IPRIVASK- desirudin
Marathon Pharmaceuticals, LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use IPRIVASK safely and effectively. See full prescribing information for IPRIVASK.
IPRIVASK® 15 mg (desirudin for injection), for subcutaneous injection Initial U.S. Approval: 2003 WARNING: SPINAL/EPIDURAL HEMATOMASSee Full Prescribing Information for complete boxed warning.When neuraxial anesthesia (epidural/spinal anesthesia) or spinal puncture is employed, patients anticoagulated or scheduled to be anticoagulated with selective inhibitors of thrombin such as Iprivask may be at risk of developing an epidural or spinal hematoma which can result in long-term or permanent paralysis (5.1). INDICATIONS AND USAGEIPRIVASK is a direct thrombin inhibitor and is indicated for the prophylaxis of deep vein thrombosis, which may lead to pulmonary embolism, in patients undergoing elective hip replacement surgery (1). DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSSingle use vials containing (15.75 mg) lyophilized powder with an accompanying sterile, non-pyrogenic diluent [0.6 mL of Mannitol USP (3%) in Water for Injection (3)]. Reconstituted solution delivers 15 mg of Iprivask. CONTRAINDICATIONSPatients with known hypersensitivity to natural or recombinant hirudins, and in patients with active bleeding and/or irreversible coagulation disorders (4). WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reaction was hemorrhage (33%). Other adverse reactions (incidence ≥2%) injection site mass, wound secretion, anemia, deep thrombophlebitis and nausea (6.1). To report SUSPECTED ADVERSE REACTIONS, contact Marathon Pharmaceuticals, LLC at 1-866-562-4620 or drugsafety@prosarcorp.com or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONSAvoid use with other drugs that inhibit or modify platelet function or affect blood clotting (e.g., anticoagulants, antiplatelet agents, NSAIDs, SSRIs) as coadministration may potentiate bleeding (7). See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 11/2014 |
FULL PRESCRIBING INFORMATION
WARNING: SPINAL/EPIDURAL HEMATOMAS
When neuraxial anesthesia (epidural/spinal anesthesia) or spinal puncture is employed, patients anticoagulated or scheduled to be anticoagulated with selective inhibitors of thrombin such as Iprivask may be at risk of developing an epidural or spinal hematoma which can result in long-term or permanent paralysis.
The risk of these events may be increased by the use of indwelling spinal catheters for administration of analgesia or by the concomitant use of drugs affecting hemostasis such as non- steroidal anti-inflammatory drugs (NSAIDs), platelet inhibitors, or other anticoagulants. Likewise with such agents, the risk appears to be increased by traumatic or repeated epidural or spinal puncture.
Patients should be frequently monitored for signs and symptoms of neurological impairment. If neurological compromise is noted, urgent treatment is necessary.
The physician should consider the potential benefit versus risk before neuraxial intervention, in patients anticoagulated or to be anticoagulated for thromboprophylaxis [see Warnings and Precautions (5.1)].
1 INDICATIONS AND USAGE
Iprivask is indicated for the prophylaxis of deep vein thrombosis, which may lead to pulmonary embolism, in patients undergoing elective hip replacement surgery.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
All patients should be evaluated for bleeding disorder risk before prophylactic administration of Iprivask [see DRUG INTERACTIONS (7)].
Initial Dosage: In patients undergoing hip replacement surgery, the recommended dose of Iprivask is 15 mg every 12 hours administered by subcutaneous injection with the initial dose given up to 5 to 15 minutes prior to surgery, but after induction of regional block anesthesia, if used [see WARNINGS AND PRECAUTIONS (5.1)]. Up to 12 days administration (average duration 9 to 12 days) of Iprivask has been well tolerated in controlled clinical trials.
2.2 Dose Adjustment in Renal Impairment
|
Degree of Renal
|
Creatinine
|
aPTT Monitoring & Dosing Instructions |
|
Moderate |
≥31 to 60 |
Initiate therapy at 5 mg every 12 hours by subcutaneous injection. Monitor aPTT and serum creatinine at least daily. If aPTT exceeds 2 times control: |
|
Severea |
<31 |
Initiate therapy at 1.7 mg every 12 hours by subcutaneous injection. Monitor aPTT and serum creatinine at least daily. If aPTT exceeds 2 times control: |
a See CLINICAL PHARMACOLOGY (12.3) and WARNINGS AND PRECAUTIONS(5.2).
2.3 Clinical Monitoring
Activated partial thromboplastin time (aPTT) should be monitored daily in patients with increased risk of bleeding and/or renal impairment. Serum creatinine should be monitored daily in patients with renal impairment. Adjust Iprivask dosage according to creatinine levels [see Dosage and Administration (2.2)].
