARXIGA should be considered.
6 ADVERSE REACTIONS
The following important adverse reactions are described below and elsewhere in the labeling:
- •
- Hypotension [see Warnings and Precautions (5.1)]
- •
- Impairment in Renal Function [see Warnings and Precautions (5.2)]
- •
- Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues [see Warnings and Precautions (5.3)]
- •
- Genital Mycotic Infections [see Warnings and Precautions (5.4)]
- •
- Increases in Low-Density Lipoprotein Cholesterol (LDL‑C) [see Warnings and Precautions (5.5)]
- •
- Bladder Cancer [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Pool of 12 Placebo-Controlled Studies for FARXIGA 5 and 10 mg
The data in Table 1 is derived from 12 placebo-controlled studies ranging from 12 to 24 weeks. In 4 studies FARXIGA was used as monotherapy, and in 8 studies FARXIGA was used as add-on to background antidiabetic therapy or as combination therapy with metformin [see Clinical Studies (14)].
These data reflect exposure of 2338 patients to FARXIGA with a mean exposure duration of 21 weeks. Patients received placebo (N=1393), FARXIGA 5 mg (N=1145), or FARXIGA 10 mg (N=1193) once daily. The mean age of the population was 55 years and 2% were older than 75 years of age. Fifty percent (50%) of the population were male; 81% were White, 14% were Asian, and 3% were Black or African American. At baseline, the population had diabetes for an average of 6 years, had a mean hemoglobin A1c (HbA1c) of 8.3%, and 21% had established microvascular complications of diabetes. Baseline renal function was normal or mildly impaired in 92% of patients and moderately impaired in 8% of patients (mean eGFR 86 mL/min/1.73 m2).
Table 1 shows common adverse reactions associated with the use of FARXIGA. These adverse reactions were not present at baseline, occurred more commonly on FARXIGA than on placebo, and occurred in at least 2% of patients treated with either FARXIGA 5 mg or FARXIGA 10 mg.
| Adverse Reaction | % of Patients | ||
|---|---|---|---|
| Pool of 12 Placebo-Controlled Studies | |||
| Placebo
N=1393 | FARXIGA 5 mg
N=1145 | FARXIGA 10 mg
N=1193 |
|
| * Genital mycotic infections include the following adverse reactions, listed in order of frequency reported for females: vulvovaginal mycotic infection, vaginal infection, vulvovaginal candidiasis, vulvovaginitis, genital infection, genital candidiasis, fungal genital infection, vulvitis, genitourinary tract infection, vulval abscess, and vaginitis bacterial. (N for females: Placebo=677, FARXIGA 5 mg=581, FARXIGA 10 mg=598). † Urinary tract infections include the following adverse reactions, listed in order of frequency reported: urinary tract infection, cystitis, Escherichia urinary tract infection, genitourinary tract infection, pyelonephritis, trigonitis, urethritis, kidney infection, and prostatitis. ‡ Increased urination includes the following adverse reactions, listed in order of frequency reported: pollakiuria, polyuria, and urine output increased. § Genital mycotic infections include the following adverse reactions, listed in order of frequency reported for males: balanitis, fungal genital infection, balanitis candida, genital candidiasis, genital infection male, penile infection, balanoposthitis, balanoposthitis infective, genital infection, posthitis. (N for males: Placebo=716, FARXIGA 5 mg=564, FARXIGA 10 mg=595). |
|||
|
Female genital mycotic infections* |
1.5 |
8.4 |
6.9 |
|
Nasopharyngitis |
6.2 |
6.6 |
6.3 |
|
Urinary tract infections† |
3.7 |
5.7 |
4.3 |
|
Back pain |
3.2 |
3.1 |
4.2 |
|
Increased urination‡ |
1.7 |
2.9 |
3.8 |
|
Male genital mycotic infections§ |
0.3 |
2.8 |
2.7 |
|
Nausea |
2.4 |
2.8 |
2.5 |
|
Influenza |
2.3 |
2.7 |
2.3 |
|
Dyslipidemia |
1.5 |
2.1 |
2.5 |
|
Constipation |
1.5 |
2.2 |
1.9 |
|
Discomfort with urination |
0.7 |
1.6 |
2.1 |
|
Pain in extremity |
1.4 |
2.0 |
1.7 |
Pool of 13 Placebo-Controlled Studies for FARXIGA 10 mg
The safety and tolerability of FARXIGA 10 mg was also evaluated in a larger placebo-controlled study pool. This pool combined 13 placebo-controlled studies, including 3 monotherapy studies, 9 add-on to background antidiabetic therapy studies, and an initial combination with metformin study. Across these 13 studies, 2360 patients were treated once daily with FARXIGA 10 mg for a mean duration of exposure of 22 weeks. The mean age of the population was 59 years and 4% were older than 75 years. Fifty-eight percent (58%) of the population were male; 84% were White, 9% were Asian, and 3% were Black or African American. At baseline, the population had diabetes for an average of 9 years, had a mean HbA1c of 8.2%, and 30% had established microvascular disease. Baseline renal function was normal or mildly impaired in 88% of patients and moderately impaired in 11% of patients (mean eGFR 82 mL/min/1.73 m2).
Volume Depletion
FARXIGA causes an osmotic diuresis, which may lead to reductions in intravascular volume. Adverse reactions related to volume depletion (including reports of dehydration, hypovolemia, orthostatic hypotension, or hypotension) are shown in Table 2 for the 12-study and 13-study, short-term, placebo-controlled pools [see Warnings and Precautions (5.1)].
| Pool of 12 Placebo-Controlled
Studies | Pool of 13 Placebo-Controlled
Studies |
||||
|---|---|---|---|---|---|
| Placebo | FARXIGA
5 mg | FARXIGA
10 mg | Placebo | FARXIGA
10 mg |
|
| * Volume depletion includes reports of dehydration, hypovolemia, orthostatic hypotension, or hypotension. | |||||
|
Overall population N (%) |
N=1393 5 (0.4%) |
N=1145 7 (0.6%) |
N=1193 9 (0.8%) |
N=2295 17 (0.7%) |
N=2360 27 (1.1%) |
|
Patient Subgroup n (%) |
|||||
|
Patients on loop diuretics |
n=55 |
n=40 |
n=31 |
n=267 |
n=236 |
|
Patients with moderate renal impairment with eGFR ≥30 and <60 mL/min/1.73 m2 |
n=107 |
n=107 |
n=89 |
n=268 |
n=265 |
|
Patients ≥65 years of age |
n=276 |
n=216 |
n=204 |
n=711 |
n=665 |
Impairment of Renal Function
Use of FARXIGA was associated with increases in serum creatinine and decreases in eGFR (see Table 3). In patients with normal or mildly impaired renal function at baseline, serum creatinine and eGFR returned to baseline values at Week 24. Renal-related adverse reactions, including renal failure and blood creatinine increase, were more frequent in patients treated with FARXIGA (see Table 4). Elderly patients and patients with impaired renal function were more susceptible to these adverse reactions (see Table 4). Sustained decreases in eGFR were seen in patients with moderate renal impairment (eGFR 30 to less than 60 mL/min/1.73 m2).
|
Pool of 12 Placebo-Controlled Studies |
||||
|
Placebo
|
FARXIGA 5 mg
|
FARXIGA 10 mg
|
||
|
Baseline Mean |
Serum Creatinine (mg/dL) |
0.853 |
0.860 |
0.847 |
|
eGFR (mL/min/1.73 m2) |
86.0 |
85.3 |
86.7 |
|
|
Week 1 Change |
Serum Creatinine (mg/dL) |
−0.003 |
0.029 |
0.041 |
|
eGFR (mL/min/1.73 m2) |
0.4 |
−2.9 |
−4.1 |
|
|
Week 24 Change |
Serum Creatinine (mg/dL) |
−0.005 |
−0.001 |
0.001 |
|
eGFR (mL/min/1.73 m2) |
0.8 |
0.8 |
0.3 |
|
|
Moderate Renal Impairment Study |
||||
|
Placebo
|
FARXIGA 5 mg
|
FARXIGA 10 mg
|
||
|
Baseline Mean |
Serum Creatinine (mg/dL) |
1.46 |
1.53 |
1.52 |
|
eGFR (mL/min/1.73 m2) |
45.6 |
44.2 |
43.9 |
|
|
Week 1 Change |
Serum Creatinine (mg/dL) |
0.01 |
0.13 |
0.18 |
|
eGFR (mL/min/1.73 m2) |
0.5 |
−3.8 |
−5.5 |
|
|
Week 24 Change |
Serum Creatinine (mg/dL) |
0.02 |
0.08 |
0.16 |
|
eGFR (mL/min/1.73 m2) |
0.03 |
−4.0 |
−7.4 |
|
|
Week 52 Change |
Serum Creatinine (mg/dL) |
0.10 |
0.06 |
0.15 |
|
eGFR (mL/min/1.73 m2) |
−2.6 |
−4.2 |
−7.3 |
|
| Pool of 6 Placebo-Controlled Studies
(up to 104 weeks)* | Pool of 9 Placebo-Controlled Studies
(up to 104 weeks)† |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline Characteristic | Placebo | FARXIGA
5 mg | FARXIGA
10 mg | Placebo | FARXIGA
10 mg |
|||||
| * Subset of patients from the pool of 12 placebo-controlled studies with long-term extensions. † Subset of patients from the pool of 13 placebo-controlled studies with long-term extensions. |
||||||||||
|
Overall population |
n=785 |
n=767 |
n=859 |
n=1956 |
n=2026 |
|||||
|
65 years of age and older |
n=190 |
n=162 |
n=159 |
n=655 |
n=620 |
|||||
|
eGFR ≥30 and <60 mL/min/1.73 m2
|
n=77 |
n=88 |
n=75 |
n=249 |
n=251 |
|||||
|
65 years of age and older and eGFR ≥30 and <60 mL/min/1.73 m2
|
n=41 |
n=43 |
n=35 |
n=141 |
n=134 |
|||||
The safety of FARXIGA was evaluated in a study of patients with moderate renal impairment (eGFR 30 to less than 60 mL/min/1.73 m2) [see Clinical Studies (14)]. In this study 13 patients experienced bone fractures for treatment durations up to 104 weeks. No fractures occurred in the placebo group, 5 occurred in the FARXIGA 5 mg group, and 8 occurred in the FARXIGA 10 mg group. Eight of these 13 fractures were in patients who had a baseline eGFR of 30 to 45 mL/min/1.73 m2. Eleven of the 13 fractures were reported within the first 52 weeks. There was no apparent pattern with respect to the anatomic site of fracture.
