VICTRELIS- boceprevir capsule
Merck Sharp & Dohme Corp.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use VICTRELIS safely and effectively. See full prescribing information for VICTRELIS.
VICTRELIS® (boceprevir) capsules, for oral use Initial U.S. Approval: 2011 RECENT MAJOR CHANGES
INDICATIONS AND USAGEVICTRELIS is a hepatitis C virus (HCV) NS3/4A protease inhibitor indicated for the treatment of chronic hepatitis C (CHC) genotype 1 infection, in combination with peginterferon alfa and ribavirin, in adult patients with compensated liver disease, including cirrhosis, who are previously untreated or who have failed previous interferon and ribavirin therapy, including prior null responders, partial responders, and relapsers. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSCapsules: 200 mg (3) CONTRAINDICATIONS
WARNINGS AND PRECAUTIONSUse of VICTRELIS with Ribavirin and Peginterferon alfa:
ADVERSE REACTIONSThe most commonly reported adverse reactions (greater than 35% of subjects) in clinical trials in adult subjects receiving the combination of VICTRELIS with PegIntron and REBETOL were fatigue, anemia, nausea, headache and dysgeusia. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., at 1-877-888-4231 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONSUSE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 1/2017 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
VICTRELIS® (boceprevir) is indicated for the treatment of chronic hepatitis C genotype 1 infection, in combination with peginterferon alfa and ribavirin, in adult patients with compensated liver disease, including cirrhosis, who are previously untreated or who have failed previous interferon and ribavirin therapy, including prior null responders, partial responders, and relapsers [see Clinical Studies (14)].
The following points should be considered when initiating VICTRELIS for treatment of chronic hepatitis C infection:
- VICTRELIS must not be used as monotherapy and should only be used in combination with peginterferon alfa and ribavirin.
- The efficacy of VICTRELIS has not been studied in patients who have previously failed therapy with a treatment regimen that includes VICTRELIS or other HCV NS3/4A protease inhibitors.
- Poorly interferon responsive patients who were treated with VICTRELIS in combination with peginterferon alfa and ribavirin have a lower likelihood of achieving a sustained virologic response (SVR), and a higher rate of detection of resistance-associated substitutions upon treatment failure, compared to patients with a greater response to peginterferon alfa and ribavirin [see Microbiology (12.4) and Clinical Studies (14)].
2 DOSAGE AND ADMINISTRATION
VICTRELIS must be administered in combination with peginterferon alfa and ribavirin. The dose of VICTRELIS is 800 mg (four 200-mg capsules) three times daily (every 7 to 9 hours) with food [a meal or light snack] (see Table 1). Refer to the prescribing information for peginterferon alfa and ribavirin for instructions on dosing.
The following dosing recommendations differ for some subgroups from the dosing studied in the Phase 3 trials [see Clinical Studies (14)]. Response-Guided Therapy (RGT) is recommended for most individuals, but longer dosing is recommended in targeted subgroups (e.g., patients with cirrhosis).
2.1 VICTRELIS/Peginterferon alfa/Ribavirin Combination Therapy: Patients Without Cirrhosis Who Are Previously Untreated or Who Previously Failed Interferon and Ribavirin Therapy
- Initiate therapy with peginterferon alfa and ribavirin for 4 weeks (Treatment Weeks 1–4).
- Add VICTRELIS 800 mg (four 200-mg capsules) orally three times daily (every 7 to 9 hours) to peginterferon alfa and ribavirin regimen after 4 weeks of treatment. Based on the patient's HCV-RNA levels at Treatment Week (TW) 8, TW12 and TW24, use the following guidelines to determine duration of treatment (see Table 1).
| ASSESSMENT*
(HCV-RNA Results†) | RECOMMENDATION | ||
|---|---|---|---|
| At Treatment Week 8 | At Treatment Week 24 | ||
|
|||
| Previously Untreated Patients | Not Detected | Not Detected | Complete three-medicine regimen at TW28. |
| Detected | Not Detected |
|
|
| Previous Partial Responders or Relapsers‡ | Not Detected | Not Detected | Complete three-medicine regimen at TW36. |
| Detected | Not Detected |
|
|
| Previous Null Responders‡ | Detected or Not Detected | Not Detected | Continue all three medicines and finish through TW48. |
Consideration should be given to treating previously untreated patients who are poorly interferon responsive (as determined at TW4) with 4 weeks peginterferon alfa and ribavirin followed by 44 weeks of VICTRELIS 800 mg orally three times daily (every 7 to 9 hours) in combination with peginterferon alfa and ribavirin in order to maximize rates of SVR.
2.2 VICTRELIS/Peginterferon alfa/Ribavirin Combination Therapy: Patients with Cirrhosis
Prior to initiating therapy in patients with compensated cirrhosis, see Use in Specific Populations (8.7) for additional information.
Patients with compensated cirrhosis should receive 4 weeks peginterferon alfa and ribavirin followed by 44 weeks VICTRELIS 800 mg (four 200-mg capsules) three times daily (every 7 to 9 hours) in combination with peginterferon alfa and ribavirin.
2.3 Dose Modification
Dose reduction of VICTRELIS is not recommended.
If a patient has a serious adverse reaction potentially related to peginterferon alfa and/or ribavirin, the peginterferon alfa and/or ribavirin dose should be reduced or discontinued. Refer to the prescribing information for peginterferon alfa and ribavirin for additional information about how to reduce and/or discontinue the peginterferon alfa and/or ribavirin dose. VICTRELIS must not be administered in the absence of peginterferon alfa and ribavirin. If peginterferon alfa or ribavirin is permanently discontinued, VICTRELIS must also be discontinued.
2.4 Discontinuation of Dosing Based on Treatment Futility
Discontinuation of therapy is recommended in all patients with 1) HCV-RNA levels of greater than or equal to 1000 IU per mL at TW8; or 2) HCV-RNA levels of greater than or equal to 100 IU per mL at TW12; or 3) confirmed detectable HCV-RNA levels at TW24.
3 DOSAGE FORMS AND STRENGTHS
VICTRELIS 200 mg Capsules, red-colored cap with the Merck logo printed in yellow ink, and a yellow-colored body with "314" printed in red ink.
4 CONTRAINDICATIONS
Contraindications to peginterferon alfa and ribavirin also apply to VICTRELIS combination treatment. Refer to the respective prescribing information for a list of the contraindications for peginterferon alfa and ribavirin.
VICTRELIS in combination with peginterferon alfa and ribavirin is contraindicated in:
- Pregnant women and men whose female partners are pregnant because of the risks for birth defects and fetal death associated with ribavirin [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1)].
- Patients with a history of a hypersensitivity reaction to boceprevir [see Warnings and Precautions (5.5)].
Coadministration with drugs that are highly dependent on CYP3A4/5 for clearance, and for which elevated plasma concentrations are associated with serious and/or life-threatening events, including those in Table 2, is contraindicated [see Drug Interactions (7)].
Coadministration with potent CYP3A4/5 inducers, where significantly reduced boceprevir plasma concentrations may be associated with reduced efficacy, including those in Table 2, is contraindicated [see Drug Interactions (7)].
| Drug Class | Drugs Within Class that are Contraindicated With VICTRELIS | Clinical Comments |
|---|---|---|
|
||
| Alpha 1-Adrenoreceptor antagonists | Alfuzosin, doxazosin, silodosin, tamsulosin | Potential for alpha 1-adrenoreceptor antagonist-associated adverse events, such as hypotension and priapism |
| Anticonvulsants | Carbamazepine, phenobarbital, phenytoin | May lead to loss of virologic response to VICTRELIS |
| Antimycobacterial Agents | Rifampin | May lead to loss of virologic response to VICTRELIS. |
| Antipsychotics | Lurasidone | Potential for serious and/or life-threatening reactions. |
| Pimozide | Potential for cardiac arrhythmias. | |
| Ergot Derivatives | Dihydroergotamine, ergonovine, ergotamine, methylergonovine | Potential for acute ergot toxicity characterized by peripheral vasospasm and ischemia of the extremities and other tissues. |
| GI Motility Agent | Cisapride | Potential for cardiac arrhythmias. |
| Herbal Products | St. John's wort (Hypericum perforatum) | May lead to loss of virologic response to VICTRELIS. |
| HMG-CoA Reductase Inhibitors | Lovastatin, simvastatin | Potential for myopathy, including rhabdomyolysis. |
| Oral Contraceptives | Drospirenone | Potential for hyperkalemia. |
| PDE5 enzyme Inhibitor | REVATIO® (sildenafil) or ADCIRCA® (tadalafil) when used for the treatment of pulmonary arterial hypertension* | Potential for PDE5 inhibitor-associated adverse events, including visual abnormalities, hypotension, prolonged erection, and syncope. |
| Sedative/Hypnotics | Triazolam; orally administered midazolam† | Prolonged or increased sedation or respiratory depression. |
5 WARNINGS AND PRECAUTIONS
5.1 Embryofetal Toxicity (Use with Ribavirin and Peginterferon Alfa)
Ribavirin may cause birth defects and/or death of the exposed fetus. Extreme care must be taken to avoid pregnancy in female patients and in female partners of male patients. Ribavirin therapy should not be started unless a report of a negative pregnancy test has been obtained immediately prior to initiation of therapy. Refer to the prescribing information for ribavirin for additional information.
Women of childbearing potential and men must use at least two forms of effective contraception during treatment and for at least 6 months after treatment has concluded. One of these forms of contraception can be a combined oral contraceptive product containing at least 1 mg of norethindrone. Oral contraceptives containing lower doses of norethindrone and other forms of hormonal contraception have not been studied or are contraindicated. Routine monthly pregnancy tests must be performed during this time [see Contraindications (4) and Drug Interactions (7)].
5.2 Anemia (Use with Ribavirin and Peginterferon Alfa)
Anemia has been reported with peginterferon alfa and ribavirin therapy. The addition of VICTRELIS to peginterferon alfa and ribavirin is associated with an additional decrease in hemoglobin concentrations. Complete blood counts (with white blood cell differential counts) should be obtained pretreatment, and at Treatment Weeks 2, 4, 8, and 12, and should be monitored closely at other time points, as clinically appropriate. If hemoglobin is less than 10 g per dL, a decrease in dosage of ribavirin is recommended; and if hemoglobin is less than 8.5 g per dL, discontinuation of ribavirin is recommended [see Adverse Reactions (6.1) and Clinical Studies (14)]. If ribavirin is permanently discontinued for management of anemia, then peginterferon alfa and VICTRELIS must also be discontinued [see Dosage and Administration (2.3)].
Refer to the prescribing information for ribavirin for additional information regarding dose reduction and/or discontinuation.
In clinical trials with VICTRELIS, the proportion of subjects who experienced hemoglobin values less than 10 g per dL and less than 8.5 g per dL was higher in subjects treated with the combination of VICTRELIS with PegIntron®/REBETOL® than in those treated with PegIntron/REBETOL alone (see Table 4). With the interventions used for anemia management in the clinical trials, the average additional decrease of hemoglobin was approximately 1 g per dL.
In clinical trials, the median time to onset of hemoglobin less than 10 g per dL from the initiation of therapy was similar among subjects treated with the combination of VICTRELIS and PegIntron/REBETOL (71 days with a range of 15-337 days), compared to those who received PegIntron/REBETOL (71 days with a range of 8-337 days). Certain adverse reactions consistent with symptoms of anemia, such as dyspnea, exertional dyspnea, dizziness and syncope were reported more frequently in subjects who received the combination of VICTRELIS with PegIntron/REBETOL than in those treated with PegIntron/REBETOL alone [see Adverse Reactions (6.1)].