Peak aPTT should not exceed two times control. Should peak aPTT exceed this level, reduce the dosage of Iprivask based on the degree of aPTT abnormality [see Dosage and Administration (2.1)]. If necessary, interrupt therapy with desirudin until aPTT falls to less than two times control, at which time treatment with desirudin can be resumed at a reduced dose. Thrombin time (TT) is not a suitable test for routine monitoring of Iprivask therapy [see Clinical Pharmacology (12.1)].
2.4 Conversion from Other Anticoagulants
If a patient is switched from oral anticoagulants to Iprivask therapy or from Iprivask to oral anticoagulants, the anticoagulant activity should continue to be closely monitored with appropriate methods [see Drug Interactions (7.2)].
2.5 Instructions for Administration
Directions on Preparation
Use Iprivask before the expiration date given on the carton and container.
1. To prepare the reconstituted aqueous solution, 0.5 mL of the mannitol diluent is added under aseptic conditions to the vial containing the sterile powder.
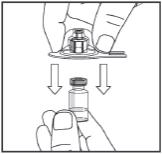
2. Remove plastic flip-top cap from Iprivask Vial. Remove back cover of Vial Adapter package. Attach Vial Adapter to Vial by using the outer package to handle Adapter. Push Adapter down onto Vial until spike pierces rubber stopper and snaps into place. Discard Vial Adapter package.
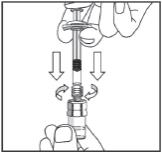
3. Remove Syringe cap by twisting and pulling gently. Attach Syringe to Adapter on Vial by twisting. Slowly push plunger down to completely transfer solution into Vial. Leaving Syringe connected to Vial, gently swirl. Lyophilized powder in Vial will dissolve within 10 seconds.
4. With Syringe still connected to Vial, turn Vial upside down and withdraw entire contents of Vial back into Syringe. Remove Syringe from Vial and hold it with plunger-end down.
5. You must use enclosed Eclipse™ needle. Attach Needle to Syringe by twisting. Pull pink lever down and uncap needle. You are ready to inject Iprivask. After injection, flip up pink lever to cover needle until it snaps into place. Dispose of the used Syringe in a Sharps® container.
Iprivask should not be mixed with other injections, solvents, or infusions. Iprivask is administered by subcutaneous injection. It must not be administered by intramuscular injection.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Subcutaneous Injection Technique:
Patients should be sitting or lying down and Iprivask injection administered by deep subcutaneous injection. Administration should be alternated between the left and right anterolateral and left and right posterolateral thigh or abdominal wall. The whole length of the needle should be introduced into a skin fold held between the thumb and forefinger; the skin fold should be held throughout the injection. To minimize bruising, do not rub the injection site after completion of the injection.
3 DOSAGE FORMS AND STRENGTHS
Iprivask (desirudin for injection) is supplied as a single dose (15.75 mg) lyophilized powder with an accompanying sterile, non-pyrogenic diluent [0.6 mL of Mannitol USP (3%) in Water for Injection]. The reconstituted solution delivers 15 mg of Iprivask in 0.5 mL.
4 CONTRAINDICATIONS
Iprivask is contraindicated in patients with known hypersensitivity to natural or recombinant hirudins due to risk of anaphylaxis, and in patients with active bleeding and/or irreversible coagulation disorders due to risk of hemorrhage.
5 WARNINGS AND PRECAUTIONS
5.1 Spinal/Epidural Hematoma
There is a risk of neuraxial hematoma formation with the concurrent use of desirudin and spinal/epidural anesthesia, which has the potential to result in long term or permanent paralysis. The risk may be greater with the use of post-operative indwelling catheters or the concomitant use of additional drugs affecting hemostasis such as NSAIDs (Non-Steroidal Anti-Inflammatory Drugs), platelet inhibitors or other anticoagulants [see Drug Interactions (7.1)]. The risk may also be increased by traumatic or repeated neuraxial puncture.
To reduce the potential risk of bleeding associated with the concurrent use of desirudin and epidural or spinal anesthesia/analgesia, the pharmacokinetic profile of the drug should be considered [see Clinical Pharmacology (12.3)] when scheduling or using epidural or spinal anesthesia in proximity to desirudin administration. The physician should consider placement of the catheter prior to initiating desirudin and removal of the catheter when the anticoagulant effect of desirudin is low [see Dosage and Administration (2.1)].
Should the physician decide to administer anticoagulation in the context of epidural/spinal anesthesia, extreme vigilance and frequent monitoring must be exercised to detect any signs and symptoms of neurological impairment such as midline back pain, sensory and motor deficits (numbness or weakness in lower limbs), bowel and/or bladder dysfunction. Patients should be instructed to inform their physician immediately if they experience any of the above signs or symptoms. If signs or symptoms of spinal hematoma are suspected, urgent diagnosis and treatment including spinal cord decompression should be initiated.