Hypoglycemia
The frequency of hypoglycemia by study [see Clinical Studies (14)] is shown in Table 5. Hypoglycemia was more frequent when FARXIGA was added to sulfonylurea or insulin [see Warnings and Precautions (5.3)].
| Placebo | FARXIGA 5 mg | FARXIGA 10 mg | |
|---|---|---|---|
| * Major episodes of hypoglycemia were defined as symptomatic episodes requiring external (third party) assistance due to severe impairment in consciousness or behavior with a capillary or plasma glucose value <54 mg/dL and prompt recovery after glucose or glucagon administration. † Minor episodes of hypoglycemia were defined as either a symptomatic episode with a capillary or plasma glucose measurement <63 mg/dL regardless of need for external assistance, or an asymptomatic capillary or plasma glucose measurement <63 mg/dL that does not qualify as a major episode. ‡ OAD = oral antidiabetic therapy. |
|||
|
Monotherapy* (24 weeks) |
N=75 |
N=64 |
N=70 |
|
Major [n (%)] |
0 |
0 |
0 |
|
Minor [n (%)] |
0 |
0 |
0 |
|
Add-on to Metformin* (24 weeks) |
N=137 |
N=137 |
N=135 |
|
Major [n (%)] |
0 |
0 |
0 |
|
Minor [n (%)] |
0 |
2 (1.5) |
1 (0.7) |
|
Active Control Add-on to Metformin versus Glipizide (52 weeks) |
N=408 |
– |
N=406 |
|
Major [n (%)] |
3 (0.7) |
– |
0 |
|
Minor [n (%)] |
147 (36.0) |
– |
7 (1.7) |
|
Add-on to Glimepiride* (24 weeks) |
N=146 |
N=145 |
N=151 |
|
Major [n (%)] |
0 |
0 |
0 |
|
Minor [n (%)] |
3 (2.1) |
8 (5.5) |
9 (6.0) |
|
Add-on to Pioglitazone* (24 weeks) |
N=139 |
N=141 |
N=140 |
|
Major [n (%)] |
0 |
0 |
0 |
|
Minor [n (%)] |
0 |
3 (2.1) |
0 |
|
Add-on to DPP4 inhibitor (24 weeks) |
N=226 |
– |
N=225 |
|
Major [n (%)] |
0 |
– |
1 (0.4) |
|
Minor [n (%)] |
3 (1.3) |
– |
4 (1.8) |
|
Add-on to Insulin with or without other OADs‡ (24 weeks) |
N=197 |
N=212 |
N=196 |
|
Major [n (%)] |
1 (0.5) |
1 (0.5) |
1 (0.5) |
|
Minor [n (%)] |
67 (34.0) |
92 (43.4) |
79 (40.3) |
Genital Mycotic Infections
Genital mycotic infections were more frequent with FARXIGA treatment. Genital mycotic infections were reported in 0.9% of patients on placebo, 5.7% on FARXIGA 5 mg, and 4.8% on FARXIGA 10 mg, in the 12-study placebo-controlled pool. Discontinuation from study due to genital infection occurred in 0% of placebo-treated patients and 0.2% of patients treated with FARXIGA 10 mg. Infections were more frequently reported in females than in males (see Table 1). The most frequently reported genital mycotic infections were vulvovaginal mycotic infections in females and balanitis in males. Patients with a history of genital mycotic infections were more likely to have a genital mycotic infection during the study than those with no prior history (10.0%, 23.1%, and 25.0% versus 0.8%, 5.9%, and 5.0% on placebo, FARXIGA 5 mg, and FARXIGA 10 mg, respectively).
Hypersensitivity Reactions
Hypersensitivity reactions (e.g., angioedema, urticaria, hypersensitivity) were reported with FARXIGA treatment. Across the clinical program, serious anaphylactic reactions and severe cutaneous adverse reactions and angioedema were reported in 0.2% of comparator-treated patients and 0.3% of FARXIGA-treated patients. If hypersensitivity reactions occur, discontinue use of FARXIGA; treat per standard of care and monitor until signs and symptoms resolve.
Laboratory Tests
Increase in Hematocrit
In the pool of 13 placebo-controlled studies, increases from baseline in mean hematocrit values were observed in FARXIGA-treated patients starting at Week 1 and continuing up to Week 16, when the maximum mean difference from baseline was observed. At Week 24, the mean changes from baseline in hematocrit were −0.33% in the placebo group and 2.30% in the FARXIGA 10 mg group. By Week 24, hematocrit values >55% were reported in 0.4% of placebo-treated patients and 1.3% of FARXIGA 10 mg–treated patients.
Increase in Serum Inorganic Phosphorus
In the pool of 13 placebo-controlled studies, increases from baseline in mean serum phosphorus levels were reported at Week 24 in FARXIGA-treated patients compared with placebo-treated patients (mean increase of 0.13 versus −0.04 mg/dL, respectively). Higher proportions of patients with marked laboratory abnormalities of hyperphosphatemia (≥5.6 mg/dL for age 17-65 years or ≥5.1 mg/dL for age ≥66 years) were reported on FARXIGA at Week 24 (0.9% versus 1.7% for placebo and FARXIGA 10 mg, respectively).
Increase in Low-Density Lipoprotein Cholesterol
In the pool of 13 placebo-controlled studies, changes from baseline in mean lipid values were reported in FARXIGA-treated patients compared to placebo-treated patients. Mean percent changes from baseline at Week 24, were 0.0% versus 2.5% for total cholesterol and −1.0% versus 2.9% for LDL cholesterol, in the placebo and FARXIGA 10 mg groups, respectively.
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C
There are no adequate and well-controlled studies of FARXIGA in pregnant women. Based on results of reproductive and developmental toxicity studies in animals, dapagliflozin may affect renal development and maturation. In a juvenile rat study, increased incidence and/or severity of renal pelvic and tubular dilatations were evident at the lowest tested dose which was approximately 15 times clinical exposure from a 10 mg dose.
These outcomes occurred with drug exposures during periods of animal development that correlate with the late second and third trimesters of human pregnancy. During pregnancy, consider appropriate alternative therapies, especially during the second and third trimesters. FARXIGA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
In a juvenile toxicity study, when dapagliflozin was dosed directly to young rats from postnatal day (PND) 21 until PND 90 at doses of 1, 15, or 75 mg/kg/day, increased kidney weights and renal pelvic and tubular dilatations were reported at all levels. Exposure at the lowest tested dose was 15 times the maximum clinical dose, based on AUC. The renal pelvic and tubular dilatations observed in juvenile animals did not fully reverse within the approximate 1-month recovery period.
In a prenatal and postnatal development study, maternal rats were dosed from gestation day 6 through lactation day 21 at doses of 1, 15, or 75 mg/kg/day, and pups were indirectly exposed in utero and throughout lactation. Increased incidence or severity of renal pelvic dilatation was observed in adult offspring of treated dams at 75 mg/kg/day (maternal and pup dapagliflozin exposures were 1415 times and 137 times, respectively, the human values at the clinical dose). Dose-related reductions in pup body weights were observed at doses ≥1 mg/kg/day (approximately ≥19 times the clinical dose). No adverse effects on developmental endpoints were noted at 1 mg/kg/day, or approximately 19 times the clinical dose.
In embryo-fetal development studies in rats and rabbits, dapagliflozin was administered for intervals coinciding with the first trimester period of organogenesis in humans. No developmental toxicities were observed in rabbits at any dose tested. In rats, dapagliflozin was neither embryolethal nor teratogenic at doses up to 75 mg/kg/day or 1441 times the maximum clinical dose of 10 mg. At higher doses in rats, malformations of blood vessels, ribs, vertebra, manubria, and skeletal variations in fetuses at ≥150 mg/kg or 2344 times the 10 mg clinical dose were observed.