In clinical trials with VICTRELIS, dose modifications (generally of PegIntron/REBETOL) due to anemia occurred twice as often in subjects treated with the combination of VICTRELIS with PegIntron/REBETOL (26%) compared to PegIntron/REBETOL (13%). The proportion of subjects who discontinued study drug due to anemia was 1% in subjects treated with the combination of VICTRELIS with PegIntron/REBETOL and 1% in subjects who received PegIntron/REBETOL. The use of erythropoiesis stimulating agents (ESAs) was permitted for management of anemia, at the investigator's discretion, with or without ribavirin dose reduction in the Phase 2 and 3 clinical trials. The proportion of subjects who received an ESA was 43% in those treated with the combination of VICTRELIS with PegIntron/REBETOL compared to 24% in those treated with PegIntron/REBETOL alone. The proportion of subjects who received a transfusion for the management of anemia was 3% of subjects treated with the combination of VICTRELIS with PegIntron/REBETOL compared to less than 1% in subjects who received PegIntron/REBETOL alone.
Thromboembolic events have been associated with ESA use in other disease states; and have also been reported with peginterferon alfa use in hepatitis C patients. Thromboembolic events were reported in clinical trials with VICTRELIS among subjects receiving the combination of VICTRELIS with PegIntron/REBETOL, and among those receiving PegIntron/REBETOL alone, regardless of ESA use. No definite causality assessment or benefit risk assessment could be made for these events due to the presence of confounding factors and lack of randomization of ESA use.
A randomized, parallel-arm, open-label clinical trial was conducted in previously untreated CHC subjects with genotype 1 infection to compare use of an ESA versus ribavirin dose reduction for initial management of anemia during therapy with VICTRELIS in combination with peginterferon alfa-2b and ribavirin. Similar SVR rates were reported in subjects who were randomized to receive ribavirin dose reduction compared to subjects who were randomized to receive an ESA. In this trial, use of ESAs was associated with an increased risk of thromboembolic events including pulmonary embolism, acute myocardial infarction, cerebrovascular accident, and deep vein thrombosis compared to ribavirin dose reduction alone. The treatment discontinuation rate due to anemia was similar in subjects randomized to receive ribavirin dose reduction compared to subjects randomized to receive ESA (2% in each group). The transfusion rate was 4% in subjects randomized to receive ribavirin dose reduction and 2% in subjects randomized to receive ESA.
Ribavirin dose reduction is recommended for the initial management of anemia.
5.3 Neutropenia (Use with Ribavirin and Peginterferon Alfa)
In Phase 2 and 3 clinical trials, seven percent of subjects receiving the combination of VICTRELIS with PegIntron/REBETOL had neutrophil counts of less than 0.5 × 109 per L compared to 4% of subjects receiving PegIntron/REBETOL alone (see Table 4). Three subjects experienced severe or life-threatening infections associated with neutropenia, and two subjects experienced life-threatening neutropenia while receiving the combination of VICTRELIS with PegIntron/REBETOL. Complete blood counts (with white blood cell differential counts) should be obtained at pretreatment, and at Treatment Weeks 2, 4, 8, and 12, and should be monitored closely at other time points, as clinically appropriate. Decreases in neutrophil counts may require dose reduction or discontinuation of peginterferon alfa and ribavirin. If peginterferon alfa and ribavirin are permanently discontinued, then VICTRELIS must also be discontinued [see Dosage and Administration (2.3)].
Refer to the prescribing information for peginterferon alfa and ribavirin for additional information regarding dose reduction or discontinuation.
5.4 Pancytopenia (Use with Ribavirin and Peginterferon Alfa)
Serious cases of pancytopenia have been reported postmarketing in patients receiving VICTRELIS in combination with peginterferon alfa and ribavirin. Complete blood counts (with white blood cell differential counts) should be obtained at pretreatment, and at Treatment Weeks 2, 4, 8, and 12, and should be monitored closely at other time points, as clinically appropriate.
Refer to the prescribing information for ribavirin and peginterferon alfa for guidelines for discontinuation of therapy based on laboratory parameters.
5.5 Hypersensitivity
Serious acute hypersensitivity reactions (e.g., urticaria, angioedema) have been observed during combination therapy with VICTRELIS, peginterferon alfa and ribavirin. If such an acute reaction occurs, combination therapy should be discontinued and appropriate medical therapy immediately instituted [see Contraindications (4) and Adverse Reactions (6.2)].
5.6 Drug Interactions
See Table 2 for a listing of drugs that are contraindicated for use with VICTRELIS due to potentially life-threatening adverse events, significant drug interactions or loss of virologic activity [see Contraindications (4)]. Please refer to Table 5 for established and other potentially significant drug interactions [see Drug Interactions (7.3)].
5.7 Laboratory Tests
HCV-RNA levels should be monitored at Treatment Weeks 4, 8, 12, and 24, at the end of treatment, during treatment follow-up, and for other time points as clinically indicated. Use of a sensitive real-time reverse-transcription polymerase chain reaction (RT-PCR) assay for monitoring HCV-RNA levels during treatment is recommended. The assay should have a lower limit of HCV-RNA quantification of equal to or less than 25 IU per mL, and a limit of HCV-RNA detection of approximately 10 to 15 IU per mL. For the purposes of assessing Response-Guided Therapy milestones, a confirmed "detectable but below limit of quantification" HCV-RNA result should not be considered equivalent to an "undetectable" HCV-RNA result (reported as "Target Not Detected" or "HCV-RNA Not Detected").
Complete blood count (with white blood cell differential counts) should be obtained at pretreatment, and at Treatment Weeks 2, 4, 8, and 12, and should be monitored closely at other time points, as clinically appropriate.
Refer to the prescribing information for peginterferon alfa and ribavirin for pre-treatment, on-treatment and post-treatment laboratory testing recommendations including hematology, biochemistry (including hepatic function tests), and pregnancy testing requirements.
6 ADVERSE REACTIONS
See the peginterferon alfa and ribavirin prescribing information for description of adverse reactions associated with their use.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of VICTRELIS cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The following serious and otherwise important adverse drug reactions (ADRs) are discussed in detail in another section of the labeling:
- Anemia [see Warnings and Precautions (5.2)]
- Neutropenia [see Warnings and Precautions (5.3)]
- Pancytopenia [see Warnings and Precautions (5.4)]
- Hypersensitivity [see Contraindications (4) and Warnings and Precautions (5.5)]
The most commonly reported adverse reactions (more than 35% of subjects regardless of investigator's causality assessment) in adult subjects were fatigue, anemia, nausea, headache, and dysgeusia when VICTRELIS was used in combination with PegIntron and REBETOL.
The safety of the combination of VICTRELIS 800 mg three times daily with PegIntron/REBETOL was assessed in 2095 subjects with chronic hepatitis C in one Phase 2, open-label trial and two Phase 3, randomized, double-blind, placebo-controlled clinical trials. SPRINT-1 (subjects who were previously untreated) evaluated the use of VICTRELIS in combination with PegIntron/REBETOL with or without a four-week lead-in period with PegIntron/REBETOL compared to PegIntron/REBETOL alone. SPRINT-2 (subjects who were previously untreated) and RESPOND-2 (subjects who had failed previous therapy) evaluated the use of VICTRELIS 800 mg three times daily in combination with PegIntron/REBETOL with a four-week lead-in period with PegIntron/REBETOL compared to PegIntron/REBETOL alone [see Clinical Studies (14)]. The population studied had a mean age of 49 years (3% of subjects were older than 65 years of age), 39% were female, 82% were white and 15% were black.
During the four week lead-in period with PegIntron/REBETOL in subjects treated with the combination of VICTRELIS with PegIntron/REBETOL, 28/1263 (2%) subjects experienced adverse reactions leading to discontinuation of treatment. During the entire course of treatment, the proportion of subjects who discontinued treatment due to adverse reactions was 13% for subjects receiving the combination of VICTRELIS with PegIntron/REBETOL and 12% for subjects receiving PegIntron/REBETOL alone. Events resulting in discontinuation were similar to those seen in previous studies with PegIntron/REBETOL. Only anemia and fatigue were reported as events that led to discontinuation in more than 1% of subjects in any arm.
Adverse reactions that led to dose modifications of any drug (primarily PegIntron and REBETOL) occurred in 39% of subjects receiving the combination of VICTRELIS with PegIntron/REBETOL compared to 24% of subjects receiving PegIntron/REBETOL alone. The most common reason for dose reduction was anemia, which occurred more frequently in subjects receiving the combination of VICTRELIS with PegIntron/REBETOL than in subjects receiving PegIntron/REBETOL alone.
Serious adverse events were reported in 11% of subjects receiving the combination of VICTRELIS with PegIntron/REBETOL and in 8% of subjects receiving PegIntron/REBETOL.
Adverse events (regardless of investigator's causality assessment) reported in greater than or equal to 10% of subjects receiving the combination of VICTRELIS with PegIntron/REBETOL and reported at a rate of greater than or equal to 5% than PegIntron/REBETOL alone in SPRINT-1, SPRINT-2, and RESPOND-2 are presented in Table 3.
| Adverse Events | Previously Untreated (SPRINT-1 and SPRINT-2) | Previous Treatment Failures (RESPOND-2) |
||
|---|---|---|---|---|
| Percentage of Subjects Reporting Adverse Events | Percentage of Subjects Reporting Adverse Events | |||
| Body System Organ Class | VICTRELIS + PegIntron + REBETOL (n=1225) | PegIntron + REBETOL (n=467) | VICTRELIS + PegIntron + REBETOL (n=323) | PegIntron + REBETOL (n=80) |
| Median Exposure (days) | 197 | 216 | 253 | 104 |
| Blood and Lymphatic System Disorders | ||||
| Anemia | 50 | 30 | 45 | 20 |
| Neutropenia | 25 | 19 | 14 | 10 |
| Gastrointestinal Disorders | ||||
| Nausea | 46 | 42 | 43 | 38 |
| Dysgeusia | 35 | 16 | 44 | 11 |
| Diarrhea | 25 | 22 | 24 | 16 |
| Vomiting | 20 | 13 | 15 | 8 |
| Dry Mouth | 11 | 10 | 15 | 9 |
| General Disorders and Administration Site Conditions | ||||
| Fatigue | 58 | 59 | 55 | 50 |
| Chills | 34 | 29 | 33 | 30 |
| Asthenia | 15 | 18 | 21 | 16 |
| Metabolism and Nutrition Disorders | ||||
| Decreased Appetite | 25 | 24 | 26 | 16 |
| Musculoskeletal and Connective Tissue Disorders | ||||
| Arthralgia | 19 | 19 | 23 | 16 |
| Nervous System Disorders | ||||
| Dizziness | 19 | 16 | 16 | 10 |
| Psychiatric Disorders | ||||
| Insomnia | 34 | 34 | 30 | 24 |
| Irritability | 22 | 23 | 21 | 13 |
| Respiratory, Thoracic, and Mediastinal Disorders | ||||
| Dyspnea Exertional | 8 | 8 | 11 | 5 |
| Skin and Subcutaneous Tissue Disorders | ||||
| Alopecia | 27 | 27 | 22 | 16 |
| Dry Skin | 18 | 18 | 22 | 9 |
| Rash | 17 | 19 | 16 | 6 |
Other Important Adverse Reactions Reported in Clinical Trials
Among subjects (previously untreated subjects or those who failed previous therapy) who received VICTRELIS in combination with peginterferon alfa and ribavirin, the following adverse drug reactions were reported. These events are notable because of their seriousness, severity, or increased frequency in subjects who received VICTRELIS in combination with peginterferon alfa and ribavirin compared with subjects who received only peginterferon alfa and ribavirin.