The physician should consider the potential benefit versus risk before neuraxial intervention in patients anticoagulated or to be anticoagulated for thromboprophylaxis
Iprivask cannot be used interchangeably with other hirudins as they differ in manufacturing process and specific biological activity (ATUs). Each of these medicines has its own instructions for use.
5.2 Hemorrhagic Events
Avoid intramuscular injection of Iprivask as bleeding and local hematoma formation can occur.
Iprivask increases the risks of hemorrhage in patients with recent major surgery, organ biopsy or puncture of a non-compressible vessel within the last month; a history of hemorrhagic stroke, intracranial or intraocular bleeding including diabetic (hemorrhagic) retinopathy; recent ischemic stroke, severe uncontrolled hypertension, bacterial endocarditis, a known hemostatic disorder (congenital or acquired, e.g. hemophilia, liver disease) or a history of gastrointestinal or pulmonary bleeding within the past 3 months.
Bleeding can occur at any site during therapy with Iprivask. An unexplained fall in hematocrit or blood pressure should lead to a search for a bleeding site. In post marketing experience reports of fatal and serious bleeding events have occurred in patients treated with Iprivask. Sources of hemorrhage included bleeding from the brain, gastrointestinal tract, spleen, rectum, and vagina [see Adverse Reactions (6.2)].
Avoid use with other drugs that inhibit or modify platelet function or affect blood clotting (e.g., anticoagulants, antiplatelet agents, NSAIDs, SSRIs) as coadministration with Iprivask may potentiate bleeding. If these concomitant medications cannot be avoided, patients should be monitored for bleeding [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
5.3 Increased Risk of Bleeding with Renal Impairment
Iprivask must be used with caution in patients with renal impairment due to an increased risk for bleeding, particularly in those with moderate and severe renal impairment (creatinine clearance ≤60 mL/min) [see Clinical Pharmacology (12.3)]. Reduce dose and monitor daily aPTT and serum creatinine in patients with moderate and severe renal impairment [see Dosage and Administration (2.2, 2.3)].
5.4 Antibodies/Re-exposure
Antibodies have been reported in patients treated with hirudins. Potential for cross-sensitivity to hirudin products cannot be excluded. Irritative skin reactions were observed in 9/322 volunteers exposed to Iprivask by subcutaneous injection or intravenous bolus or infusion in single or multiple administrations of the drug. The allergic reaction in volunteers consisted of arthralgia, erythema, pruritus, or uticaria. Allergic events were reported in <2% of patients who were administered desirudin in Phase III clinical trials. Allergic events were reported in 1% of patients receiving unfractionated heparin and 1% of patients receiving enoxaparin. Hirudin-specific IgE evaluations may not be indicative of sensitivity to Iprivask as this test was not always positive in the presence of symptoms., Anti-hirudin antibodies have been detected upon re-exposure to desirudin. [see Adverse Reactions (6.1)]. Fatal anaphylactic reactions have been reported during hirudin therapy.
6 ADVERSE REACTIONS
The following serious reactions are described further in other sections of the prescribing information
- •
- Spinal/Epidural Hematoma [see Warnings and Precautions (5.1)]
- •
- Hemorrhagic Events [see Warnings and Precautions (5.2)]
- •
- Increased Risk of Bleeding with Renal Impairment [see Warnings and Precautions (5.3)]
- •
- Antibodies/Re-exposure [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In the Phase II and III clinical studies, desirudin was administered to 2159 patients undergoing elective hip replacement surgery to determine the safety and efficacy of Iprivask in preventing VTE in this population. Below is the safety profile of the Iprivask 15 mg (q12h) regimen.
Hemorrhagic Events [see Warnings and Precautions (5.2)]: The following rates of hemorrhagic events have been reported during clinical trials.
|
Dosing Regimen |
|||
|
Iprivask |
Heparin |
Enoxaparin |
|
|
15 mg q12h SC |
5000 IU q8h SC |
40 mg QD SC |
|
|
Patients with Any Hemorrhagea |
464 (30) |
111 (22) |
341 (33) |
|
Patients with Serious Hemorrhageb |
41 (3) |
15 (3) |
21 (2) |
|
Patients with Major Hemorrhagec |
13 (<1) |
0 (0) |
2 (<1) |
a Includes hematomas which occurred at an incidence of 6% in the Iprivask and enoxaparin treatment groups and 5% in the heparin treatment group [see Warnings and Precautions (5.1)].
b Bleeding complications were considered serious if perioperative transfusion requirements exceeded 5 units of whole blood or packed red cells, or if total transfusion requirements up to postoperative Day 6 inclusive exceeded 7 units of whole blood or packed red cells, or total blood loss up to postoperative Day 6 inclusive exceeded 3500 mL.
c Bleeding complications were considered major if the hemorrhage was: (1) overt and it produced a fall in hemoglobin of ≥2g/dL or if it lead to a transfusion of 2 or more units of whole or packed cells outside the perioperative period (the time from start of surgery until up to 12 hours after); (2) Retroperitoneal, intracranial, intraocular, intraspinal, or occurred in a major prosthetic joint.