8.3 Nursing Mothers
It is not known whether FARXIGA is excreted in human milk. Dapagliflozin is excreted in rat milk reaching levels 0.49 times that found in maternal plasma. Data in juvenile rats directly exposed to dapagliflozin showed risk to the developing kidney (renal pelvic and tubular dilatations) during maturation. Since human kidney maturation occurs in utero and during the first 2 years of life when lactational exposure may occur, there may be risk to the developing human kidney. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from FARXIGA, a decision should be made whether to discontinue nursing or to discontinue FARXIGA, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
Safety and effectiveness of FARXIGA in pediatric patients under 18 years of age have not been established.
8.5 Geriatric Use
No FARXIGA dosage change is recommended based on age. A total of 1424 (24%) of the 5936 FARXIGA-treated patients were 65 years and older and 207 (3.5%) patients were 75 years and older in a pool of 21 double-blind, controlled, clinical safety and efficacy studies of FARXIGA. After controlling for level of renal function (eGFR), efficacy was similar for patients under age 65 years and those 65 years and older. In patients ≥65 years of age, a higher proportion of patients treated with FARXIGA had adverse reactions related to volume depletion and renal impairment or failure compared to patients treated with placebo [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
8.6 Renal Impairment
The safety and efficacy of FARXIGA were evaluated in a study that included patients with moderate renal impairment (eGFR 30 to less than 60 mL/min/1.73 m2). Compared to placebo-treated patients, patients with moderate renal impairment treated with FARXIGA did not have improvement in glycemic control [see Clinical Studies (14.7)] and had more renal-related adverse reactions and more bone fractures [see Dosage and Administration (2.2), Warnings and Precautions (5.2), and Adverse Reactions (6.1)]; therefore, FARXIGA should not be initiated in this population.
Based on its mechanism of action, FARXIGA is not expected to be effective in patients with severe renal impairment (eGFR less than 30 mL/min/1.73 m2) or ESRD [see Contraindications (4)].
8.7 Hepatic Impairment
No dose adjustment is recommended for patients with mild, moderate, or severe hepatic impairment. However, the benefit-risk for the use of dapagliflozin in patients with severe hepatic impairment should be individually assessed since the safety and efficacy of dapagliflozin have not been specifically studied in this population [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
There were no reports of overdose during the clinical development program for FARXIGA.
In the event of an overdose, contact the Poison Control Center. It is also reasonable to employ supportive measures, as dictated by the patient’s clinical status. The removal of dapagliflozin by hemodialysis has not been studied.
11 DESCRIPTION
Dapagliflozin is described chemically as D-glucitol, 1,5-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-, (1S)-, compounded with (2S)-1,2-propanediol, hydrate (1:1:1). The empirical formula is C21H25ClO6•C3H8O2•H2O and the molecular weight is 502.98. The structural formula is:
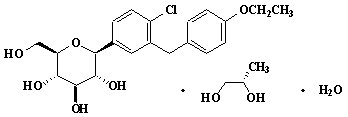
FARXIGA is available as a film-coated tablet for oral administration containing the equivalent of 5 mg dapagliflozin as dapagliflozin propanediol or the equivalent of 10 mg dapagliflozin as dapagliflozin propanediol, and the following inactive ingredients: microcrystalline cellulose, anhydrous lactose, crospovidone, silicon dioxide, and magnesium stearate. In addition, the film coating contains the following inactive ingredients: polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and yellow iron oxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Sodium-glucose cotransporter 2 (SGLT2), expressed in the proximal renal tubules, is responsible for the majority of the reabsorption of filtered glucose from the tubular lumen. Dapagliflozin is an inhibitor of SGLT2. By inhibiting SGLT2, dapagliflozin reduces reabsorption of filtered glucose and lowers the renal threshold for glucose, and thereby increases urinary glucose excretion.
12.2 Pharmacodynamics
General
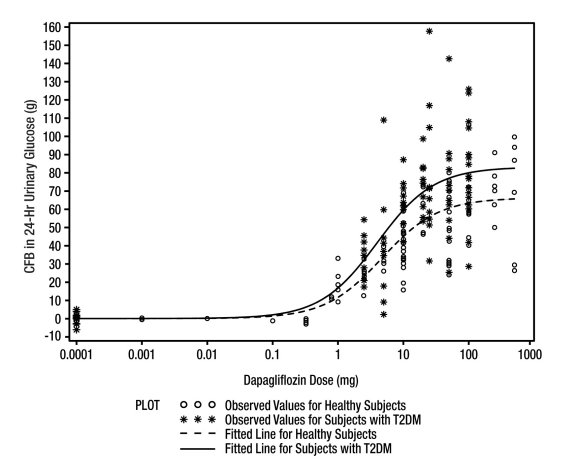
Increases in the amount of glucose excreted in the urine were observed in healthy subjects and in patients with type 2 diabetes mellitus following the administration of dapagliflozin (see Figure 1). Dapagliflozin dose of 10 mg per day in patients with type 2 diabetes mellitus for 12 weeks resulted in excretion of approximately 70 grams of glucose in the urine per day at Week 12. A near maximum glucose excretion was observed at the dapagliflozin daily dose of 20 mg. This urinary glucose excretion with dapagliflozin also results in increases in urinary volume [see Adverse Reactions (6.1)].
Figure 1: Scatter Plot and Fitted Line of Change from Baseline in 24-Hour Urinary Glucose Amount versus Dapagliflozin Dose in Healthy Subjects and Subjects with Type 2 Diabetes Mellitus (T2DM) (Semi-Log Plot)
Cardiac Electrophysiology
Dapagliflozin was not associated with clinically meaningful prolongation of QTc interval at daily doses up to 150 mg (15 times the recommended maximum dose) in a study of healthy subjects. In addition, no clinically meaningful effect on QTc interval was observed following single doses of up to 500 mg (50 times the recommended maximum dose) of dapagliflozin in healthy subjects.
12.3 Pharmacokinetics
Absorption
Following oral administration of dapagliflozin, the maximum plasma concentration (Cmax) is usually attained within 2 hours under fasting state. The Cmax and AUC values increase dose proportionally with increase in dapagliflozin dose in the therapeutic dose range. The absolute oral bioavailability of dapagliflozin following the administration of a 10 mg dose is 78%. Administration of dapagliflozin with a high-fat meal decreases its Cmax by up to 50% and prolongs Tmax by approximately 1 hour, but does not alter AUC as compared with the fasted state. These changes are not considered to be clinically meaningful and dapagliflozin can be administered with or without food.
Distribution
Dapagliflozin is approximately 91% protein bound. Protein binding is not altered in patients with renal or hepatic impairment.
Metabolism
The metabolism of dapagliflozin is primarily mediated by UGT1A9; CYP-mediated metabolism is a minor clearance pathway in humans. Dapagliflozin is extensively metabolized, primarily to yield dapagliflozin 3-O-glucuronide, which is an inactive metabolite. Dapagliflozin 3-O-glucuronide accounted for 61% of a 50 mg [14C]-dapagliflozin dose and is the predominant drug-related component in human plasma.
Elimination
Dapagliflozin and related metabolites are primarily eliminated via the renal pathway. Following a single 50 mg dose of [14C]-dapagliflozin, 75% and 21% total radioactivity is excreted in urine and feces, respectively. In urine, less than 2% of the dose is excreted as parent drug. In feces, approximately 15% of the dose is excreted as parent drug. The mean plasma terminal half-life (t½) for dapagliflozin is approximately 12.9 hours following a single oral dose of FARXIGA 10 mg.
Specific Populations
Renal Impairment
At steady state (20 mg once-daily dapagliflozin for 7 days), patients with type 2 diabetes with mild, moderate, or severe renal impairment (as determined by eGFR) had geometric mean systemic exposures of dapagliflozin that were 45%, 2.04-fold, and 3.03-fold higher, respectively, as compared to patients with type 2 diabetes with normal renal function. Higher systemic exposure of dapagliflozin in patients with type 2 diabetes mellitus with renal impairment did not result in a correspondingly higher 24-hour urinary glucose excretion. The steady-state 24-hour urinary glucose excretion in patients with type 2 diabetes and mild, moderate, and severe renal impairment was 42%, 80%, and 90% lower, respectively, than patients with type 2 diabetes with normal renal function. The impact of hemodialysis on dapagliflozin exposure is not known. [See Dosage and Administration (2.2), Warnings and Precautions (5.2), Use in Specific Populations (8.6), and Clinical Studies (14.7).]
Hepatic Impairment
In subjects with mild and moderate hepatic impairment (Child-Pugh classes A and B), mean Cmax and AUC of dapagliflozin were up to 12% and 36% higher, respectively, as compared to healthy matched control subjects following single-dose administration of 10 mg dapagliflozin. These differences were not considered to be clinically meaningful. In patients with severe hepatic impairment (Child-Pugh class C), mean Cmax and AUC of dapagliflozin were up to 40% and 67% higher, respectively, as compared to healthy matched controls [see Use in Specific Populations (8.7)].