Gastrointestinal Disorders
Dysgeusia (alteration of taste) was an adverse event reported at an increased frequency in subjects receiving VICTRELIS in combination with peginterferon alfa and ribavirin compared with subjects receiving peginterferon alfa and ribavirin alone (Table 3). Adverse events such as dry mouth, nausea, vomiting and diarrhea were also reported at an increased frequency in subjects receiving VICTRELIS in combination with peginterferon alfa and ribavirin.
Laboratory Values
Changes in selected hematological parameters during treatment of adult subjects with the combination of VICTRELIS with PegIntron and REBETOL are described in Table 4.
Hemoglobin
Decreases in hemoglobin may require a decrease in dosage or discontinuation of ribavirin [see Warnings and Precautions (5.2) and Clinical Studies (14)] [see prescribing information for ribavirin]. If ribavirin is permanently discontinued, then peginterferon alfa and VICTRELIS must also be discontinued [see Dosage and Administration (2.3)].
Neutrophils and Platelets
The proportion of subjects with decreased neutrophil and platelet counts was higher in subjects treated with VICTRELIS in combination with PegIntron/REBETOL compared to subjects receiving PegIntron/REBETOL alone. Three percent of subjects receiving the combination of VICTRELIS with PegIntron/REBETOL had platelet counts of less than 50 × 109 per L compared to 1% of subjects receiving PegIntron/REBETOL alone. Decreases in neutrophils or platelets may require a decrease in dosage or interruption of peginterferon alfa, or discontinuation of therapy [see prescribing information for peginterferon alfa and ribavirin]. If peginterferon alfa is permanently discontinued, then ribavirin and VICTRELIS must also be discontinued [see Dosage and Administration (2.3)].
| Previously Untreated (SPRINT-1 and SPRINT-2) | Previous Treatment Failures (RESPOND-2) |
|||
|---|---|---|---|---|
| Percentage of Subjects Reporting Selected Hematological Parameters | Percentage of Subjects Reporting Selected Hematological Parameters | |||
| Hematological Parameters | VICTRELIS + PegIntron + REBETOL (n=1225) | PegIntron + REBETOL (n=467) | VICTRELIS + PegIntron + REBETOL (n=323) | PegIntron + REBETOL (n=80) |
| Hemoglobin (g/dL) | ||||
| <10 | 49 | 29 | 49 | 25 |
| <8.5 | 6 | 3 | 10 | 1 |
| Neutrophils (× 109/L) | ||||
| <0.75 | 31 | 18 | 26 | 13 |
| <0.5 | 8 | 4 | 7 | 4 |
| Platelets (× 109/L) | ||||
| <50 | 3 | 1 | 4 | 0 |
| <25 | <1 | 0 | 0 | 0 |
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of VICTRELIS in combination with peginterferon alfa and ribavirin. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders: agranulocytosis, pancytopenia, thrombocytopenia [see Warnings and Precautions (5.4)]
Gastrointestinal Disorders: mouth ulceration, stomatitis
Infections and Infestations: pneumonia, sepsis
Skin and Subcutaneous Tissue Disorders: angioedema, urticaria [see Warnings and Precautions (5.5)]; drug rash with eosinophilia and systemic symptoms (DRESS) syndrome, exfoliative rash, exfoliative dermatitis, Stevens-Johnson syndrome, toxic skin eruption, toxicoderma
7 DRUG INTERACTIONS
[See Contraindications (4), Warnings and Precautions (5.6), and Clinical Pharmacology (12.3).]
7.1 Potential for VICTRELIS to Affect Other Drugs
Boceprevir is a strong inhibitor of CYP3A4/5. Drugs metabolized primarily by CYP3A4/5 may have increased exposure when administered with VICTRELIS, which could increase or prolong their therapeutic and adverse effects. Boceprevir does not inhibit CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 or CYP2E1 in vitro. In addition, boceprevir does not induce CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 or CYP3A4/5 in vitro.
Boceprevir is a potential inhibitor of p-glycoprotein (P-gp) based on in vitro studies. In a drug interaction trial conducted with digoxin, VICTRELIS had limited p-glycoprotein inhibitory potential at clinically relevant concentrations.
7.2 Potential for Other Drugs to Affect VICTRELIS
Boceprevir is primarily metabolized by aldo-ketoreductase (AKR). In drug interaction trials conducted with AKR inhibitors diflunisal and ibuprofen, boceprevir exposure did not increase to a clinically significant extent. VICTRELIS may be coadministered with AKR inhibitors.
Boceprevir is partly metabolized by CYP3A4/5. It is also a substrate for p-glycoprotein. Coadministration of VICTRELIS with drugs that induce or inhibit CYP3A4/5 could decrease or increase exposure to boceprevir.
7.3 Established and Other Potential Significant Drug Interactions
Table 5 provides recommendations based on established or potentially clinically significant drug interactions. VICTRELIS is contraindicated with drugs that are potent inducers of CYP3A4/5 and drugs that are highly dependent on CYP3A4/5 for clearance, and for which elevated plasma concentrations are associated with serious and/or life-threatening events [see Contraindications (4)].
| Concomitant Drug Class: Drug Name | Effect on Concentration of Boceprevir or Concomitant Drug | Recommendations |
|---|---|---|
|
||
| Antiarrhythmics: amiodarone, bepridil, propafenone, quinidine | ↑ antiarrhythmics | Coadministration with VICTRELIS has the potential to produce serious and/or life-threatening adverse events and has not been studied. Caution is warranted and therapeutic concentration monitoring of these drugs is recommended if they are used concomitantly with VICTRELIS. |
| digoxin* | ↑ digoxin | Digoxin concentrations increased when administered with VICTRELIS [see Clinical Pharmacology (12.3)]. Measure serum digoxin concentrations before initiating VICTRELIS. Continue monitoring digoxin concentrations; consult the digoxin prescribing information for information on titrating the digoxin dose. |
| Anticoagulant: warfarin | ↑ or ↓ warfarin | Concentrations of warfarin may be altered when co-administered with VICTRELIS. Monitor INR closely. |
| Antidepressants: trazodone, desipramine | ↑ trazodone ↑ desipramine | Plasma concentrations of trazodone and desipramine may increase when administered with VICTRELIS, resulting in adverse events such as dizziness, hypotension and syncope. Use with caution and consider a lower dose of trazodone or desipramine. |
| escitalopram* | ↓escitalopram | Exposure of escitalopram was slightly decreased when coadministered with VICTRELIS. Selective serotonin reuptake inhibitors such as escitalopram have a wide therapeutic index, but doses may need to be adjusted when combined with VICTRELIS. |
| Antifungals: ketoconazole*, itraconazole, posaconazole, voriconazole | ↑ boceprevir ↑ itraconazole ↑ ketoconazole ↑ posaconazole ↑ voriconazole | Plasma concentrations of ketoconazole, itraconazole, voriconazole or posaconazole may be increased with VICTRELIS. When coadministration is required, doses of ketoconazole and itraconazole should not exceed 200 mg/day. |
| Anti-gout: colchicine | ↑ colchicine | Significant increases in colchicine levels are expected; fatal colchicine toxicity has been reported with other strong CYP3A4 inhibitors. Patients with renal or hepatic impairment should not be given colchicine with VICTRELIS. Treatment of gout flares (during treatment with VICTRELIS): 0.6 mg (1 tablet) × 1 dose, followed by 0.3 mg (half tablet) 1 hour later. Dose to be repeated no earlier than 3 days. Prophylaxis of gout flares (during treatment with VICTRELIS): If the original regimen was 0.6 mg twice a day, reduce dose to 0.3 mg once a day. If the original regimen was 0.6 mg once a day, reduce the dose to 0.3 mg once every other day. Treatment of familial Mediterranean fever (FMF) (during treatment with VICTRELIS): Maximum daily dose of 0.6 mg (may be given as 0.3 mg twice a day). |
| Anti-infective: clarithromycin | ↑ clarithromycin | Concentrations of clarithromycin may be increased with VICTRELIS; however, no dosage adjustment is necessary for patients with normal renal function. |
| Antimycobacterial: rifabutin | ↓ boceprevir ↑ rifabutin | Increases in rifabutin exposure are anticipated, while exposure of boceprevir may be decreased. Doses have not been established for the 2 drugs when used in combination. Concomitant use is not recommended. |
| Calcium Channel Blockers such as: amlodipine, diltiazem, felodipine, nifedipine, nicardipine, nisoldipine, verapamil | ↑ calcium channel blockers | Plasma concentrations of calcium channel blockers may increase when administered with VICTRELIS. Caution is warranted and clinical monitoring is recommended. |
| Corticosteroid, systemic: dexamethasone | ↓ boceprevir | Coadministration of VICTRELIS with CYP3A4/5 inducers may decrease plasma concentrations of boceprevir, which may result in loss of therapeutic effect. Therefore, this combination should be avoided if possible and used with caution if necessary. |
| prednisone* | ↑ prednisone | Concentrations of prednisone and its active metabolite, prednisolone, increased when administered with VICTRELIS [see Clinical Pharmacology (12.3)]. No dose adjustment of prednisone is necessary when co-administered with VICTRELIS. Patients receiving prednisone and VICTRELIS should be monitored appropriately. |
| Corticosteroid, inhaled: budesonide, fluticasone | ↑ budesonide ↑ fluticasone | Concomitant use of inhaled budesonide or fluticasone with VICTRELIS may result in increased plasma concentrations of budesonide or fluticasone, resulting in significantly reduced serum cortisol concentrations. Avoid coadministration if possible, particularly for extended durations. |
| Endothelin Receptor Antagonist: bosentan | ↑ bosentan | Concentrations of bosentan may be increased when coadministered with VICTRELIS. Use with caution and monitor closely. |
| HIV Integrase Inhibitor: raltegravir* | ↔ raltegravir | No dose adjustment required for VICTRELIS or raltegravir. |
| HIV Non-Nucleoside Reverse Transcriptase Inhibitors: efavirenz* | ↓ boceprevir | Plasma trough concentrations of boceprevir were decreased when VICTRELIS was coadministered with efavirenz, which may result in loss of therapeutic effect. Avoid combination. |
| etravirine* | ↓ etravirine | Concentrations of etravirine decreased when coadministered with VICTRELIS. The clinical significance of the reductions in etravirine pharmacokinetic parameters has not been directly assessed. |
| rilpivirine* | ↑ rilpivirine | Concomitant administration of rilpivirine with VICTRELIS increased the exposure to rilpivirine. No dose adjustment of VICTRELIS or rilpivirine is recommended. |
| HIV Protease Inhibitors: atazanavir/ritonavir* | ↓ atazanavir ↓ ritonavir | Concomitant administration of boceprevir and atazanavir/ritonavir resulted in reduced steady-state exposures to atazanavir and ritonavir. Coadministration of atazanavir/ritonavir and boceprevir is not recommended. |
| darunavir/ritonavir* | ↓ darunavir ↓ ritonavir ↓ boceprevir | Concomitant administration of boceprevir and darunavir/ritonavir resulted in reduced steady-state exposures to boceprevir, darunavir and ritonavir. Coadministration of darunavir/ritonavir and boceprevir is not recommended. |
| lopinavir/ritonavir* | ↓ lopinavir ↓ ritonavir ↓ boceprevir | Concomitant administration of boceprevir and lopinavir/ritonavir resulted in reduced steady-state exposures to boceprevir, lopinavir and ritonavir. Coadministration of lopinavir/ritonavir and boceprevir is not recommended. |
| ritonavir* | ↓ boceprevir | When boceprevir is administered with ritonavir alone, boceprevir concentrations are decreased. |
| HMG-CoA Reductase Inhibitors: | For contraindicated HMG-CoA reductase inhibitors, [see Contraindications (4)]. | |
| atorvastatin* | ↑ atorvastatin | Exposure to atorvastatin was increased when administered with VICTRELIS. Use the lowest effective dose of atorvastatin, but do not exceed a daily dose of 40 mg when coadministered with VICTRELIS. |
| pravastatin* | ↑ pravastatin | Concomitant administration of pravastatin with VICTRELIS increased exposure to pravastatin. Treatment with pravastatin can be initiated at the recommended dose when coadministered with VICTRELIS. Close clinical monitoring is warranted. |
| Immunosuppressants: cyclosporine* | ↑cyclosporine | Dose adjustments of cyclosporine should be anticipated when administered with VICTRELIS and should be guided by close monitoring of cyclosporine blood concentrations, and frequent assessments of renal function and cyclosporine-related side effects. |
| tacrolimus* | ↑tacrolimus | Concomitant administration of VICTRELIS with tacrolimus requires significant dose reduction and prolongation of the dosing interval for tacrolimus, with close monitoring of tacrolimus blood concentrations and frequent assessments of renal function and tacrolimus-related side effects. |
| sirolimus* | ↑sirolimus | Concomitant administration of VICTRELIS with sirolimus requires significant dose reduction and prolongation of the dosing interval for sirolimus, with close monitoring of sirolimus blood concentrations and frequent assessments of renal function and sirolimus-related side effects. |
| Inhaled beta-agonist: salmeterol | ↑ salmeterol | Concurrent use of inhaled salmeterol and VICTRELIS is not recommended due to the risk of cardiovascular events associated with salmeterol. |
| Narcotic Analgesic/Opioid Dependence: methadone* | ↓ R-methadone | Plasma concentrations of R-methadone decreased when coadministered with VICTRELIS [see Clinical Pharmacology (12.3)]. The observed changes are not considered clinically relevant. No dose adjustment of methadone or VICTRELIS is recommended. Individual patients may require additional titration of their methadone dosage when VICTRELIS is started or stopped to ensure clinical effect of methadone. |
| buprenorphine/naloxone* | ↑ buprenorphine/naloxone | Plasma concentrations of buprenorphine and naloxone increased when coadministered with VICTRELIS [see Clinical Pharmacology (12.3)]. The observed changes are not considered clinically relevant. No dose adjustment of buprenorphine/naloxone or VICTRELIS is recommended. |
| Oral hormonal contraceptives: | For contraindicated oral contraceptives, [see Contraindications (4)].