Non-hemorrhagic Events: Non-hemorrhagic adverse events occurring at ≥2% incidence in patients treated with Iprivask 15 mg (q 12h) during elective hip replacement surgery and considered to be remotely, possibly, or probably related to desirudin are provided below.
|
Iprivask |
Heparin |
Enoxaparin |
|
|
Body System (Preferred Term) |
15 mg q12h SC |
5000 IU q8h SC |
40 mg QD SC |
|
Injection Site Mass |
56 (4) |
32 (6) |
7 (<1) |
|
Wound Secretion |
59 (4) |
23 (5) |
34 (3) |
|
Anemia |
51 (3) |
11 (2) |
37 (4) |
|
Deep Thrombophlebitis |
24 (2) |
41 (8) |
22 (2) |
|
Nausea |
24 (2) |
5 (<1) |
10 (<1) |
a Represents events reported while on treatment, excluding unrelated adverse events
b All hemorrhages that occurred are included in ADVERSE REACTIONS, Hemorrhagic Events.
Related Adverse Events with a Frequency of <2% and >0.2% (in decreasing order of frequency): thrombosis, hypotension, leg edema, fever, decreased hemoglobin, hematuria, dizziness, epistaxis, vomiting, impaired healing, cerebrovascular disorder, leg pain, hematemesis.
Allergic Reactions. In clinical studies, allergic events were reported <2% overall and in 2% of patients who were administered 15 mg desirudin. [see Warnings and Precautions (5.4)].
6.2 Post Marketing Experience
In addition to adverse events reported from clinical trials the following adverse events have been identified during post approval use of Iprivask. These events were reported voluntarily from a population of unknown size and the frequency of occurrence cannot be determined precisely: reports of major hemorrhages [see Warnings and Precautions (5.2)], some of which were fatal, and anaphylactic/anaphylactoid reactions [see Warnings and Precautions (5.4)].
7 DRUG INTERACTIONS
7.1 Drugs that Increase Bleeding Risk
Discontinue any agent which may enhance the risk of hemorrhage prior to initiation of Iprivask therapy. These agents include medications such as Dextran 40, systemic glucocorticoids, thrombolytics, and anticoagulants. If co-administration cannot be avoided, close clinical and laboratory monitoring should be conducted. During prophylaxis of venous thromboembolism, concomitant treatment with heparins (unfractionated and low-molecular weight heparins) or dextrans is not recommended. The effects of desirudin and unfractionated heparins on prolongation of aPTT are additive.
As with other anticoagulants, desirudin should be used with caution in conjunction with drugs which affect platelet function. These medications include systemic salicylates, NSAIDS including ketorolac, acetylsalicylic acid, ticlopidine, dipyridamole, sulfinpyrazone, clopidogrel, abciximab and other glycoprotein IIb/IIIa antagonists [see Warnings and Precautions (5.2)].
7.2 Coadministration with Warfarin
The concomitant administration of warfarin did not significantly affect the pharmacokinetic effects of desirudin. When warfarin and desirudin were co-administered, greater inhibition of hemostasis measured by activated partial thromboplastin time (aPTT), prothrombin time (PT), and international normalized ratio (INR) was observed [see Dosage and Administration (2.4)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C.
Risk Summary
There are no adequate and well controlled studies in pregnant women. Iprivask should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Iprivask was teratogenic in rats and rabbits when given in doses 0.3 to 4 times the human dose.
Animal Data
Teratology studies have been performed in rats at subcutaneous doses in a range of 1 to 15 mg/kg/day (about 0.3 to 4 times the recommended human dose based on body surface area) and in rabbits at intravenous doses in a range of 0.6 to 6 mg/kg/day (about 0.3 to 3 times the recommended human dose based on body surface area) and have revealed desirudin to be teratogenic. Observed teratogenic findings included: omphalocele, asymmetric and fused sternebrae, edema, and shortened hind limbs in rats; and spina bifida, malrotated hind limb, hydrocephaly, and gastroschisis in rabbits.
8.3 Nursing Mothers
It is not known whether desirudin is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Iprivask, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.5 Geriatric Use
In three clinical studies of Iprivask, the percentage of patients greater than 65 years of age treated with 15 mg of Iprivask subcutaneously every 12 hours was 58.5%, while 20.8% were 75 years of age or older. Elderly patients treated with Iprivask had a reduction in the incidence of VTE similar to that observed in the younger patients.