Drug Interactions
In Vitro Assessment of Drug Interactions
In in vitro studies, dapagliflozin and dapagliflozin 3-O-glucuronide neither inhibited CYP 1A2, 2C9, 2C19, 2D6, or 3A4, nor induced CYP 1A2, 2B6, or 3A4. Dapagliflozin is a weak substrate of the P-glycoprotein (P-gp) active transporter, and dapagliflozin 3-O-glucuronide is a substrate for the OAT3 active transporter. Dapagliflozin or dapagliflozin 3-O-glucuronide did not meaningfully inhibit P-gp, OCT2, OAT1, or OAT3 active transporters. Overall, dapagliflozin is unlikely to affect the pharmacokinetics of concurrently administered medications that are P-gp, OCT2, OAT1, or OAT3 substrates.
Effects of Other Drugs on Dapagliflozin
Table 6 shows the effect of coadministered drugs on the pharmacokinetics of dapagliflozin. No dose adjustments are recommended for dapagliflozin.
| Coadministered Drug
(Dose Regimen)* | Dapagliflozin
(Dose Regimen)* | Effect on Dapagliflozin Exposure
(% Change [90% CI]) |
|
|---|---|---|---|
| Cmax | AUC† | ||
| * Single dose unless otherwise noted. † AUC = AUC(INF) for drugs given as single dose and AUC = AUC(TAU) for drugs given in multiple doses. ↔ = no change (geometric mean ratio of test:reference within 0.80 to 1.25); ↓ or ↑ = parameter was lower or higher, respectively, with coadministration compared to dapagliflozin administered alone (geometric mean ratio of test:reference was lower than 0.80 or higher than 1.25). |
|||
|
No dosing adjustments required for the following: |
|||
|
Oral Antidiabetic Agents |
|||
|
Metformin (1000 mg) |
20 mg |
↔ |
↔ |
|
Pioglitazone (45 mg) |
50 mg |
↔ |
↔ |
|
Sitagliptin (100 mg) |
20 mg |
↔ |
↔ |
|
Glimepiride (4 mg) |
20 mg |
↔ |
↔ |
|
Voglibose (0.2 mg three times daily) |
10 mg |
↔ |
↔ |
|
Other Medications |
|||
|
Hydrochlorothiazide (25 mg) |
50 mg |
↔ |
↔ |
|
Bumetanide (1 mg) |
10 mg once daily |
↔ |
↔ |
|
Valsartan (320 mg) |
20 mg |
↓12% |
↔ |
|
Simvastatin (40 mg) |
20 mg |
↔ |
↔ |
|
Anti-infective Agent |
|||
|
Rifampin (600 mg once daily for 6 days) |
10 mg |
↓7% |
↓22% |
|
Nonsteroidal Anti-inflammatory Agent |
|||
|
Mefenamic Acid (loading dose of 500 mg followed by 14 doses of 250 mg every 6 hours) |
10 mg |
↑13% |
↑51% |
Effects of Dapagliflozin on Other Drugs
Table 7 shows the effect of dapagliflozin on other coadministered drugs. Dapagliflozin did not meaningfully affect the pharmacokinetics of the coadministered drugs.
| Coadministered Drug
(Dose Regimen)* | Dapagliflozin
(Dose Regimen)* | Effect on Coadministered Drug Exposure
(% Change [90% CI]) |
|
|---|---|---|---|
| Cmax | AUC† | ||
| * Single dose unless otherwise noted. † AUC = AUC(INF) for drugs given as single dose and AUC = AUC(TAU) for drugs given in multiple doses. ↔ = no change (geometric mean ratio of test:reference within 0.80 to 1.25); ↓ or ↑ = parameter was lower or higher, respectively, with coadministration compared to dapagliflozin administered alone (geometric mean ratio of test:reference was lower than 0.80 or higher than 1.25). |
|||
|
No dosing adjustments required for the following: |
|||
|
Oral Antidiabetic Agents |
|||
|
Metformin (1000 mg) |
20 mg |
↔ | |
|
Pioglitazone (45 mg) |
50 mg |
↓7% |
↔ |
|
Sitagliptin (100 mg) |
20 mg |
↔ |
↔ |
|
Glimepiride (4 mg) |
20 mg |
↔ |
↑13% |
|
Other Medications |
|||
|
Hydrochlorothiazide (25 mg) |
50 mg |
↔ |
↔ |
|
Bumetanide (1 mg) |
10 mg once daily |
↑13% |
↑13% |
|
Valsartan (320 mg) |
20 mg |
↓6% |
↑5% |
|
Simvastatin (40 mg) |
20 mg |
↔ |
↑19% |
|
Digoxin (0.25 mg) |
20 mg loading dose |
↔ |
↔ |
|
Warfarin (25 mg) |
20 mg loading dose |
↔ |
↔ |
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Dapagliflozin did not induce tumors in either mice or rats at any of the doses evaluated in 2-year carcinogenicity studies. Oral doses in mice consisted of 5, 15, and 40 mg/kg/day in males and 2, 10, and 20 mg/kg/day in females, and oral doses in rats were 0.5, 2, and 10 mg/kg/day for both males and females. The highest doses evaluated in mice were approximately 72 times (males) and 105 times (females) the clinical dose of 10 mg per day based on AUC exposure. In rats, the highest dose was approximately 131 times (males) and 186 times (females) the clinical dose of 10 mg per day based on AUC exposure.
Dapagliflozin was negative in the Ames mutagenicity assay and was positive in a series of in vitro clastogenicity assays in the presence of S9 activation and at concentrations ≥100 μg/mL. Dapagliflozin was negative for clastogenicity in a series of in vivo studies evaluating micronuclei or DNA repair in rats at exposure multiples >2100 times the clinical dose.
There was no carcinogenicity or mutagenicity signal in animal studies, suggesting that dapagliflozin does not represent a genotoxic risk to humans.
Dapagliflozin had no effects on mating, fertility, or early embryonic development in treated male or female rats at exposure multiples ≤1708 times and 998 times the maximum recommended human dose in males and females, respectively.
14 CLINICAL STUDIES
14.1 Overview of Clinical Studies of FARXIGA for Type 2 Diabetes
FARXIGA has been studied as monotherapy and in combination with metformin, pioglitazone, glimepiride, sitagliptin (with or without metformin), or insulin (with or without other oral antidiabetic therapy). The efficacy of FARXIGA was compared to a sulfonylurea (glipizide) added on to metformin. FARXIGA has also been studied in patients with type 2 diabetes and moderate renal impairment.
Treatment with FARXIGA as monotherapy and in combination with metformin, glimepiride, pioglitazone, sitagliptin, or insulin produced statistically significant improvements in mean change from baseline at Week 24 in HbA1c compared to control. Reductions in HbA1c were seen across subgroups including gender, age, race, duration of disease, and baseline BMI.
14.2 Monotherapy
A total of 840 treatment-naive patients with inadequately controlled type 2 diabetes participated in 2 placebo-controlled studies to evaluate the safety and efficacy of monotherapy with FARXIGA.
In 1 monotherapy study, a total of 558 treatment-naive patients with inadequately controlled diabetes participated in a 24-week study. Following a 2-week diet and exercise placebo lead-in period, 485 patients with HbA1c ≥7% and ≤10% were randomized to FARXIGA 5 mg or FARXIGA 10 mg once daily in either the morning (QAM, main cohort) or evening (QPM), or placebo.
At Week 24, treatment with FARXIGA 10 mg QAM provided significant improvements in HbA1c and FPG compared with placebo (see Table 8).
| Efficacy Parameter | FARXIGA 10 mg
N=70† | FARXIGA 5 mg
N=64† | Placebo
N=75† |
|---|---|---|---|
| * LOCF: last observation (prior to rescue for rescued patients) carried forward. † All randomized patients who took at least one dose of double-blind study medication during the short-term double-blind period. ‡ Least squares mean adjusted for baseline value. § p-value <0.0001 versus placebo. Sensitivity analyses yielded smaller estimates of treatment difference with placebo. ¶ Not evaluated for statistical significance as a result of the sequential testing procedure for the secondary endpoints. |
|||
|
HbA1c (%) |
|||
|
Baseline (mean) |
8.0 |
7.8 |
7.8 |
|
Change from baseline (adjusted mean‡) |
−0.9 |
−0.8 |
−0.2 |
|
Difference from placebo (adjusted mean‡) |
−0.7§
|
−0.5 | |
|
Percent of patients achieving HbA1c <7% |
50.8%¶ |
44.2%¶ |
31.6% |
|
FPG (mg/dL) |
|||
|
Baseline (mean) |
166.6 |
157.2 |
159.9 |
|
Change from baseline (adjusted mean‡) |
−28.8 |
−24.1 |
−4.1 |
|
Difference from placebo (adjusted mean‡) |
−24.7§
|
−19.9 | |
14.3 Initial Combination Therapy with Metformin
A total of 1241 treatment-naive patients with inadequately controlled type 2 diabetes (HbA1c ≥7.5% and ≤12%) participated in 2 active-controlled studies of 24-week duration to evaluate the safety and efficacy of initial therapy with FARXIGA 5 mg or 10 mg in combination with metformin extended-release (XR) formulation.