|
|
| drospirenone/ethinyl estradiol* | ↑ drospirenone ↓ ethinyl estradiol | Concentrations of drospirenone increased in the presence of boceprevir. Thus, the use of drospirenone-containing products is contraindicated during treatment with VICTRELIS due to potential for hyperkalemia [see Contraindications (4)]. |
| norethindrone/ethinyl estradiol* | ↓ ethinyl estradiol ↔ norethindrone | Concentrations of ethinyl estradiol decreased in the presence of boceprevir. Norethindrone Cmax decreased 17% in the presence of boceprevir [see Clinical Pharmacology (12.3)]. Coadministration of VICTRELIS with a combined oral contraceptive containing ethinyl estradiol and at least 1 mg of norethindrone is not likely to alter the effectiveness of this combined oral contraceptive [see Use in Specific Populations (8.1)].
Patients using estrogens as hormone replacement therapy should be clinically monitored for signs of estrogen deficiency. |
| PDE5 inhibitors: | For contraindicated PDE5 enzyme inhibitors, [see Contraindications (4)]. | |
| sildenafil, tadalafil, vardenafil | ↑ sildenafil ↑ tadalafil ↑ vardenafil | Increases in PDE5 inhibitor concentrations are expected, and may result in an increase in adverse events, including hypotension, syncope, visual disturbances, and priapism. Use of REVATIO® (sildenafil) or ADCIRCA® (tadalafil) for the treatment of pulmonary arterial hypertension (PAH) is contraindicated with VICTRELIS [see Contraindications (4)]. Use of PDE5 inhibitors for erectile dysfunction: Use with caution in combination with VICTRELIS with increased monitoring for PDE5 inhibitor-associated adverse events. Do not exceed the following doses: Sildenafil: 25 mg every 48 hours Tadalafil: 10 mg every 72 hours Vardenafil: 2.5 mg every 24 hours |
| Proton Pump Inhibitor: omeprazole* | ↔ omeprazole | No dose adjustment of omeprazole or VICTRELIS is recommended. |
| Sedative/hypnotics: | For contraindicated sedatives/hypnotics, [see Contraindications (4)]. | |
| alprazolam; IV midazolam | ↑ midazolam ↑ alprazolam | Close clinical monitoring for respiratory depression and/or prolonged sedation should be exercised during coadministration of VICTRELIS. A lower dose of IV midazolam or alprazolam should be considered. |
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
VICTRELIS must be administered in combination with peginterferon alfa and ribavirin [see Dosage and Administration (2)].
Pregnancy Category X: Use with Ribavirin and Peginterferon Alfa
Significant teratogenic and/or embryocidal effects have been demonstrated in all animal species exposed to ribavirin; and therefore ribavirin is contraindicated in women who are pregnant and in the male partners of women who are pregnant [see Contraindications (4) and Warnings and Precautions (5.1)] [see prescribing information for ribavirin]. Interferons have abortifacient effects in animals and should be assumed to have abortifacient potential in humans [see prescribing information for peginterferon alfa].
Extreme caution must be taken to avoid pregnancy in female patients and female partners of male patients while taking this combination. Women of childbearing potential and their male partners should not receive ribavirin unless they are using effective contraception (two reliable forms) during treatment with ribavirin and for 6 months after treatment. One of these reliable forms of contraception can be a combined oral contraceptive product containing at least 1 mg of norethindrone. Oral contraceptives containing lower doses of norethindrone and other forms of hormonal contraception have not been studied or are contraindicated [see Contraindications (4) and Warnings and Precautions (5.1)].
In case of exposure during pregnancy, a Ribavirin Pregnancy Registry has been established to monitor maternal-fetal outcomes of pregnancies in female patients and female partners of male patients exposed to ribavirin during treatment and for 6 months following cessation of treatment. Physicians and patients are encouraged to report such cases by calling 1-800-593-2214.
Pregnancy Category B: VICTRELIS
VICTRELIS must not be used as a monotherapy [see Indications and Usage (1)]. There are no adequate and well-controlled studies with VICTRELIS in pregnant women.
No effects on fetal development have been observed in rats and rabbits at boceprevir AUC exposures approximately 11.8- and 2.0-fold higher, respectively, than those in humans at the recommended dose of 800 mg three times daily [see Nonclinical Toxicology (13.1)].
8.3 Nursing Mothers
It is not known whether VICTRELIS is excreted into human breast milk. Levels of boceprevir and/or metabolites in the milk of lactating rats were slightly higher than levels observed in maternal blood. Peak blood concentrations of boceprevir and/or metabolites in nursing pups were less than 1% of those of maternal blood concentrations. Because of the potential for adverse reactions from the drug in nursing infants, a decision must be made whether to discontinue nursing or discontinue treatment with VICTRELIS, taking into account the importance of the therapy to the mother.
8.4 Pediatric Use
The safety, efficacy, and pharmacokinetic profile of VICTRELIS in pediatric patients have not been studied.
8.5 Geriatric Use
Clinical studies of VICTRELIS did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, caution should be exercised in the administration and monitoring of VICTRELIS in geriatric patients due to the greater frequency of decreased hepatic function, concomitant diseases and other drug therapy [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
No dosage adjustment of VICTRELIS is required for patients with any degree of renal impairment [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment of VICTRELIS is required for patients with mild, moderate or severe hepatic impairment [see Clinical Pharmacology (12.3)]. Safety and efficacy of VICTRELIS have not been studied in patients with decompensated cirrhosis.
In published observational studies of patients with compensated cirrhosis treated with first generation HCV protease inhibitors, including boceprevir, in combination with peginterferon alfa and ribavirin, platelet count < 100,000/mm3 and serum albumin < 3.5 g/dL were baseline characteristics that were identified as predictors of death or serious complications (severe infection or hepatic decompensation) during therapy.
The potential risks and benefits of VICTRELIS in combination with peginterferon alfa and ribavirin should be carefully considered before initiating therapy in patients with compensated cirrhosis who have platelet count < 100,000/mm3 and serum albumin < 3.5 g/dL at baseline. If therapy is initiated, close monitoring for signs of infections and worsening liver function is warranted.
[See the prescribing information for peginterferon alfa for use in patients with hepatic decompensation.]
8.8 Organ Transplantation
The safety and efficacy of VICTRELIS alone or in combination with peginterferon alfa and ribavirin for the treatment of chronic hepatitis C genotype 1 infection in liver or other organ transplant recipients have not been studied. For data regarding drug-drug interactions with immunosuppressants, see Drug Interactions (7.3) and Clinical Pharmacology (12.3).
10 OVERDOSAGE
Daily doses of 3600 mg have been taken by healthy volunteers for 5 days without untoward symptomatic effects.
There is no specific antidote for overdose with VICTRELIS. Treatment of overdosage with VICTRELIS should consist of general supportive measures, including monitoring of vital signs, and observation of the patient's clinical status.
11 DESCRIPTION
VICTRELIS (boceprevir) is an inhibitor of the hepatitis C virus (HCV) non-structural protein 3 (NS3) serine protease.
Boceprevir has the following chemical name: (1R,5S)-N-[3-Amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide. The molecular formula is C27H45N5O5 and its molecular weight is 519.7. Boceprevir has the following structural formula:
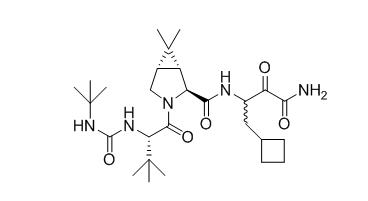
Boceprevir is manufactured as an approximately equal mixture of two diastereomers. Boceprevir is a white to off-white amorphous powder. It is freely soluble in methanol, ethanol and isopropanol and slightly soluble in water.
VICTRELIS 200 mg capsules are available as hard gelatin capsules for oral administration. Each capsule contains 200 mg of boceprevir and the following inactive ingredients: sodium lauryl sulfate, microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, pre-gelatinized starch, and magnesium stearate. The red capsule cap consists of gelatin, titanium dioxide, D&C Yellow #10, FD&C Blue #1, and FD&C Red #40. The yellow capsule body contains gelatin, titanium dioxide, D&C Yellow #10, FD&C Red #40, and FD&C Yellow #6. The capsule is printed with red and yellow ink. The red ink contains shellac and red iron oxide, while the yellow ink consists of shellac, titanium dioxide, povidone and D&C Yellow #10 Aluminum Lake.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
VICTRELIS is a direct acting antiviral drug against the hepatitis C virus [see Microbiology (12.4)].