Regarding safety, in the clinical studies the incidence of hemorrhage (major or otherwise) in patients 65 years of age or older was similar to that in patients less than 65 years of age. In addition, the elderly had a similar incidence of total, treatment-related, or serious adverse events compared to those patients less than 65 years of age. Serious adverse events occurred more frequently in patients 75 years of age or older as compared to those less than 65 years of age. [see Clinical Pharmacology (12.3) and Dosage and Administration (2.2)].
8.6 Renal Impairment
Iprivask is primarily eliminated and metabolized by the kidney. Patients with moderate and severe renal impairment had significant increases in exposure and aPPT prolongation compared to individuals with normal renal function [see Clinical Pharmacology (12.3.)]. Reduce dose and monitor daily aPTT and serum creatinine in patients with renal impairment [see Dosage and Administration (2.2, 2.3)].
8.7 Hepatic Impairment
No information is available about the use of desirudin in patients with hepatic impairment. Although Iprivask is not significantly metabolized by the liver, hepatic impairment or serious liver injury (e.g., liver cirrhosis) may alter the anticoagulant effect of Iprivask due to coagulation defects secondary to reduced generation of vitamin K-dependent coagulation factors. Iprivask bleeding risk may be increased.
10 OVERDOSAGE
In case of overdose, most likely reflected in hemorrhagic complications or suggested by excessively high aPTT values, Iprivask therapy should be discontinued. Emergency procedures should be instituted as appropriate (for example, determination of aPTT and other coagulation levels, hemoglobin, the use of blood transfusion or plasma expanders).
No specific antidote for Iprivask is available; however, the anticoagulant effect of desirudin may be partially reversible using thrombin-rich plasma concentrates while aPTT levels can be reduced by the intravenous administration of 0.3 µg/kg DDAVP (desmopressin). The clinical effectiveness of DDAVP in treating bleeding due to desirudin overdose has not been studied. In an open, pilot, dose-ascending study to assess safety, the highest dose of desirudin (40 mg every 12 hours) caused excessive hemorrhage.
11 DESCRIPTION
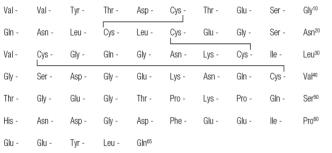
Iprivask® (desirudin for injection) is a direct inhibitor of human thrombin. It has a protein structure that is similar to that of hirudin, the naturally occurring anticoagulant present in the peripharyngeal glands in the medicinal leech, Hirudo medicinalis. Hirudin is a single polypeptide chain of 65 amino acids residues and contains three disulfide bridges. Desirudin has a chemical formula of C287H440N80O110S6 with a molecular weight of 6963.52. Desirudin, which is expressed in yeast (Saccharomyces cerevisiae, strain TR 1456) by recombinant DNA technology differs from the natural hirudin by lack of a sulfate group on Tyr-63. The biological activity of desirudin is determined through a chromogenic assay which measures the ability of desirudin to inhibit the hydrolysis of a chromogenic peptidic substrate by thrombin in comparison to a desirudin standard. One vial of desirudin contains 15.75 mg desirudin corresponding to approximately 315,000 antithrombin units (ATU) or 20,000 ATU per milligram of desirudin with reference to the WHO International Standard (prepared 1991) for alphathrombin.
Iprivask 15 mg is supplied as a sterile, white, freeze dried powder for injection. Each vial contains 15.75 mg desirudin and the following inactive ingredients: 1.31 mg anhydrous magnesium chloride USP, sodium hydroxide for injection USP. Each prefilled syringe of diluent for Iprivask contains 0.6 mL sterile Mannitol USP (3%) in Water for Injection and is preservative free. The reconstituted solution has a pH of 7.4.
STRUCTURAL FORMULA
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Desirudin is a direct inhibitor of free circulating and clot-bound thrombin. The anticoagulant properties of desirudin are demonstrated by its ability to prolong the clotting time of human plasma. One molecule of desirudin binds to one molecule of thrombin and thereby blocks the thrombogenic activity of thrombin. As a result, all thrombin-dependent coagulation assays are affected. Activated partial thromboplastin time (aPTT) is a measure of the anticoagulant activity of desirudin and increases in a dose-dependent fashion.
12.2 Pharmacodynamics
The pharmacodynamic effect of desirudin on proteolytic activity of thrombin was assessed as an increase in aPTT. A mean peak aPTT prolongation of about 1.38 times baseline value (range 0.58 to 3.41) was observed following subcutaneous b.i.d. injections of 15 mg desirudin. Thrombin time (TT) frequently exceeds 200 seconds even at low plasma concentrations of desirudin, which renders this test unsuitable for routine monitoring of Iprivask therapy. At therapeutic serum concentrations, desirudin has no effect on other enzymes of the hemostatic system such as factors IXa, Xa, kallikrein, plasmin, tissue plasminogen activator, or activated protein C. In addition, it does not display any effect on other serine proteases, such as the digestive enzymes trypsin, chymotrypsin, or on complement activation by the classical or alternative pathways.