In 1 study, 638 patients were randomized to 1 of 3 treatment arms following a 1-week lead-in period: FARXIGA 10 mg plus metformin XR (up to 2000 mg per day), FARXIGA 10 mg plus placebo, or metformin XR (up to 2000 mg per day) plus placebo. Metformin XR dose was up-titrated weekly in 500 mg increments, as tolerated, with a median dose achieved of 2000 mg.
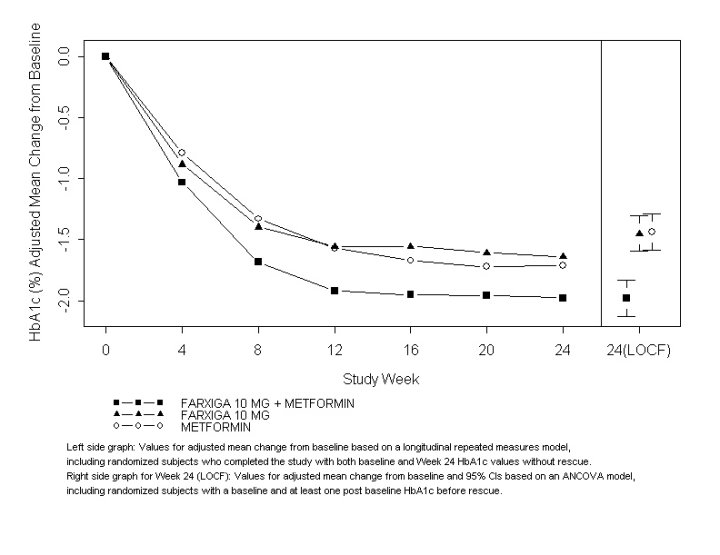
The combination treatment of FARXIGA 10 mg plus metformin XR provided statistically significant improvements in HbA1c and FPG compared with either of the monotherapy treatments and statistically significant reduction in body weight compared with metformin XR alone (see Table 9 and Figure 2). FARXIGA 10 mg as monotherapy also provided statistically significant improvements in FPG and statistically significant reduction in body weight compared with metformin alone and was noninferior to metformin XR monotherapy in lowering HbA1c.
| Efficacy Parameter | FARXIGA
10 mg + Metformin XR | FARXIGA
10 mg | Metformin XR |
|---|---|---|---|
| N=211† | N=219† | N=208† | |
| * LOCF: last observation (prior to rescue for rescued patients) carried forward. † All randomized patients who took at least one dose of double-blind study medication during the short-term double-blind period. ‡ Least squares mean adjusted for baseline value. § p-value <0.0001. ¶ Noninferior versus metformin XR. # p-value <0.05. |
|||
|
HbA1c (%) |
|||
|
Baseline (mean) |
9.1 |
9.0 |
9.0 |
|
Change from baseline (adjusted mean‡) |
−2.0 |
−1.5 |
−1.4 |
|
Difference from FARXIGA (adjusted mean‡) |
−0.5§
| ||
|
Difference from metformin XR (adjusted mean‡) |
−0.5§
|
0.0¶
| |
|
Percent of patients achieving HbA1c <7% |
46.6%# |
31.7% |
35.2% |
|
FPG (mg/dL) |
|||
|
Baseline (mean) |
189.6 |
197.5 |
189.9 |
|
Change from baseline (adjusted mean‡) |
−60.4 |
−46.4 |
−34.8 |
|
Difference from FARXIGA (adjusted mean‡) |
−13.9§
| ||
|
Difference from metformin XR (adjusted mean‡) |
−25.5§
|
−11.6#
| |
|
Body Weight (kg) |
|||
|
Baseline (mean) |
88.6 |
88.5 |
87.2 |
|
Change from baseline (adjusted mean‡) |
−3.3 |
−2.7 |
−1.4 |
|
Difference from metformin XR (adjusted mean‡) |
−2.0§
|
−1.4§
| |
Figure 2: Adjusted Mean Change from Baseline Over Time in HbA1c (%) in a 24-Week Active-Controlled Study of FARXIGA Initial Combination Therapy with Metformin XR
In a second study, 603 patients were randomized to 1 of 3 treatment arms following a 1-week lead-in period: FARXIGA 5 mg plus metformin XR (up to 2000 mg per day), FARXIGA 5 mg plus placebo, or metformin XR (up to 2000 mg per day) plus placebo. Metformin XR dose was up-titrated weekly in 500 mg increments, as tolerated, with a median dose achieved of 2000 mg.
The combination treatment of FARXIGA 5 mg plus metformin XR provided statistically significant improvements in HbA1c and FPG compared with either of the monotherapy treatments and statistically significant reduction in body weight compared with metformin XR alone (see Table 10).
| Efficacy Parameter | FARXIGA
5 mg + Metformin XR | FARXIGA
5 mg | Metformin XR | ||||
|---|---|---|---|---|---|---|---|
| N=194† | N=203† | N=201† | |||||
| * LOCF: last observation (prior to rescue for rescued patients) carried forward. † All randomized patients who took at least one dose of double-blind study medication during the short-term double-blind period. ‡ Least squares mean adjusted for baseline value. § p-value <0.0001. ¶ p-value <0.05. |
|||||||
|
HbA1c (%) |
|||||||
|
Baseline (mean) |
9.2 |
9.1 |
9.1 |
||||
|
Change from baseline (adjusted mean‡) |
−2.1 |
−1.2 |
−1.4 |
||||
|
Difference from FARXIGA (adjusted mean‡) |
−0.9§
| ||||||
|
Difference from metformin XR (adjusted mean‡) |
−0.7§
| ||||||
|
Percent of patients achieving HbA1c <7% |
52.4%¶ |
22.5% |
34.6% |
||||
|
FPG (mg/dL) |
|||||||
|
Baseline (mean) |
193.4 |
190.8 |
196.7 |
||||
|
Change from baseline (adjusted mean‡) |
−61.0 |
−42.0 |
−33.6 |
||||
|
Difference from FARXIGA (adjusted mean‡) |
−19.1§
| ||||||
|
Difference from metformin XR (adjusted mean‡) |
−27.5§
| ||||||
|
Body Weight (kg) |
|||||||
|
Baseline (mean) |
84.2 |
86.2 |
85.8 |
||||
|
Change from baseline (adjusted mean‡) |
−2.7 |
−2.6 |
−1.3 |
||||
|
Difference from metformin XR (adjusted mean‡) |
−1.4§
| ||||||
14.4 Add-On to Metformin
A total of 546 patients with type 2 diabetes with inadequate glycemic control (HbA1c ≥7% and ≤10%) participated in a 24-week, placebo-controlled study to evaluate FARXIGA in combination with metformin. Patients on metformin at a dose of at least 1500 mg per day were randomized after completing a 2-week, single-blind, placebo lead-in period. Following the lead-in period, eligible patients were randomized to FARXIGA 5 mg, FARXIGA 10 mg, or placebo in addition to their current dose of metformin.
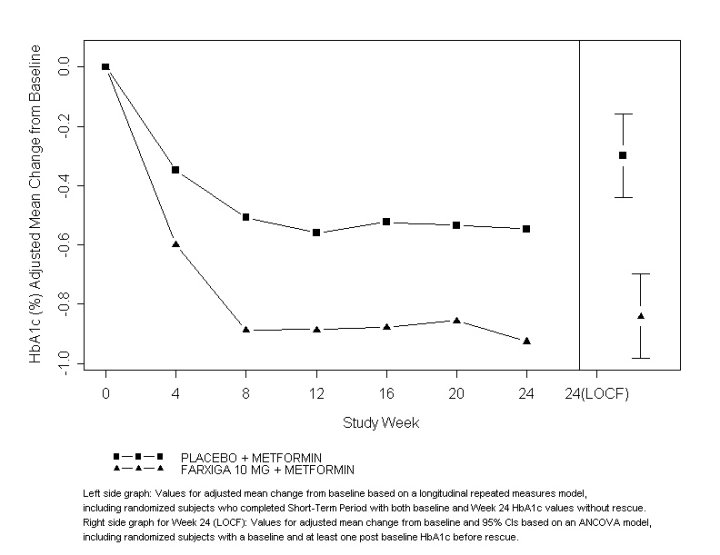
As add-on treatment to metformin, FARXIGA 10 mg provided statistically significant improvements in HbA1c and FPG, and statistically significant reduction in body weight compared with placebo at Week 24 (see Table 11 and Figure 3). Statistically significant (p<0.05 for both doses) mean changes from baseline in systolic blood pressure relative to placebo plus metformin were −4.5 mmHg and −5.3 mmHg with FARXIGA 5 mg and 10 mg plus metformin, respectively.