12.2 Pharmacodynamics
Evaluation of Effect of VICTRELIS on QTc Interval
The effect of boceprevir 800 mg and 1200 mg on QTc interval was evaluated in a randomized, multiple-dose, placebo-, and active-controlled (moxifloxacin 400 mg) 4-way crossover thorough QT study in 36 healthy subjects. In the study with demonstrated ability to detect small effects, the upper bound of the one-sided 95% confidence interval for the largest placebo-adjusted, baseline-corrected QTc based on individual correction method (QTcI) was below 10 ms, the threshold for regulatory concern. The dose of 1200 mg yields a boceprevir maximum exposure increase of approximately 15% which may not cover exposures due to coadministration with strong CYP3A4 inhibitors or use in patients with severe hepatic impairment. However, at the doses studied in the thorough QT study, no apparent concentration-QT relationship was identified. Thus, there is no expectation of a QTc effect under a higher exposure scenario.
12.3 Pharmacokinetics
VICTRELIS capsules contain a 1:1 mixture of two diastereomers, SCH534128 and SCH534129. In plasma the diastereomer ratio changes to 2:1, favoring the active diastereomer, SCH534128. Plasma concentrations of boceprevir described below consist of both diastereomers SCH534128 and SCH534129, unless otherwise specified.
In healthy subjects who received 800 mg three times daily alone, boceprevir drug exposure was characterized by AUC(т) of 5408 ng × hr per mL (n=71), Cmax of 1723 ng per mL (n=71), and Cmin of 88 ng per mL (n=71). Pharmacokinetic results were similar between healthy subjects and HCV-infected subjects.
Absorption
Boceprevir was absorbed following oral administration with a median Tmax of 2 hours. Steady state AUC, Cmax, and Cmin increased in a less-than-dose-proportional manner and individual exposures overlapped substantially at 800 mg and 1200 mg, suggesting diminished absorption at higher doses. Accumulation is minimal (0.8- to 1.5-fold) and pharmacokinetic steady state is achieved after approximately 1 day of three times daily dosing.
The absolute bioavailability of boceprevir has not been studied.
Effects of Food on Oral Absorption
VICTRELIS should be administered with food. Food enhanced the exposure of boceprevir by up to 65% at the 800 mg three times daily dose, relative to the fasting state. The bioavailability of boceprevir was similar regardless of meal type (e.g., high-fat vs. low-fat) or whether taken 5 minutes prior to eating, during a meal, or immediately following completion of the meal. Therefore, VICTRELIS may be taken without regard to either meal type or timing of the meal.
Distribution
Boceprevir has a mean apparent volume of distribution (Vd/F) of approximately 772 L at steady state in healthy subjects. Human plasma protein binding is approximately 75% following a single dose of boceprevir 800 mg. Boceprevir is administered as an approximately equal mixture of two diastereomers, SCH534128 and SCH534129, which rapidly interconvert in plasma. The predominant diastereomer, SCH534128, is pharmacologically active and the other diastereomer is inactive.
Metabolism
Studies in vitro indicate that boceprevir primarily undergoes metabolism through the aldo-keto reductase (AKR)-mediated pathway to ketone-reduced metabolites that are inactive against HCV. After a single 800-mg oral dose of 14C-boceprevir, the most abundant circulating metabolites were a diastereomeric mixture of ketone-reduced metabolites with a mean exposure approximately 4-fold greater than that of boceprevir. Boceprevir also undergoes, to a lesser extent, oxidative metabolism mediated by CYP3A4/5.
Drug Interactions
Drug interaction studies were performed with boceprevir and drugs likely to be coadministered or drugs commonly used as probes for pharmacokinetic interactions. The effects of coadministration of boceprevir on AUC, Cmax and Cmin are summarized in Table 6 (effects of coadministered drugs on boceprevir) and Table 7 (effects of boceprevir on coadministered drugs).
| Co-administered Drug | Co-administered Drug Dose/Schedule | Boceprevir Dose/Schedule | Ratio Estimate of Boceprevir Pharmacokinetic Parameters (in Combination vs. Alone) (90% CI of the Ratio Estimate) * |
||
|---|---|---|---|---|---|
| Change in mean Cmax | Change in mean AUC | Change in mean Cmin | |||
| N/A = not available | |||||
| Atazanavir/Ritonavir | 300 mg/100 mg daily × 22 days | 800 mg three times daily × 6 days | 0.93 (0.80-1.08) | 0.95 (0.87-1.05) | 0.82 (0.68-0.98) |
| Atorvastatin | 40 mg single dose | 800 mg three times daily × 7 days | 1.04 (0.89-1.21) | 0.95 (0.90-1.01) | N/A |
| Buprenorphine/ Naloxone | Buprenorphine: 8-24 mg + Naloxone: 2-6 mg daily × 6 days | 800 mg three times daily × 6 days | 0.82 (0.71-0.94) | 0.88 (0.76-1.02) | 0.95 (0.70-1.28) |
| Cyclosporine | 100 mg single dose | 800 mg single dose | 1.08 (0.97-1.20) | 1.16 (1.06-1.26) | N/A |
| Darunavir/Ritonavir | 600 mg/100 mg two times daily × 22 days | 800 mg three times daily × 6 days | 0.75 (0.67-0.85) | 0.68 (0.65-0.72) | 0.65 (0.56-0.76) |
| Diflunisal | 250 mg two times daily × 7 days | 800 mg three times daily × 12 days | 0.86 (0.56-1.32) | 0.96 (0.79-1.17) | 1.31 (1.04-1.65) |
| Efavirenz | 600 mg daily × 16 days | 800 mg three times daily × 6 days | 0.92 (0.78-1.08) | 0.81 (0.75-0.89) | 0.56 (0.42-0.74) |
| Escitalopram | 10 mg single dose | 800 mg three times daily × 11 days | 0.91 (0.81-1.02) | 1.02 (0.96-1.08) | N/A |
| Etravirine | 200 mg two times daily × 11-14 days | 800 mg three times daily × 11-14 days | 1.10 (0.94-1.29) | 1.10 (0.94-1.28) | 0.88†
(0.66-1.17) |
| Ibuprofen | 600 mg three times daily × 6 days | 400 mg single oral dose | 0.94 (0.67-1.32) | 1.04 (0.90-1.20) | N/A |
| Ketoconazole | 400 mg two times daily × 6 days | 400 mg single oral dose | 1.41 (1.00-1.97) | 2.31 (2.00-2.67) | N/A |
| Lopinavir/Ritonavir | 400 mg/100 mg two times daily × 22 days | 800 mg three times daily × 6 days | 0.50 (0.45-0.55) | 0.55 (0.49-0.61) | 0.43 (0.36-0.53) |
| Methadone | 20-150 mg daily × 6 days | 800 mg three times daily × 6 days | 0.62 (0.53-0.72) | 0.80 (0.69-0.93) | 1.03 (0.75-1.42) |
| Omeprazole | 40 mg daily × 5 days | 800 mg three times daily × 5 days | 0.94 (0.86-1.02) | 0.92 (0.87-0.97) | 1.17†
(0.97-1.42) |
| Peginterferon alfa-2b | 1.5 mcg/kg subcutaneous weekly × 2 weeks | 400 mg three times daily × 1 week | 0.88 (0.66-1.18) | 1.00*
(0.89-1.13) | N/A |
| Pravastatin | 40 mg single dose | 800 mg three times daily × 6 days | 0.93 (0.83-1.04) | 0.94 (0.88-1.01) | N/A |
| Raltegravir | 400 mg every 12 hours × 6 days | 800 mg every 8 hours × 6 days | 0.96 (0.88, 1.05) | 0.98‡
(0.90, 1.08) | 0.74†
(0.47, 1.16) |
| Rilpivirine | 25 mg every 24 hours × 11 days | 800 mg three times daily × 11 days | 0.98 (0.89, 1.08) | 0.94‡
(0.88, 1.00) | 1.04†
(0.93, 1.16) |
| Ritonavir | 100 mg daily × 12 days | 400 mg three times daily × 15 days | 0.73 (0.57-0.93) | 0.81 (0.73-0.91) | 1.04 (0.62-1.75) |
| Sirolimus | 2 mg single dose | 800 mg three times daily × 9 days | 0.94 (0.82, 1.07) | 0.95‡
(0.89, 1.01) | 1.21†
(1.00, 1.47) |
| Tacrolimus | 0.5 mg single dose | 800 mg single dose | 0.97 (0.84-1.13) | 1.00*
(0.95-1.06) | N/A |
| Tenofovir | 300 mg daily × 7 days | 800 mg three times daily × 7 days | 1.05 (0.98-1.12) | 1.08 (1.02-1.14) | 1.08 (0.97-1.20) |
| Co-administered Drug | Co-administered Drug Dose/Schedule | Boceprevir Dose/Schedule | Ratio Estimate of Co-administered Pharmacokinetic Parameters (in Combination vs. Alone) (90% CI of the Ratio Estimate) * |
||
|---|---|---|---|---|---|
| Change in mean Cmax | Change in mean AUC(τ) | Change in mean Cmin | |||
| N/A = not available | |||||
| Atazanavir/Ritonavir | 300 mg/100 mg daily × 22 days | 800 mg three times daily × 6 days | Atazanavir: 0.75 (0.64-0.88) Ritonavir: 0.73 (0.64-0.83) | Atazanavir: 0.65†
(0.55-0.78) Ritonavir: 0.64 (0.58-0.72) | Atazanavir: 0.51 (0.44-0.61) Ritonavir: 0.55 (0.45-0.67) |
| Atorvastatin | 40 mg single dose | 800 mg three times daily × 7 days | 2.66 (1.81-3.90) | 2.30‡
(1.84-2.88) | N/A |
| Buprenorphine/ Naloxone | Buprenorphine: 8-24 mg + Naloxone: 2-6 mg daily × 6 days | 800 mg three times daily × 6 days | Buprenorphine: 1.18 (0.93-1.50) | Buprenorphine: 1.19 (0.91-1.57) | Buprenorphine: 1.31 (0.95-1.79) |
| Naloxone: 1.09 (0.79-1.51) | Naloxone: 1.33 (0.90-1.98) | Naloxone: N/A |
|||
| Cyclosporine | 100 mg single dose | 800 mg three times daily × 7 days | 2.01 (1.69-2.40) | 2.68‡
(2.38-3.03) | N/A |
| Darunavir/Ritonavir | 600 mg/100 mg two times daily × 22 days | 800 mg three times daily × 6 days | Darunavir: 0.64 (0.58-0.71) Ritonavir: 0.87 (0.76-1.00) | Darunavir: 0.56†
(0.51-0.61) Ritonavir: 0.73 (0.68-0.79) | Darunavir: 0.41 (0.38-0.45) Ritonavir: 0.55 (0.52-0.59) |
| Digoxin | 0.25 mg single dose | 800 mg three times daily × 10 days | 1.18 (1.07-1.31) | 1.19‡
(1.12-1.27) | N/A |
| Drospirenone/ Ethinyl estradiol | Drospirenone: 3 mg + Ethinyl estradiol: 0.02 mg daily × 14 days | 800 mg three times daily × 7 days | Drospirenone: 1.57 (1.46-1.70) Ethinyl estradiol: 1.00 (0.91-1.10) | Drospirenone: 1.99 (1.87-2.11) Ethinyl estradiol: 0.76 (0.73-0.79) | N/A |
| Efavirenz | 600 mg daily × 16 days | 800 mg three times daily × 6 days | 1.11 (1.02-1.20) | 1.20 (1.15-1.26) | N/A |
| Escitalopram | 10 mg single dose | 800 mg three times daily × 11 days | 0.81 (0.76-0.87) | 0.79‡
(0.71-0.87) | N/A |
| Etravirine | 200 mg two times daily × 11-14 days | 800 mg three times daily × 11-14 days | 0.76 (0.68-0.85) | 0.77 (0.66-0.91) | 0.71 (0.54-0.95) |
| Lopinavir/Ritonavir | 400 mg/100 mg two times daily × 22 days | 800 mg three times daily × 6 days | Lopinavir: 0.70 (0.65-0.77) Ritonavir: 0.88 (0.72-1.07) | Lopinavir: 0.66†
(0.60-0.72) Ritonavir: 0.78 (0.71-0.87) | Lopinavir: 0.57 (0.49-0.65) Ritonavir: 0.58 (0.52-0.65) |
| Methadone | 20-150 mg daily × 6 days | 800 mg three times daily × 6 days | R-methadone: 0.90 (0.71-1.13) | R-methadone: 0.85 (0.74-0.96) | R-methadone: 0.81 (0.66-1.00) |
| S-methadone: 0.83 (0.64-1.09) | S-methadone: 0.78 (0.66-0.93) | S-methadone: 0.74 (0.58-0.95) |
|||
| Midazolam | 4 mg single oral dose | 800 mg three times daily × 6 days | 2.77 (2.36-3.25) | 5.30 (4.66-6.03) | N/A |