12.3 Pharmacokinetics
Properties
Pharmacokinetic parameters were calculated based on plasma concentration data obtained by a non-specific ELISA method that does not discriminate between native desirudin and its metabolites. It is not known if the metabolites are pharmacologically active.
Absorption
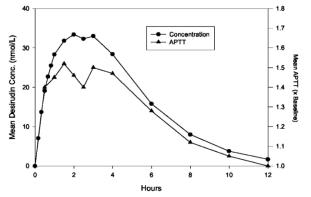
The absorption of desirudin is complete when subcutaneously administered at doses of 0.3 mg/kg or 0.5 mg/kg. Following subcutaneous administration of single doses of 0.1 to 0.75 mg/kg, plasma concentrations of desirudin increased to a maximum level (Cmax) between 1 and 3 hours. Both Cmax and area-under-the-curve (AUC) values are dose proportional.
Mean Desirudin Concentrations and Changes in APTT
After A Single 15 mg Subcutaneous Dose in 12 Healthy Subjects
Distribution
The pharmacokinetic properties of desirudin following intravenous administration are well described by a two- or three- compartment disposition model. Desirudin is distributed in the extracellular space with a volume of distribution at steady state of 0.25 L/kg, independent of the dose. Desirudin binds specifically and directly to thrombin, forming an extremely tight, non-covalent complex with an inhibition constant of approximately 2.6 x 10-13 M. Thus, free or protein bound desirudin immediately binds circulating thrombin. The pharmacological effect of desirudin is not modified when co-administered with highly protein bound drugs (>99%).
Metabolism
Human and animal data suggest that desirudin is primarily eliminated and metabolized by the kidney. The total urinary excretion of unchanged desirudin amounts to 40 to 50% of the administered dose. Metabolites lacking one or two C-terminal amino acids constitute a minor proportion of the material recovered from urine (< 7%). There is no evidence for the presence of other metabolites. This indicates that desirudin is metabolized by stepwise degradation from the C-terminus probably catalyzed by carboxypeptidase(s) such as carboxypeptidase A, originating from the pancreas. Total clearance of desirudin is approximately 1.5 to 2.7 mL/min/kg following either subcutaneous or intravenous administration and is independent of dose. This clearance value is close to the glomerular filtration rate.
Elimination
The elimination of desirudin from plasma is rapid after intravenous administration, with approximately 90% of the dose disappearing from the plasma within 2 hours of the injection. Plasma concentrations of desirudin then decline with a mean terminal elimination half-life of 2 to 3 hours. After subcutaneous administration, the mean terminal elimination half-life is also approximately 2 hours.
Specific Populations
Renal Impairment
In a pharmacokinetic study of renally impaired subjects, subjects with mild [creatinine clearance (CLcr) between 61 and 90 mL/min], moderate (CLcr between 31 and 60 mL/min), and severe (CLcr below 31 mL/min) renal impairment, were administered a single intravenous dose of 0.5, 0.25, or 0.125 mg/kg desirudin, respectively. This resulted in mean dose-normalized AUCeffect (AUC0-60th for aPTT prolongation) increases of approximately 3-, and 9-fold for the moderate and severe renally impaired subjects, respectively, compared with healthy individuals. In subjects with mild renal impairment, there was no increase in AUCeffect compared with healthy individuals. In subjects with severe renal impairment, terminal elimination half-lives were prolonged up to 12 hours compared with 2 to 4 hours in normal volunteers or subjects with mild to moderate renal impairment. Dose adjustments are recommended in certain circumstances in relation to the degree of impairment or degree of aPTT abnormality [see Dosage and Administration (2.2, 2.3), and Warnings and Precautions (5.1)].
Hepatic Impairment
No pharmacokinetic studies have been conducted to investigate the effects of Iprivask in hepatic impairment
Age and Gender
The mean plasma clearance of desirudin in patients ≥65 years of age (n=12; 110 mL/min) is approximately 28% lower than in patients <65 years of age (n=8; 153 mL/min). Population pharmacokinetics conducted in 301 patients undergoing elective total hip replacement indicate that age or gender do not affect the systemic clearance of desirudin when renal creatinine clearance is considered. This drug is substantially excreted by the kidney, and the risk of adverse events due to it may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. Dosage adjustment in the case of moderate and severe renal impairment is necessary. [see Dosage and Administration (2.2, 2.3)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term studies in animals have been conducted to evaluate the carcinogenic potential of desirudin.
Desirudin was not genotoxic in the Ames test, the Chinese hamster lung cell (V79/HGPRT) forward mutation test or the rat micronucleus test. It was, however, equivocal in its genotoxic effect in Chinese hamster ovarian cell (CCL 61) chromosome aberration tests.