| Efficacy Parameter | FARXIGA 10 mg
+ Metformin N=135† | FARXIGA 5 mg
+ Metformin N=137† | Placebo
+ Metformin N=137† |
|---|---|---|---|
| * LOCF: last observation (prior to rescue for rescued patients) carried forward. † All randomized patients who took at least one dose of double-blind study medication during the short-term double-blind period. ‡ Least squares mean adjusted for baseline value. § p-value <0.0001 versus placebo + metformin. ¶ p-value <0.05 versus placebo + metformin. |
|||
|
HbA1c (%) |
|||
|
Baseline (mean) |
7.9 |
8.2 |
8.1 |
|
Change from baseline (adjusted mean‡) |
−0.8 |
−0.7 |
−0.3 |
|
Difference from placebo (adjusted mean‡) |
−0.5§
|
−0.4§
| |
|
Percent of patients achieving HbA1c <7% |
40.6%¶
|
37.5%¶
|
25.9% |
|
FPG (mg/dL) |
|||
|
Baseline (mean) |
156.0 |
169.2 |
165.6 |
|
Change from baseline at Week 24 (adjusted mean‡) |
−23.5 |
−21.5 |
−6.0 |
|
Difference from placebo (adjusted mean‡) |
−17.5§
|
−15.5§
| |
|
Change from baseline at Week 1 (adjusted mean‡) |
−16.5§
|
−12.0§
|
1.2 |
|
Body Weight (kg) |
|||
|
Baseline (mean) |
86.3 |
84.7 |
87.7 |
|
Change from baseline (adjusted mean‡) |
−2.9 |
−3.0 |
−0.9 |
|
Difference from placebo (adjusted mean‡) |
−2.0§
|
−2.2§
| |
Figure 3: Adjusted Mean Change from Baseline Over Time in HbA1c (%) in a 24-Week Placebo-Controlled Study of FARXIGA in Combination with Metformin
14.5 Active Glipizide-Controlled Study Add-On to Metformin
A total of 816 patients with type 2 diabetes with inadequate glycemic control (HbA1c >6.5% and ≤10%) were randomized in a 52-week, glipizide-controlled, noninferiority study to evaluate FARXIGA as add-on therapy to metformin. Patients on metformin at a dose of at least 1500 mg per day were randomized following a 2-week placebo lead-in period to glipizide or dapagliflozin (5 mg or 2.5 mg, respectively) and were up-titrated over 18 weeks to optimal glycemic effect (FPG <110 mg/dL, <6.1 mmol/L) or to the highest dose level (up to glipizide 20 mg and FARXIGA 10 mg) as tolerated by patients. Thereafter, doses were kept constant, except for down-titration to prevent hypoglycemia.
At the end of the titration period, 87% of patients treated with FARXIGA had been titrated to the maximum study dose (10 mg) versus 73% treated with glipizide (20 mg). FARXIGA led to a similar mean reduction in HbA1c from baseline at Week 52 (LOCF), compared with glipizide, thus demonstrating noninferiority (see Table 12). FARXIGA treatment led to a statistically significant mean reduction in body weight from baseline at Week 52 (LOCF) compared with a mean increase in body weight in the glipizide group. Statistically significant (p<0.0001) mean change from baseline in systolic blood pressure relative to glipizide plus metformin was −5.0 mmHg with FARXIGA plus metformin.
| Efficacy Parameter | FARXIGA
+ Metformin N=400† | Glipizide
+ Metformin N=401† |
|---|---|---|
| * LOCF: last observation carried forward. † Randomized and treated patients with baseline and at least 1 postbaseline efficacy measurement. ‡ Least squares mean adjusted for baseline value. § Noninferior to glipizide + metformin. ¶ p-value <0.0001. |
||
|
HbA1c (%) |
||
|
Baseline (mean) |
7.7 |
7.7 |
|
Change from baseline (adjusted mean‡) |
−0.5 |
−0.5 |
|
Difference from glipizide + metformin (adjusted mean‡) |
0.0§
| |
|
Body Weight (kg) |
||
|
Baseline (mean) |
88.4 |
87.6 |
|
Change from baseline (adjusted mean‡) |
−3.2 |
1.4 |
|
Difference from glipizide + metformin (adjusted mean‡) |
−4.7¶
| |
14.6 Add-On Combination Therapy with Other Antidiabetic Agents
Add-On Combination Therapy with a Sulfonylurea
A total of 597 patients with type 2 diabetes and inadequate glycemic control (HbA1c ≥7% and ≤10%) were randomized in this 24-week, placebo-controlled study to evaluate FARXIGA in combination with glimepiride (a sulfonylurea).
Patients on at least half the maximum recommended dose of glimepiride as monotherapy (4 mg) for at least 8 weeks lead-in were randomized to FARXIGA 5 mg, FARXIGA 10 mg, or placebo in addition to glimepiride 4 mg per day. Down-titration of glimepiride to 2 mg or 0 mg was allowed for hypoglycemia during the treatment period; no up-titration of glimepiride was allowed.
In combination with glimepiride, FARXIGA 10 mg provided statistically significant improvement in HbA1c, FPG, and 2-hour PPG, and statistically significant reduction in body weight compared with placebo plus glimepiride at Week 24 (see Table 13). Statistically significant (p<0.05 for both doses) mean changes from baseline in systolic blood pressure relative to placebo plus glimepiride were −2.8 mmHg and −3.8 mmHg with FARXIGA 5 mg and 10 mg plus glimepiride, respectively.
Add-On Combination Therapy with a Thiazolidinedione
A total of 420 patients with type 2 diabetes with inadequate glycemic control (HbA1c ≥7% and ≤10.5%) participated in a 24-week, placebo-controlled study to evaluate FARXIGA in combination with pioglitazone (a thiazolidinedione [TZD]) alone. Patients on a stable dose of pioglitazone of 45 mg per day (or 30 mg per day, if 45 mg per day was not tolerated) for 12 weeks were randomized after a 2-week lead-in period to 5 or 10 mg of FARXIGA or placebo in addition to their current dose of pioglitazone. Dose titration of FARXIGA or pioglitazone was not permitted during the study.
In combination with pioglitazone, treatment with FARXIGA 10 mg provided statistically significant improvements in HbA1c, 2-hour PPG, FPG, the proportion of patients achieving HbA1c <7%, and a statistically significant reduction in body weight compared with the placebo plus pioglitazone treatment groups (see Table 13) at Week 24. A statistically significant (p<0.05) mean change from baseline in systolic blood pressure relative to placebo in combination with pioglitazone was −4.5 mmHg with FARXIGA 10 mg in combination with pioglitazone.
Add-On Combination Therapy with a DPP4 Inhibitor
A total of 452 patients with type 2 diabetes who were drug naive, or who were treated at entry with metformin or a DPP4 inhibitor alone or in combination, and had inadequate glycemic control (HbA1c ≥7.0% and ≤10.0% at randomization), participated in a 24-week, placebo-controlled study to evaluate FARXIGA in combination with sitagliptin (a DPP4 inhibitor) with or without metformin.
Eligible patients were stratified based on the presence or absence of background metformin (≥1500 mg per day), and within each stratum were randomized to either FARXIGA 10 mg plus sitagliptin 100 mg once daily, or placebo plus sitagliptin 100 mg once daily. Endpoints were tested for FARXIGA 10 mg versus placebo for the total study group (sitagliptin with and without metformin) and for each stratum (sitagliptin alone or sitagliptin with metformin). Thirty-seven percent (37%) of patients were drug naive, 32% were on metformin alone, 13% were on a DPP4 inhibitor alone, and 18% were on a DPP4 inhibitor plus metformin. Dose titration of FARXIGA, sitagliptin, or metformin was not permitted during the study.
In combination with sitagliptin (with or without metformin), FARXIGA 10 mg provided statistically significant improvements in HbA1c, FPG, and a statistically significant reduction in body weight compared with the placebo plus sitagliptin (with or without metformin) group at Week 24 (see Table 13). These improvements were also seen in the stratum of patients who received FARXIGA 10 mg plus sitagliptin alone (placebo-corrected mean change for HbA1c −0.56%; n=110) compared with placebo plus sitagliptin alone (n=111), and the stratum of patients who received FARXIGA 10 mg plus sitagliptin and metformin (placebo-corrected mean change for HbA1c −0.40; n=113) compared with placebo plus sitagliptin with metformin (n=113).
Add-On Combination Therapy with Insulin
A total of 808 patients with type 2 diabetes who had inadequate glycemic control (HbA1c ≥7.5% and ≤10.5%) were randomized in a 24-week, placebo-controlled study to evaluate FARXIGA as add-on therapy to insulin. Patients on a stable insulin regimen, with a mean dose of at least 30 IU of injectable insulin per day, for a period of at least 8 weeks prior to enrollment and on a maximum of 2 oral antidiabetic medications (OADs), including metformin, were randomized after completing a 2-week enrollment period to receive either FARXIGA 5 mg, FARXIGA 10 mg, or placebo in addition to their current dose of insulin and other OADs, if applicable. Patients were stratified according to the presence or absence of background OADs. Up- or down-titration of insulin was only permitted during the treatment phase in patients who failed to meet specific glycemic goals. Dose modifications of blinded study medication or OAD(s) were not allowed during the treatment phase, with the exception of decreasing OAD(s) where there were concerns over hypoglycemia after cessation of insulin therapy.