| Norethindrone/ Ethinyl estradiol | Norethindrone: 1 mg + Ethinyl estradiol : 0.035 mg daily × 21 days | 800 mg three times daily × 28 days | Norethindrone: 0.83 (0.76-0.90) Ethinyl estradiol: 0.79 (0.75 -0.84) | Norethindrone: 0.96 (0.87-1.06) Ethinyl estradiol: 0.74 (0.68-0.80) | N/A |
| Omeprazole | 40 mg daily × 5 days | 800 mg three times daily × 5 days | 1.03 (0.85-1.26) | 1.06 (0.90-1.25) | 1.12 §
(0.75-1.67) |
| Peginterferon alfa-2b | 1.5 mcg/kg subcutaneous weekly × 2 weeks | 200 mg or 400 mg three times daily × 1 week | N/A | 0.99¶,#
(0.83-1.17) | N/A |
| Pravastatin | 40 mg single dose | 800 mg three times daily × 6 days | 1.49 (1.03-2.14) | 1.63‡
(1.01-2.62) | N/A |
| Prednisone | 40 mg single dose | 800 mg three times daily × 6 days | Prednisone: 0.99 (0.94-1.04) | Prednisone: 1.22 (1.16-1.28) | Prednisone: N/A |
| Prednisolone: 1.16 (1.09-1.24) | Prednisolone: 1.37 (1.31-1.44) | Prednisolone: N/A |
|||
| Raltegravir | 400 mg single dose | 800 mg three times daily × 10 days | 1.11 (0.91-1.36) | 1.04 (0.88-1.22) | 0.75Þ
(0.45-1.23) |
| Rilpivirine | 25 mg every 24 hours × 11 days | 800 mg three times daily × 11 days | 1.15 (1.04, 1.28) | 1.39†
(1.27, 1.52) | 1.51 (1.36, 1.68) |
| Sirolimus | 2 mg single dose | 800 mg every 8 hours × 9 days | 4.84 (3.99, 5.88) | 8.12‡
(7.08, 9.32) | N/A |
| Tacrolimus | 0.5 mg single dose | 800 mg three times daily × 11 days | 9.90 (7.96-12.3) | 17.1‡
(14.0-20.8) | N/A |
| Tenofovir
| 300 mg daily × 7 days | 800 mg three times daily × 7 days | 1.32 (1.19-1.45) | 1.05 (1.01-1.09) | N/A |
Elimination
Boceprevir is eliminated with a mean plasma half-life (t½) of approximately 3.4 hours. Boceprevir has a mean total body clearance (CL/F) of approximately 161 L per hr. Following a single 800 mg oral dose of 14C-boceprevir, approximately 79% and 9% of the dose was excreted in feces and urine, respectively, with approximately 8% and 3% of the dosed radiocarbon eliminated as boceprevir in feces and urine. The data indicate that boceprevir is eliminated primarily by the liver.
Special Populations
Hepatic Impairment
The pharmacokinetics of boceprevir was studied in adult non-HCV infected subjects with normal, mild (Child-Pugh score 5 to 6), moderate (Child-Pugh score 7 to 9), and severe (Child-Pugh score 10 to 12) hepatic impairment following a single 400 mg dose of VICTRELIS. The mean AUC of the active diastereomer of boceprevir (SCH534128) was 32% and 45% higher in subjects with moderate and severe hepatic impairment, respectively, relative to subjects with normal hepatic function. Mean Cmax values for SCH534128 were 28% and 62% higher in moderate and severe hepatic impairment, respectively. Subjects with mild hepatic impairment had similar SCH534128 exposure as subjects with normal hepatic function. A similar magnitude of effect is anticipated for boceprevir. No dosage adjustment of VICTRELIS is recommended for patients with hepatic impairment. For additional information in patients with compensated cirrhosis, see Use in Specific Populations (8.7). [see the prescribing information for peginterferon alfa for use in patients with hepatic decompensation.]
Renal Impairment
The pharmacokinetics of boceprevir was studied in non-HCV-infected subjects with end-stage renal disease (ESRD) requiring hemodialysis following a single 800 mg dose of VICTRELIS. The mean AUC of boceprevir was 10% lower in subjects with ESRD requiring hemodialysis relative to subjects with normal renal function. Hemodialysis removed less than 1% of the boceprevir dose. No dosage adjustment of VICTRELIS is required in patients with any degree of renal impairment.
Gender
Population pharmacokinetic analysis of VICTRELIS indicated that gender had no apparent effect on exposure.
12.4 Microbiology
Mechanism of Action
Boceprevir is an inhibitor of the HCV NS3/4A protease that is necessary for the proteolytic cleavage of the HCV encoded polyprotein into mature forms of the NS4A, NS4B, NS5A and NS5B proteins. Boceprevir covalently, yet reversibly, binds to the NS3 protease active site serine (S139) through an (alpha)-ketoamide functional group to inhibit viral replication in HCV-infected host cells. In a biochemical assay, boceprevir inhibited the activity of recombinant HCV genotype 1a and 1b NS3/4A protease enzymes, with Ki values of 14 nM for each subtype.
Activity in Cell Culture
The EC50 and EC90 values for boceprevir against an HCV replicon constructed from a single genotype 1b isolate were approximately 200 nM and 400 nM, respectively, in a 72-hour cell culture assay. Boceprevir cell culture anti-HCV activity was approximately 2-fold lower for an HCV replicon derived from a single genotype 1a isolate, relative to the 1b isolate-derived replicon. In replicon assays, boceprevir had approximately 2-fold reduced activity against a genotype 2a isolate relative to genotype 1a and 1b replicon isolates. In a biochemical assay, boceprevir had approximately 3- and 2-fold reduced activity against NS3/4A proteases derived from single isolates representative of HCV genotypes 2 and 3a, respectively, relative to a genotype 1b-derived NS3/4A protease. The presence of 50% human serum reduced the cell culture anti-HCV activity of boceprevir by approximately 3-fold.
Evaluation of varying combinations of boceprevir and interferon alfa-2b that produced 90% suppression of replicon RNA in cell culture showed additivity of effect without evidence of antagonism.
Resistance
In HCV Replicon Cell Culture and Biochemical Studies
The activity of boceprevir against the HCV genotype 1a replicon was reduced (2- to 6-fold) by the following amino acid substitutions in the NS3 protease domain: V36A/L/M, Q41R, T54A/S, V55A, R155K and V158I. A greater than 10-fold reduction in boceprevir susceptibility was conferred by the amino acid substitutions R155T and A156S. The V55I and D168N single substitutions did not reduce sensitivity to boceprevir. The following double amino acid substitutions conferred more than 10-fold reduced sensitivity to boceprevir: V55A+I170V, T54S+R155K, R155K+D168N, R155T+D168N and V36M+R155K.
The activity of boceprevir against the HCV genotype 1b replicon was reduced (2- to 8-fold) by the following amino acid substitutions in the NS3 protease domain: V36A/M, Q41R, F43S, T54A/G/S, V55A/I, R155K, V158I, V170M and M175L. A greater than 10-fold reduction in boceprevir susceptibility was conferred by the amino acid substitutions A156S/T/V, V170A and V36M+R155K. The D168V single substitution did not reduce sensitivity to boceprevir.
Additional NS3 protease domain substitutions that have not been evaluated in the HCV replicon but have been shown to reduce boceprevir activity against the HCV NS3/4A protease in a biochemical assay include F43C and R155G/I/M/Q.
Resistance-associated amino acid substitutions for HCV genotype 1a and 1b observed in clinical trials are presented in Table 8.
In Clinical Studies
An as-treated, pooled genotypic resistance analysis was conducted for subjects who received four weeks of PegIntron/REBETOL followed by VICTRELIS 800 mg three times daily in combination with PegIntron/REBETOL in two Phase 3 studies, SPRINT-2 and RESPOND-2. Among subjects treated with VICTRELIS who did not achieve a sustained virologic response, and for whom samples were analyzed, 53% had one or more specific post-baseline, treatment-emergent NS3 protease domain amino acid substitutions detected by a population-based sequencing assay (Table 8). Similar patterns of treatment-emergent substitutions were observed in P06086, a Phase 3 clinical trial in previously untreated CHC subjects with genotype 1 infection comparing the use of ESA to ribavirin dose reduction for initial management of anemia during therapy with VICTRELIS in combination with PegIntron/REBETOL. Nearly all of these substitutions have been shown to reduce boceprevir anti-HCV activity in cell culture or biochemical assays. Among subjects treated with VICTRELIS in SPRINT-2 and RESPOND-2 who did not achieve SVR and for whom post-baseline samples were analyzed, 31% of PegIntron/REBETOL-responsive subjects, as defined by greater than or equal to 1-log10 decline in viral load at Treatment Week 4 (end of 4-week PegIntron/REBETOL lead-in period), had detectable treatment-emergent substitutions, compared to 68% of subjects with less than 1-log10 decline in viral load at Treatment Week 4. Clear patterns of boceprevir treatment-emergent substitutions in the NS3 helicase domain or NS4A coding regions of the HCV genome were not observed.
| Subjects Infected with HCV Genotype 1a | Subjects Infected with HCV Genotype 1b | |
|---|---|---|
| >10% of subjects treated with VICTRELIS who did not achieve SVR | V36M, T54S, R155K | T54A, T54S, V55A, A156S, V170A |
| <1% to 10% of subjects treated with VICTRELIS who did not achieve SVR | V36A, T54A, V55A, V55I, V107I, R155T, A156S, A156T, V158I, D168N, I170F, I170T, I170V | V36A, V36M, T54C, T54G, V107I, R155C, R155K, A156T, A156V, V158I, I/V170T, M175L |
Persistence of Resistance-Associated Substitutions
Data from an ongoing, long-term follow-up study of subjects who did not achieve SVR in Phase 2 trials with VICTRELIS, with a median duration of follow-up of approximately 2 years, indicate that HCV populations harboring certain post-baseline, treatment-emergent substitutions may decline in relative abundance over time. However, among those subjects with available data, one or more treatment-emergent substitutions remained detectable with a population-based sequencing assay in 25% of subjects after 2.5 years of follow-up. The most common NS3 substitutions detected after 2.5 years of follow-up were T54S and R155K. The lack of detection of a substitution based on a population-based assay does not necessarily indicate that viral populations carrying that substitution have declined to a background level that may have existed prior to treatment. The long-term clinical impact of the emergence or persistence of boceprevir-resistance-associated substitutions is unknown. No data are available regarding the efficacy of VICTRELIS among subjects who were previously exposed to VICTRELIS, or who previously failed treatment with a regimen containing VICTRELIS.