Desirudin at subcutaneous doses up to 10mg/kg/day (about 2.7 times the recommended human dose based on body surface area) had no effect on fertility and reproductive function of male and female rats.
14 CLINICAL STUDIES
Iprivask was evaluated in two controlled, randomized, multicenter, clinical efficacy trials and a controlled, double-blind, dose-finding study. In the efficacy studies, Iprivask was compared to subcutaneously administered unfractionated heparin or enoxaparin sodium for the reduction of the risk of venous thromboembolic events (VTE) in patients undergoing total hip replacement surgery. In all studies Iprivask was initiated prior to surgery and continued for 8 to 12 days postoperatively (median duration 10 days).
In the first study, Iprivask 15 mg subcutaneously administered every 12 hours was compared to unfractionated heparin 5000 IU subcutaneously administered every 8 hours. A total of 445 patients were randomized in the study, 436 patients were treated, and 85 of the treated patients were excluded from efficacy analysis, mainly because of no phlebography or inadequate reading of phlebography. Patients ranged in age from 34 to 89 years (mean age 68.4 years) with 41.8% men and 58.2% women. All enrolled patients were Caucasian. Iprivask significantly reduced the number of total VTE compared to unfractionated heparin: Evaluable population: Iprivask, 13/174 (7.5%) vs. heparin, 41/177 (23.2%); p value <0.001; Intent-to-Treat population: Iprivask 13/225 (5.8%) vs. heparin 42/220 (19.1%); p value <0.0001.]. Significantly fewer patients in the group treated with Iprivask experienced proximal DVT than those patients treated with heparin: Evaluable population: Iprivask 6/174 (3.4%) vs. heparin 29/177 (16.4%); p value <0.001: Intent-to-Treat population: Iprivask 6/225 (2.7%) vs. heparin 30/220 (13.6%); p value <0.0001.
In a second study, Iprivask 15 mg subcutaneously administered every 12 hours was compared to enoxaparin sodium 40 mg subcutaneously administered every 24 hours. A total of 2079 patients were randomized in the study, 2049 patients were treated, and 508 of the treated patients were excluded from efficacy analysis mainly because of no phlebography or inadequate reading of phlebography. Patients ranged in age from 18 to 90 years (mean age 68.5 years) with 41.7% men and 58.5% women. All enrolled patients were Caucasian. In both the evaluable patient population and the intent-to-treat population, patients who received Iprivask had a lower incidence of major VTE, total VTE, and proximal DVT than did patients who received enoxaparin (see table below).
|
Efficacy of Iprivask in Hip Replacement Surgery Patients |
|||
|
Dosing Regimen |
|||
|
Iprivask a
|
Enoxaparina
| ||
|
Evaluable Hip Replacement
|
n=773 |
n=768 | |
|
|
n (%) |
n (%) |
p Value |
|
Treatment failures
|
|
|
|
|
Total VTEe |
145 (18.8) |
197 (25.7) |
p<0.001 |
|
Proximal DVT |
36 (4.5) |
59 (7.7) |
p=0.012 |
|
| |||
|
Intent-to-Treat Hip Replacement
|
n=1043 |
n=1036 | |
|
n (%) |
n (%) |
p Value |
|
|
Treatment Failures | |||
|
Major VTEb |
39 (3.7)f |
61 (5.9) |
p=0.024 |
|
Total VTEe |
145 (13.9) |
199 (19.2) |
p=0.001 |
|
Proximal DVT |
36 (3.5) |
59 (5.7) |
p=0.012 |
a Treatment was initiated no more than 30 minutes preoperatively, but after induction of regional block anesthesia, if used.
b Major VTE included proximal DVT, PE, or death.
c Total number of patients in this evaluation: Iprivask 802; Enoxaparin 785.
d Odds ratio 0.61 with 95% Confidence Interval of: 0.40; 0.92
e Total VTE = Venous thromboembolic events which included DVT (including proximal events), PE, or death considered to be thromboembolic in origin.
f Odds ratio 0.62 with 95% Confidence Interval of 0.41; 0.94
In a multicenter, double-blind, dose-finding study, Iprivask 10 mg, 15 mg, and 20 mg subcutaneously administered every 12 hours was compared to unfractionated heparin 5,000 IU administered every 8 hours subcutaneously in patients undergoing hip replacement surgery. A dose response was seen with regard to both effectiveness and bleeding complications. The 15-mg and 20-mg doses were superior to heparin and the 10-mg dose. In a smaller, open-labeled, dose-finding study of Iprivask 10 mg, 15 mg, 20 mg, and 40 mg subcutaneously administered every 12 hours in patients undergoing hip replacement surgery, the 40-mg dose was associated with unacceptable major bleeding.
16 HOW SUPPLIED/STORAGE AND HANDLING
Iprivask (desirudin for injection) is supplied as a single dose (15.75 mg) lyophilized powder with an accompanying sterile, non-pyrogenic diluent [0.6 mL of Mannitol USP (3%) in Water for Injection].