In this study, 50% of patients were on insulin monotherapy at baseline, while 50% were on 1 or 2 OADs in addition to insulin. At Week 24, FARXIGA 10 mg dose provided statistically significant improvement in HbA1c and reduction in mean insulin dose, and a statistically significant reduction in body weight compared with placebo in combination with insulin, with or without up to 2 OADs (see Table 13); the effect of FARXIGA on HbA1c was similar in patients treated with insulin alone and patients treated with insulin plus OAD. Statistically significant (p<0.05) mean change from baseline in systolic blood pressure relative to placebo in combination with insulin was −3.0 mmHg with FARXIGA 10 mg in combination with insulin.
At Week 24, FARXIGA 5 mg (−5.7 IU, difference from placebo) and 10 mg (−6.2 IU, difference from placebo) once daily resulted in a statistically significant reduction in mean daily insulin dose (p<0.0001 for both doses) compared to placebo in combination with insulin, and a statistically significantly higher proportion of patients on FARXIGA 10 mg (19.6%) reduced their insulin dose by at least 10% compared to placebo (11.0%).
| Efficacy Parameter | FARXIGA 10 mg | FARXIGA 5 mg | Placebo
|
|---|---|---|---|
| * LOCF: last observation (prior to rescue for rescued patients) carried forward. † Randomized and treated patients with baseline and at least 1 postbaseline efficacy measurement. ‡ Least squares mean adjusted for baseline value. § p-value <0.0001 versus placebo. ¶ 2-hour PPG level as a response to a 75-gram oral glucose tolerance test (OGTT). # All randomized patients who took at least one dose of double-blind study medication during the short-term, double-blind period. ** p-value <0.05 versus placebo. †† NT: Not formally tested because of failing to achieve a statistically significant difference in an endpoint that was earlier in the testing sequence. |
|||
|
In Combination with Sulfonylurea (Glimepiride) |
|||
|
Intent-to-Treat Population |
N=151† |
N=142† |
N=145† |
|
HbA1c (%) |
|||
|
Baseline (mean) |
8.1 |
8.1 |
8.2 |
|
Change from baseline (adjusted mean‡) |
−0.8 |
−0.6 |
−0.1 |
|
Difference from placebo + glimepiride (adjusted mean‡) |
−0.7§
|
−0.5§
| |
|
Percent of patients achieving HbA1c <7% |
31.7%§
|
30.3%§
|
13.0% |
|
FPG (mg/dL) |
|||
|
Baseline (mean) |
172.4 |
174.5 |
172.7 |
|
Change from baseline (adjusted mean‡) |
−28.5 |
−21.2 |
−2.0 |
|
Difference from placebo + glimepiride (adjusted mean‡) (95% CI) |
−26.5§
|
−19.3§
| |
|
2-hour PPG¶ (mg/dL) |
|||
|
Baseline (mean) |
329.6 |
322.8 |
324.1 |
|
Change from baseline (adjusted mean‡) |
−60.6 |
–54.5 |
−11.5 |
|
Difference from placebo + glimepiride (adjusted mean‡) (95% CI) |
−49.1§
|
−43.0§
| |
|
Body Weight (kg) |
|||
|
Baseline (mean) |
80.6 |
81.0 |
80.9 |
|
Change from baseline (adjusted mean‡) |
−2.3 |
−1.6 |
−0.7 |
|
Difference from placebo + glimepiride (adjusted mean‡) (95% CI) |
−1.5§
|
−0.8§
| |
|
In Combination with Thiazolidinedione (Pioglitazone) |
|||
|
Intent-to-Treat Population |
N=140# |
N=141# |
N=139# |
|
HbA1c (%) |
|||
|
Baseline (mean) |
8.4 |
8.4 |
8.3 |
|
Change from baseline (adjusted mean‡) |
−1.0 |
−0.8 |
−0.4 |
|
Difference from placebo (adjusted mean‡) |
−0.6§
|
−0.4§
| |
|
Percent of patients achieving HbA1c <7% |
38.8%** |
32.5%** |
22.4% |
|
FPG (mg/dL) |
|||
|
Baseline (mean) |
164.9 |
168.3 |
160.7 |
|
Change from baseline (adjusted mean‡) |
−29.6 |
−24.9 |
−5.5 |
|
Difference from placebo (adjusted mean‡) |
−24.1§
|
−19.5§
| |
|
2-hour PPG¶ (mg/dL) |
|||
|
Baseline (mean) |
308.0 |
284.8 |
293.6 |
|
Change from baseline (adjusted mean‡) |
−67.5 |
−65.1 |
−14.1 |
|
Difference from placebo (adjusted mean‡) |
−53.3§
|
−51.0§
| |
|
Body Weight (kg) |
|||
|
Baseline (mean) |
84.8 |
87.8 |
86.4 |
|
Change from baseline (adjusted mean‡) |
−0.1 |
0.1 |
1.6 |
|
Difference from placebo (adjusted mean‡) |
−1.8§
|
−1.6§
| |
|
In Combination with DPP4 Inhibitor (Sitagliptin) with or without Metformin |
|||
|
Intent-to-Treat Population |
N=223† |
– |
N=224† |
|
HbA1c (%) |
|||
|
Baseline (mean) |
7.90 |
– |
7.97 |
|
Change from baseline (adjusted mean‡) |
−0.45 |
– |
0.04 |
|
Difference from placebo (adjusted mean‡) |
−0.48§
|
– | |
|
Patients with HbA1c decrease ≥0.7% (adjusted percent) |
35.4% |
– |
16.6% |
|
FPG (mg/dL) |
|||
|
Baseline (mean) |
161.7 |
– |
163.1 |
|
Change from baseline at Week 24 (adjusted mean‡) |
−24.1 |
– |
3.8 |
|
Difference from placebo (adjusted mean‡) |
−27.9§
|
– | |
|
Body Weight (kg) |
|||
|
Baseline (mean) |
91.02 |
– |
89.23 |
|
Change from baseline (adjusted mean‡) |
−2.14 |
– |
−0.26 |
|
Difference from placebo (adjusted mean‡) |
−1.89§
|
– | |
|
In Combination with Insulin with or without up to 2 Oral Antidiabetic Therapies |
|||
|
Intent-to-Treat Population |
N=194† |
N=211† |
N=193† |
|
HbA1c (%) |
|||
|
Baseline (mean) |
8.6 |
8.6 |
8.5 |
|
Change from baseline (adjusted mean‡) |
−0.9 |
−0.8 |
−0.3 |
|
Difference from placebo (adjusted mean‡) |
−0.6§
|
−0.5§
| |
|
FPG (mg/dL) |
|||
|
Baseline (mean) |
173.7 |
NT†† |
170.0 |
|
Change from baseline (adjusted mean‡) |
−21.7 |
NT†† |
3.3 |
|
Difference from placebo (adjusted mean‡) |
−25.0§
|
NT†† | |
|
Body Weight (kg) |
|||
|
Baseline (mean) |
94.6 |
93.2 |
94.2 |
|
Change from baseline (adjusted mean‡) |
−1.7 |
−1.0 |
0.0 |
|
Difference from placebo (adjusted mean‡) |
−1.7§
|
−1.0§
| |
14.7 Use in Patients with Type 2 Diabetes and Renal Impairment
The efficacy of FARXIGA was assessed in a study of diabetic patients with moderate renal impairment (252 patients with mean eGFR 45 mL/min/1.73 m2). FARXIGA did not show efficacy in this study. The placebo-corrected mean HbA1c change at 24 weeks was −0.1% (95% CI [−0.4%, 0.2%]) for both FARXIGA 5 mg (n=83) and 10 mg (n=82).
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
FARXIGA (dapagliflozin) tablets have markings on both sides and are available in the strengths and packages listed in Table 14.
| Tablet Strength | Film-Coated Tablet
Color/Shape | Tablet
Markings | Package Size | NDC Code |
|---|---|---|---|---|
|
5 mg |
yellow, |
“5” engraved on one |
Bottles of 30 |
0003-1427-11 |
|
10 mg |
yellow, |
“10” engraved on one side and “1428” engraved on the other side |
Bottles of 30 |
0003-1428-11 |
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Medication Guide).
Instructions
Instruct patients to read the Medication Guide before starting treatment with FARXIGA and to reread it each time the prescription is renewed.
Inform patients of the potential risks and benefits of FARXIGA and of alternative modes of therapy. Also inform patients about the importance of adherence to dietary instructions, regular physical activity, periodic blood glucose monitoring and HbA1c testing, recognition and management of hypoglycemia and hyperglycemia, and assessment of diabetes complications. Advise patients to seek medical advice promptly during periods of stress such as fever, trauma, infection, or surgery, as medication requirements may change.
Instruct patients to take FARXIGA only as prescribed. If a dose is missed, advise patients to take it as soon as it is remembered unless it is almost time for the next dose, in which case patients should skip the missed dose and take the medicine at the next regularly scheduled time. Advise patients not to take two doses of FARXIGA at the same time.
Inform patients that the most common adverse reactions associated with use of FARXIGA are genital mycotic infections, nasopharyngitis, and urinary tract infections.