Effect of Baseline HCV Polymorphisms on Treatment Response
A pooled analysis was conducted to explore the association between the detection of baseline NS3/4A amino acid polymorphisms and treatment outcome in the two Phase 3 studies, SPRINT-2 and RESPOND-2.
Baseline resistance associated polymorphisms were detected in 7% of subjects by a population-based sequencing method. Overall, the presence of these polymorphisms alone did not impact SVR rates in subjects treated with VICTRELIS. However, among subjects with a relatively poor response to PegIntron/REBETOL during the 4-week lead-in period, the efficacy of VICTRELIS appeared to be reduced for those who had V36M, T54A, T54S, V55A or R155K detected at baseline. Subjects with these baseline polymorphisms and reduced response to PegIntron/REBETOL represented approximately 1% of the total number of subjects treated with VICTRELIS.
Cross-Resistance
Many of the treatment-emergent NS3 amino acid substitutions detected in subjects treated with VICTRELIS who did not achieve SVR in the Phase 3 clinical trials have been demonstrated to reduce the anti-HCV activity of other HCV NS3/4A protease inhibitors. The impact of prior exposure to VICTRELIS or treatment failure on the efficacy of other HCV NS3/4A protease inhibitors has not been studied. The efficacy of VICTRELIS has not been established for patients with a history of exposure to other NS3/4A protease inhibitors. Cross-resistance is not expected between VICTRELIS and interferons, or VICTRELIS and ribavirin.
12.5 Pharmacogenomics
A genetic variant near the gene encoding interferon-lambda-3 (IL28B rs12979860, a C to T change) is a strong predictor of response to PegIntron/REBETOL. IL28B rs12979860 was genotyped in 653 of 1048 (62%) subjects in SPRINT-2 (previously untreated) and 259 of 394 (66%) subjects in RESPOND-2 (previous partial responders and relapsers) [see Clinical Studies (14) for trial descriptions]. Among subjects that received at least one dose of placebo or VICTRELIS (Modified-Intent-to-Treat population), SVR rates tended to be lower in subjects with the C/T and T/T genotypes compared to those with the C/C genotype, particularly among previously untreated subjects receiving 48 weeks of PegIntron and REBETOL (see Table 9). Among previous treatment failures, subjects of all genotypes appeared to have higher SVR rates with regimens containing VICTRELIS. The results of this retrospective subgroup analysis should be viewed with caution because of the small sample size and potential differences in demographic or clinical characteristics of the substudy population relative to the overall trial population.
| SVR, % (n/N) | ||||
|---|---|---|---|---|
| Clinical Study | IL28B rs12979860 Genotype | PR48* | Boceprevir-RGT* | Boceprevir-PR48* |
|
||||
| SPRINT-2 (Previously Untreated Subjects) | ||||
| C/C | 78 (50/64) | 82 (63/77) | 80 (44/55) | |
| C/T | 28 (33/116) | 65 (67/103) | 71 (82/115) | |
| T/T | 27 (10/37) | 55 (23/42) | 59 (26/44) | |
| RESPOND-2 (Previous Partial Responders and Relapsers) | ||||
| C/C | 46 (6/13) | 79 (22/28) | 77 (17/22) | |
| C/T | 17 (5/29) | 61 (38/62) | 73 (48/66) | |
| T/T | 50 (5/10) | 55 (6/11) | 72 (13/18) | |
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
Use with Ribavirin and Peginterferon alfa: Ribavirin is genotoxic in in vitro and in vivo assays. Ribavirin was not oncogenic in mouse and rat carcinogenicity studies at doses less than the maximum recommended daily human dose. Please refer to the prescribing information for ribavirin for additional information.
Two-year carcinogenicity studies in mice and rats were conducted with boceprevir. Mice were administered doses of up to 500 mg per kg in males and 650 mg per kg in females, and rats were administered doses of up to 125 mg per kg in males and 100 mg per kg in females. In mice, no significant increases in the incidence of drug-related neoplasms were observed at the highest doses tested resulting in boceprevir AUC exposures approximately 2.3- and 6.0-fold higher in males and females, respectively, than those in humans at the recommended dose of 800 mg three times daily. In rats, no increases in the incidence of drug-related neoplasms were observed at the highest doses tested resulting in boceprevir AUC exposures similar to those in humans at the recommended dose of 800 mg three times daily.
Boceprevir was not genotoxic in a battery of in vitro or in vivo assays, including bacterial mutagenicity, chromosomal aberration in human peripheral blood lymphocytes and mouse micronucleus assays.
Impairment of Fertility
Use with Ribavirin and Peginterferon alfa: In fertility studies in male animals, ribavirin induced reversible testicular toxicity; while peginterferon alfa may impair fertility in females. Please refer to the prescribing information for ribavirin and peginterferon alfa for additional information.
Boceprevir-induced reversible effects on fertility and early embryonic development in female rats, with no effects observed at a 75 mg per kg dose level. At this dose, boceprevir AUC exposures are approximately 1.3-fold higher than those in humans at the recommended dose of 800 mg three times daily. Decreased fertility was also observed in male rats, most likely as a consequence of testicular degeneration. No testicular degeneration was observed at a 15 mg per kg dose level resulting in boceprevir AUC exposures of less than those in humans at the recommended dose of 800 mg three times daily. Testicular degeneration was not observed in mice or monkeys administered boceprevir for 3 months at doses of up to 900 or 1000 mg per kg, respectively. At these doses, boceprevir AUC exposures are approximately 6.8- and 4.4-fold higher in mice and monkeys, respectively, than those in humans at the recommended dose of 800 mg three times daily. Additionally, limited clinical monitoring has revealed no evidence of testicular toxicity in human subjects.
14 CLINICAL STUDIES
The efficacy of VICTRELIS as a treatment for chronic hepatitis C (genotype 1) infection was assessed in approximately 1500 adult subjects who were previously untreated (SPRINT-2) or who had failed previous peginterferon alfa and ribavirin therapy (RESPOND-2) in Phase 3 clinical studies.
Previously Untreated Subjects
SPRINT-2 was a randomized, double-blind, placebo-controlled study comparing two therapeutic regimens of VICTRELIS 800 mg orally three times daily in combination with PR [PegIntron 1.5 micrograms per kg per week subcutaneously and weight-based dosing with REBETOL (600–1400 mg per day orally divided twice daily)] to PR alone in adult subjects who had chronic hepatitis C (HCV genotype 1) infection with detectable levels of HCV-RNA and were not previously treated with interferon alfa therapy. Subjects were randomized in a 1:1:1 ratio within two separate cohorts (Cohort 1/non-Black and Cohort 2/Black) and were stratified by HCV genotype (1a or 1b) and by HCV-RNA viral load (less than or equal to 400,000 IU per mL vs. more than 400,000 IU per mL) to one of the following three treatment arms:
- PegIntron + REBETOL for 48 weeks (PR48).
- PegIntron + REBETOL for four weeks followed by VICTRELIS 800 mg three times daily + PegIntron + REBETOL for 24 weeks. The subjects were then continued on different regimens based on Treatment Week (TW) 8 through TW24 response-guided therapy (boceprevir-RGT). All subjects in this treatment arm were limited to 24 weeks of therapy with VICTRELIS.
- Subjects with undetectable HCV-RNA (Target Not Detected) at TW8 (early responders) and remained undetectable through TW24 discontinued therapy and entered follow-up at the TW28 visit.
- Subjects with detectable HCV-RNA at TW8 or any subsequent treatment week but subsequently achieving undetectable HCV-RNA (Target Not Detected) at TW24 (late responders) were changed in a blinded fashion to placebo at the TW28 visit and continued therapy with PegIntron + REBETOL for an additional 20 weeks, for a total treatment duration of 48 weeks.
- PegIntron + REBETOL for four weeks followed by VICTRELIS 800 mg three times daily + PegIntron + REBETOL for 44 weeks (boceprevir-PR48).
All subjects with detectable HCV-RNA in plasma at TW24 were discontinued from treatment. Sustained Virologic Response (SVR) was defined as plasma HCV-RNA less than 25 IU/mL at Follow-up Week 24. Plasma HCV-RNA results at Follow-up Week 12 were used if plasma HCV-RNA results at Follow-up Week 24 were missing.
Mean age of subjects randomized was 49 years. The racial distribution of subjects was as follows: 82% White, 14% Black, and 4% others. The distribution of subjects by gender was 60% men and 40% women.
The addition of VICTRELIS to PegIntron and REBETOL significantly increased the SVR rates compared to PegIntron and REBETOL alone in the combined cohort (63% to 66% in arms containing VICTRELIS vs. 38% PR48 control) for randomized subjects who received at least one dose of any study medication (Full-Analysis-Set population). SVR rates for Blacks who received the combination of VICTRELIS with PegIntron and REBETOL were 42% to 53% in a predefined analysis (see Table 10).
| Study Cohorts | Boceprevir-RGT | Boceprevir-PR48 | PR48 |
|---|---|---|---|
|
|||
| Cohort 1 Plus Cohort 2 (all subjects) | n=368 | n=366 | n=363 |
| SVR† % | 63 | 66 | 38 |
| Relapse‡ % | 9 | 9 | 22 |
| (n/N) | (24/257) | (24/265) | (39/176) |
| Cohort 1 Plus Cohort 2 (subjects without cirrhosis) | n=352 | n=342 | n=350 |
| SVR†,§ % | 65 | 68 | 38 |
| (n/N) | (228/352) | (232/342) | (132/350) |
| Cohort 1 (non-Black) | n=316 | n=311 | n=311 |
| SVR† % | 67 | 68 | 40 |
| Relapse‡ % | 9 | 8 | 23 |
| (n/N) | (21/232) | (18/230) | (37/162) |
| Cohort 2 (Black) | n=52 | n=55 | n=52 |
| SVR† % | 42 | 53 | 23 |
| Relapse‡ % | 12 | 17 | 14 |
| (n/N) | (3/25) | (6/35) | (2/14) |
In subjects with cirrhosis at baseline, sustained virologic response was higher in those who received treatment with the combination of VICTRELIS with PegIntron and REBETOL for 44 weeks after lead-in therapy with PegIntron and REBETOL (10/24, 42%) compared to those who received RGT (5/16 , 31%).
Sustained Virologic Response (SVR) Based on TW8 HCV-RNA Results
Table 11 presents sustained virologic response based on TW8 HCV-RNA results in previously untreated subjects. Fifty-seven percent (208/368) of subjects in the boceprevir-RGT arm and 56% (204/366) of subjects in the boceprevir-PR48 arm had undetectable HCV-RNA (Target Not Detected) at TW8 (early responders) compared with 17% (60/363) of subjects in the PR48 arm.
| Boceprevir-RGT | Boceprevir-PR48 | PR48 | |
|---|---|---|---|
|
|||
| SVR by TW8 Detectability, % (n/N)* | N=337 | N=335 | N=331 |
| Undetectable (Target Not Detected) | 88 (184/208) | 90 (184/204) | 85 (51/60) |
| Detectable | 36 (46/129) | 40 (52/131) | 30 (82/271) |
Among subjects with detectable HCV-RNA at TW8 who had attained undetectable HCV-RNA (Target Not Detected) at TW24 and completed at least 28 weeks of treatment, the SVR rates were 66% (45/68) in boceprevir-RGT arm (4 weeks of PegIntron and REBETOL then 24 weeks of VICTRELIS with PegIntron and REBETOL followed by 20 weeks of PegIntron and REBETOL alone) and 75% (55/73) in boceprevir-PR48 arms (4 weeks of PegIntron and REBETOL then 44 weeks of VICTRELIS with PegIntron and REBETOL).