Each Iprivask Vial contains 15.75 mg desirudin and the following inactive ingredients: 1.31 mg anhydrous magnesium chloride USP, sodium hydroxide for injection USP.
Each carton (NDC 42998-715-10) of Iprivask (desirudin for injection) contains 10 individual doses of Iprivask, each in a separate tray.
Each tray of Iprivask (desirudin for injection) contains:
- •
- One (1) x 15.75 mg Single Dose Vial
- •
- One (1) x 0.6 mL Prefilled syringe of Diluent
- •
- One (1) Eclipse™ needle
- •
- One (1) Vial Adapter
Each prefilled syringe of diluent contains 0.6 mL Mannitol USP (3% w/v) in Water for Injection provided for reconstitution of the desirudin lyophilized powder.
Storage: Protect from light.
Unopened vials or prefilled syringes: Store at 25°C (77°F); excursions permitted to 15–30°C (59-86°F). [See USP Controlled Room Temperature.]
17 PATIENT COUNSELING INFORMATION
Advise patients to watch carefully for any signs of bleeding or bruising and to report these to their health care provider immediately. Inform patients that it might take them longer than usual to stop bleeding, and that they may bruise and/or bleed more easily when they are treated with Iprivask. If patients have had neuraxial anesthesia or spinal puncture, and particularly, if they are taking concomitant NSAIDs or platelet inhibitors, advise patients to watch for signs and symptoms of spinal or epidural hematoma, such as back pain, tingling, numbness (especially in the lower limbs), muscle weakness, and stool or urine incontinence. If any of these symptoms occur, advise the patient to contact his or her physician immediately.
Advise patients that allergic reactions, including anaphylaxis, can occur during treatment with Iprivask. Inform patients to immediately notify their health care provider if the patient experiences symptoms of an allergic reaction while taking Iprivask.
Advise patients to discuss with their health care provider their use of any other medications, including over-the-counter medications or herbal products, prior to Iprivask use. Examples of other medications that should not be taken with Iprivask are aspirin, non-steroidal anti-inflammatory drugs including ketorolac, acetylsalicylic acid ticlopidine, dipyridamole, sulfinpyrazone, clopidogrel, abciximab and other glycoprotein IIb/IIIa antagonists.
Manufactured for:
Marathon Pharmaceuticals, LLC.

Northbrook, IL 60062 USA
Made in Germany
Product Assembly Iprivask®
Instructions for Use (desirudin for injection)
Patients are encouraged to refer to complete Patient Instruction Sheet for additional information on Iprivask assembly and administration.
Step 1:
Remove plastic flip-top cap from Iprivask Vial. Remove back cover of Vial Adapter package. Attach Vial Adapter to Vial by using the outer package to handle Adapter. Push Adapter down onto Vial until spike pierces rubber stopper and snaps into place. Discard Vial Adapter package.
Step 2:
Remove Syringe cap by twisting and pulling gently. Attach Syringe to Adapter on Vial by twisting. Slowly push plunger down to completely transfer solution into Vial. Leaving Syringe connected to Vial, gently swirl; round tablet in Vial will dissolve within 10 seconds.
Step 3:
With Syringe still connected to Vial, turn Vial upside down and withdraw entire contents of Vial back into Syringe. Remove Syringe from Vial and hold it with plunger-end down.
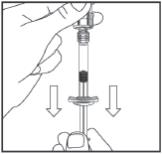
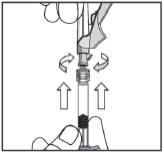
Step 4:
You must use enclosed Eclipse™ needle. Attach Needle to Syringe by twisting. Pull pink lever down and uncap needle. You are ready to inject Iprivask.
After injection, flip up pink lever to cover needle until it snaps into place. Dispose of the used Syringe in a Sharps® container.
Eclipse is a registered trademark of Becton, Dickinson and Company.
Sharps is a registered trademark of Sharps Compliance Corporation.
© Marathon Pharmaceuticals, LLC., Northbrook, IL 60062 USA
PRINCIPAL DISPLAY PANEL
Rx only
NDC 42998-715-01
Iprivask
(desirudin for injection)
FOR SUBCUTANEOUS INJECTION
15 mg*
Marathon Pharmaceuticals LLC

PRINCIPAL DISPLAY PANEL
Rx only
NDC 42998-715-10
Iprivask
(desirudin for injection)
FOR SUBCUTANEOUS INJECTION
15 mg*
Marathon Pharmaceuticals LLC



| IPRIVASK
desirudin kit |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Marathon Pharmaceuticals, LLC (828905336) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Wasserburger Arzneimittel Werk, GMBH | 326482247 | MANUFACTURE(42998-715, 42998-806) | |