Instruct patient to immediately inform her healthcare provider if she is pregnant or plans to become pregnant. Based on animal data, FARXIGA may cause fetal harm in the second and third trimesters of pregnancy.
Instruct patient to immediately inform her healthcare provider if she is breastfeeding or planning to breastfeed. It is not known if FARXIGA is excreted in breast milk; however, based on animal data, FARXIGA may cause harm to nursing infants.
Hypotension
Inform patients that symptomatic hypotension may occur with FARXIGA and advise them to contact their healthcare provider if they experience such symptoms [see Warnings and Precautions (5.1)]. Inform patients that dehydration may increase the risk for hypotension, and to have adequate fluid intake.
Genital Mycotic Infections in Females (e.g., Vulvovaginitis)
Inform female patients that vaginal yeast infections may occur and provide them with information on the signs and symptoms of vaginal yeast infections. Advise them of treatment options and when to seek medical advice [see Warnings and Precautions (5.4)].
Genital Mycotic Infections in Males (e.g., Balanitis)
Inform male patients that yeast infections of the penis (e.g., balanitis or balanoposthitis) may occur, especially in patients with prior history. Provide them with information on the signs and symptoms of balanitis and balanoposthitis (rash or redness of the glans or foreskin of the penis). Advise them of treatment options and when to seek medical advice [see Warnings and Precautions (5.4)].
Hypersensitivity Reactions
Inform patients that serious hypersensitivity reactions (e.g., urticaria and angioedema) have been reported with FARXIGA. Advise patients to immediately report any signs or symptoms suggesting allergic reaction or angioedema, and to take no more of the drug until they have consulted prescribing physicians.
Urinary Tract Infections
Inform patients of the potential for urinary tract infections. Provide them with information on the symptoms of urinary tract infections. Advise them to seek medical advice if such symptoms occur.
Manufactured by:
Bristol-Myers Squibb Company
Princeton, NJ 08543 USA
Marketed by:
Bristol-Myers Squibb Company
Princeton, NJ 08543
and
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
Product of Ireland
Revised: August 2014
MEDICATION GUIDE
FARXIGA (far-SEE-guh)
(dapagliflozin)
tablets
What is the most important information I should know about FARXIGA?
FARXIGA can cause serious side effects, including:
- •
-
Dehydration. FARXIGA can cause some people to have dehydration (the loss of body water and salt). Dehydration may cause you to feel dizzy, faint, lightheaded, or weak, especially when you stand up (orthostatic hypotension). You may be at a higher risk of dehydration if you:
- •
- have low blood pressure
- •
- take medicines to lower your blood pressure, including water pills (diuretics)
- •
- are 65 years of age or older
- •
- are on a low salt diet
- •
- have kidney problems
- •
-
Vaginal yeast infection. Women who take FARXIGA may get vaginal yeast infections. Symptoms of a vaginal yeast infection include:
- •
- vaginal odor
- •
- white or yellowish vaginal discharge (discharge may be lumpy or look like cottage cheese)
- •
- vaginal itching
- •
-
Yeast infection of the penis (balanitis). Men who take FARXIGA may get a yeast infection of the skin around the penis. Certain men who are not circumcised may have swelling of the penis that makes it difficult to pull back the skin around the tip of the penis. Other symptoms of yeast infection of the penis include:
- •
- redness, itching, or swelling of the penis
- •
- rash of the penis
- •
- foul smelling discharge from the penis
- •
- pain in the skin around the penis
Talk to your healthcare provider about what to do if you get symptoms of a yeast infection of the vagina or penis. Your healthcare provider may suggest you use an over-the-counter antifungal medicine. Talk to your healthcare provider right away if you use an over-the-counter antifungal medication and your symptoms do not go away.
- •
-
Bladder cancer. In studies of FARXIGA in people with diabetes, bladder cancer occurred in a few more people who were taking FARXIGA than in people who were taking other diabetes medications. There were too few cases to know if bladder cancer was related to FARXIGA. You should not take FARXIGA if you have bladder cancer. Tell your healthcare provider right away if you have any of the following symptoms:
- •
- blood or a red color in your urine
- •
- pain while you urinate
What is FARXIGA?
FARXIGA is a prescription medicine used along with diet and exercise to lower blood sugar in adults with type 2 diabetes.
FARXIGA is not for people with type 1 diabetes.
FARXIGA is not for people with diabetic ketoacidosis (increased ketones in your blood or urine).
It is not known if FARXIGA is safe and effective in children younger than 18 years of age.
Who should not take FARXIGA?
Do not take FARXIGA if you:
- •
- are allergic to dapagliflozin or any of the ingredients in FARXIGA. See the end of this Medication Guide for a list of ingredients in FARXIGA. Symptoms of a serious allergic reaction to FARXIGA may include:
- •
- skin rash
- •
- raised red patches on your skin (hives)
- •
- swelling of the face, lips, tongue, and throat that may cause difficulty in breathing or swallowing
- If you have any of these symptoms, stop taking FARXIGA and contact your healthcare provider or go to the nearest hospital emergency room right away.
- •
- have severe kidney problems or are on dialysis.
What should I tell my healthcare provider before taking FARXIGA?
Before you take FARXIGA, tell your healthcare provider if you:
- •
- have type 1 diabetes or have had diabetic ketoacidosis.
- •
- have kidney problems.
- •
- have or have had bladder cancer.
- •
- are pregnant or plan to become pregnant. FARXIGA may harm your unborn baby. If you are pregnant or plan to become pregnant, talk to your healthcare provider about the best way to control your blood sugar.
- •
- are breastfeeding or plan to breastfeed. It is not known if FARXIGA passes into your breast milk. Talk with your healthcare provider about the best way to feed your baby if you are taking FARXIGA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How should I take FARXIGA?
- •
- Take FARXIGA exactly as your healthcare provider tells you to take it.
- •
- Do not change your dose of FARXIGA without talking to your healthcare provider.
- •
- Take FARXIGA by mouth 1 time each day, with or without food.
- •
- When your body is under some types of stress, such as fever, trauma (such as a car accident), infection, or surgery, the amount of diabetes medicine you need may change. Tell your healthcare provider right away if you have any of these conditions and follow your healthcare provider’s instructions.
- •
- Stay on your prescribed diet and exercise program while taking FARXIGA.
- •
- Your healthcare provider may do certain blood tests before you start FARXIGA and during your treatment.
- •
- Your healthcare provider will check your diabetes with regular blood tests, including your blood sugar levels and your A1c.
- •
- Follow your healthcare provider’s instructions for treating low blood sugar (hypoglycemia). Talk to your healthcare provider if low blood sugar is a problem for you.
- •
- If you miss a dose, take it as soon as you remember. If it is almost time for your next dose, skip the missed dose and take the medicine at the next regularly scheduled time. Do not take 2 doses of FARXIGA at the same time.
- •
- If you take too much FARXIGA, call your healthcare provider or go to the nearest emergency room right away.
What are the possible side effects of FARXIGA?
FARXIGA may cause serious side effects, including:
See “What is the most important information I should know about FARXIGA?”
- •
-
Low blood sugar (hypoglycemia). If you take FARXIGA with another medicine that can cause low blood sugar, such as a sulfonylurea or insulin, your risk of getting low blood sugar is higher. The dose of your sulfonylurea medicine or insulin may need to be lowered while you take FARXIGA. Signs and symptoms of low blood sugar may include:
- •
- headache
- •
- weakness
- •
- confusion
- •
- shaking or feeling jittery
- •
- drowsiness
- •
- dizziness
- •
- irritability
- •
- sweating
- •
- hunger
- •
- fast heartbeat
- •
- Kidney problems
- •
- Increased fats in your blood (bad cholesterol or LDL)
The most common side effects of FARXIGA include:
- •
- vaginal yeast infections and yeast infections of the penis
- •
- stuffy or runny nose and sore throat
- •
- urinary tract infections
- •
- changes in urination, including urgent need to urinate more often, in larger amounts, or at night
These are not all the possible side effects of FARXIGA. For more information, ask your healthcare provider or pharmacist.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store FARXIGA?
Store FARXIGA at room temperature between 68°F and 77°F (20°C and 25°C).
General information about the safe and effective use of FARXIGA
This Medication Guide summarizes the most important information about FARXIGA. If you would like more information, talk to your healthcare provider. You can ask your pharmacist or healthcare provider for information about FARXIGA that is written for healthcare professionals.
For more information about FARXIGA, go to www.farxiga.com or call 1-800-321-1335.
What are the ingredients in FARXIGA?
Active ingredient: dapagliflozin.
Inactive ingredients: microcrystalline cellulose, anhydrous lactose, crospovidone, silicon dioxide, and magnesium stearate. The film coating contains: polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and yellow iron oxide.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Bristol-Myers Squibb Company
Princeton, NJ 08543 USA
Marketed by:
Bristol-Myers Squibb Company
Princeton, NJ 08543
and
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
Product of Ireland
Revised: August 2014


| FARXIGA
dapagliflozin tablet, film coated |
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
| FARXIGA
dapagliflozin tablet, film coated |
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
| Labeler - E.R. Squibb & Sons, L.L.C. (011550092) |