Previous Partial Responders and Relapsers to Interferon and Ribavirin Therapy
RESPOND-2 was a randomized, parallel-group, double-blind study comparing two therapeutic regimens of VICTRELIS 800 mg orally three times daily in combination with PR [PegIntron 1.5 micrograms per kg per week subcutaneously and weight-based ribavirin (600–1400 mg per day orally divided twice daily)] compared to PR alone in adult subjects with chronic hepatitis C (HCV genotype 1) infection with demonstrated interferon responsiveness (as defined historically by a decrease in HCV-RNA viral load greater than or equal to 2-log10 by Week 12, but never achieved SVR [partial responders] or undetectable HCV-RNA at end of prior treatment with a subsequent detectable HCV-RNA in plasma [relapsers]). Subjects with less than 2-log10 decrease in HCV-RNA by week 12 of previous treatment (prior null responders) were not eligible for enrollment in this trial. Subjects were randomized in a 1:2:2 ratio and stratified based on response to their previous qualifying regimen (relapsers vs. partial responders) and by HCV subtype (1a vs. 1b) to one of the following treatment arms:
- PegIntron + REBETOL for 48 weeks (PR48)
- PegIntron + REBETOL for 4 weeks followed by VICTRELIS 800 mg three times daily + PegIntron + REBETOL for 32 weeks. The subjects were then continued on different treatment regimens based on TW8 and TW12 response-guided therapy (boceprevir-RGT). All subjects in this treatment arm were limited to 32 weeks of VICTRELIS.
- Subjects with undetectable HCV-RNA (Target Not Detected) at TW8 (early responders) and TW12 completed therapy at TW36 visit.
- Subjects with a detectable HCV-RNA at TW8 but subsequently undetectable (Target Not Detected) at TW12 (late responders) were changed in a blinded fashion to placebo at the TW36 visit and continued treatment with PegIntron + REBETOL for an additional 12 weeks, for a total treatment duration of 48 weeks.
- PegIntron + REBETOL for 4 weeks followed by VICTRELIS 800 mg three times daily + PegIntron + REBETOL for 44 weeks (boceprevir-PR48).
All subjects with detectable HCV-RNA in plasma at TW12 were discontinued from treatment. Sustained Virologic Response (SVR) was defined as plasma HCV-RNA less than 25 IU/mL at Follow-up Week 24. Plasma HCV-RNA results at Follow-up Week 12 were used if plasma HCV-RNA results at Follow-up Week 24 were missing.
Mean age of subjects randomized was 53 years. The racial distribution of subjects was as follows: 85% White, 12% Black, and 3% others. The distribution of subjects by gender was 67% men and 33% women.
The addition of VICTRELIS to the PegIntron and REBETOL therapy significantly increased the SVR rates compared to PegIntron/REBETOL alone (59% to 66% in arms containing VICTRELIS vs. 23% PR48 control) for randomized subjects who received at least one dose of any study medication (Full-Analysis-Set population) (see Table 12).
| Boceprevir-RGT | Boceprevir-PR48 | PR48 | ||
|---|---|---|---|---|
| Previous Partial Responder = subject who failed to achieve SVR after at least 12 weeks of previous treatment with peginterferon alfa and ribavirin, but demonstrated a ≥2-log10 reduction in HCV-RNA by Week 12 and had detectable HCV-RNA at End of Treatment (EOT). | ||||
| Previous Relapser = subject who failed to achieve SVR after at least 12 weeks of previous treatment with peginterferon alfa and ribavirin, but had undetectable HCV-RNA at the end of treatment. | ||||
|
||||
| N=162 | N=161 | N=80 | ||
| SVR† % | 59 | 66 | 23 | |
| Relapse‡ % | 14 | 12 | 28 | |
| (n/N) | (16/111) | (14/121) | (7/25) | |
| SVR (subjects without cirrhosis) § | 62 | 65 | 26 | |
| (n/N) | (90/145) | (90/139) | (18/70) | |
| SVR by Response to Previous Peginterferon and Ribavirin Therapy | ||||
| Previous Response | Relapser, % (n/N) | 70 (73/105) | 75 (77/103) | 31 (16/51) |
| Partial responder, % (n/N) | 40 (23/57) | 52 (30/58) | 7 (2/29) | |
In subjects with cirrhosis at baseline, sustained virologic response was higher in those who received treatment with the combination of VICTRELIS with PegIntron and REBETOL for 44 weeks after 4 weeks of lead-in therapy with PegIntron and REBETOL (17/22, 77%) compared to those who received RGT (6/17, 35%).
Sustained Virologic Response (SVR) Based on TW8 HCV-RNA Results
Table 13 presents sustained virologic response based on TW8 HCV-RNA results in subjects who were relapsers or partial responders to previous interferon and ribavirin therapy. Forty-six percent (74/162) of subjects in the boceprevir-RGT arm and 52% (84/161) in the boceprevir-PR48 had undetectable HCV-RNA (Target Not Detected) at TW8 (early responders) compared with 9% (7/80) in the PR48 arm.
| Boceprevir-RGT | Boceprevir-PR48 | PR48 | |
|---|---|---|---|
|
|||
| SVR by TW8 Detectability, % (n/N)* | N=146 | N=154 | N=72 |
| Undetectable (Target Not Detected) | 88 (65/74) | 88 (74/84) | 100 (7/7) |
| Detectable | 40 (29/72) | 43 (30/70) | 14 (9/65) |
Among subjects with detectable HCV-RNA at TW8 who attained an undetectable HCV-RNA (Target Not Detected) at TW12 and completed at least 36 weeks of treatment, the SVR rates were 79% (27/34) in boceprevir-RGT arm (4 weeks of PegIntron and REBETOL then 32 weeks of VICTRELIS with PegIntron and REBETOL followed by 12 weeks of PegIntron and REBETOL alone) and 72% (29/40) in boceprevir-PR48 arm (4 weeks of PegIntron and REBETOL then 44 weeks of VICTRELIS with PegIntron and REBETOL).
Interferon Responsiveness during Lead-In Therapy with Peginterferon alfa and Ribavirin
Previously Untreated Subjects
In previously untreated subjects evaluated in SPRINT-2, interferon-responsiveness (defined as greater than or equal to 1-log10 decline in viral load at TW4) was predictive of SVR. Subjects treated with VICTRELIS who demonstrated interferon responsiveness at TW4 achieved SVR rates of 81% (203/252) in boceprevir-RGT arm and 79% (200/254) in boceprevir-PR48 arm, compared to 52% (134/260) in subjects treated with PegIntron/REBETOL.
Subjects treated with VICTRELIS who demonstrated poor interferon responsiveness (defined as less than 1-log10 decline in viral load at TW4), achieved SVR rates of 28% (27/97) in boceprevir-RGT arm and 38% (36/95) in boceprevir-PR48 arm, compared to 4% (3/83) in subjects treated with PegIntron/REBETOL. Subjects with less than a 0.5-log10 decline in viral load at TW4 achieved SVR rates of 28% (13/47) in boceprevir-RGT arm and 30% (11/37) in boceprevir-PR48 arm, compared to 0% (0/25) in subjects treated with PegIntron/REBETOL. Subjects with less than a 0.5-log10 decline in viral load at TW4 with peginterferon alfa plus ribavirin therapy alone are predicted to have a null response (less than 2-log10 viral load decline at TW12) to peginterferon alfa and ribavirin.
Previous Partial Responders and Relapsers to Interferon and Ribavirin Therapy
In subjects who were previous relapsers and partial responders evaluated in RESPOND-2, interferon-responsiveness (defined as greater than or equal to 1-log10 decline in viral load at TW4) was predictive of SVR. Subjects treated with VICTRELIS who demonstrated interferon responsiveness at TW4 achieved SVR rates of 74% (81/110) in boceprevir-RGT arm and 79% (90/114) in boceprevir-PR48 arm, compared to 27% (18/67) in subjects treated with PegIntron/REBETOL. Subjects treated with VICTRELIS who demonstrated poor interferon responsiveness (defined as less than 1-log10 decline in viral load at TW4) achieved SVR rates of 33% (15/46) in boceprevir-RGT arm and 34% (15/44) in boceprevir-PR48 arm, compared to 0% (0/12) in subjects treated with PegIntron/REBETOL.
Prior Null Responders to Interferon and Ribavirin Therapy
PROVIDE was an open-label, single-arm trial of VICTRELIS 800 mg orally three times daily in combination with peginterferon alfa-2b 1.5 micrograms per kg per week subcutaneously and weight-based ribavirin (600 – 1,400 mg per day orally divided twice daily) in adult subjects with chronic hepatitis C (HCV) genotype 1 infection who did not achieve SVR while in the peginterferon alfa/ribavirin control arms of previous Phase 2 and 3 trials of combination therapy with VICTRELIS. Subjects who enrolled in PROVIDE within 2 weeks after the last dose of peginterferon alfa/ribavirin in the prior trial received VICTRELIS 800 mg three times daily + peginterferon alfa-2b + ribavirin for 44 weeks. Subjects who were not able to enroll in this trial within 2 weeks received PegIntron/REBETOL lead-in for 4 weeks followed by VICTRELIS 800 mg three times daily + peginterferon alfa-2b + ribavirin for 44 weeks.
Among subjects who were null responders in the peginterferon alfa/ribavirin control arm of the prior trial, SVR (reported as plasma HCV-RNA <25 IU/mL at follow-up week 24) was 38% (20/52) and the relapse rate was 13% (3/23).
Use of Ribavirin Dose Reduction versus Erythropoiesis Stimulating Agent (ESA) in the Management of Anemia in Previously Untreated Subjects
A randomized, parallel-arm, open-label study was conducted to compare two strategies for the management of anemia (use of ESA versus ribavirin dose reduction) in 687 subjects with previously untreated CHC genotype 1 infection who became anemic during therapy with VICTRELIS 800 mg orally three times daily plus peginterferon alfa-2b 1.5 micrograms per kg per week subcutaneously and weight-based ribavirin (600 – 1,400 mg orally per day divided twice daily). The study enrolled subjects with serum hemoglobin concentrations of less than 15 g per dL. Subjects were treated for 4 weeks with peginterferon alfa-2b and ribavirin followed by up to 44 weeks of VICTRELIS plus peginterferon alfa-2b and ribavirin. If a subject became anemic (serum hemoglobin of approximately less than or equal to 10 g per dL within the treatment period), the subject was randomized in a 1:1 ratio to either ribavirin dose reduction (N=249) or use of erythropoietin 40,000 units subcutaneously once weekly for the management of the anemia (N=251). If serum hemoglobin concentrations continued to decrease to less than or equal to 8.5 g per dL, subjects could be treated with additional anemia interventions, including the addition of erythropoietin (18% of those in the ribavirin dose reduction arm) or ribavirin dose reduction (37% of those in the ESA arm).
Mean age of subjects randomized was 49 years. The racial distribution of subjects was as follows: 77% White, 19% Black, and 4% other. The distribution of subjects by gender was 37% men and 63% women.
The overall intent-to-treat SVR rate for all enrolled subjects (including those subjects who were not randomized to RBV dose reduction or ESA for the management of anemia) was 63% (431/687). The SVR rate in subjects randomized who received ribavirin dose reduction was 71% (178/249), similar to the SVR rate of 71% (178/251) in subjects randomized to receive an ESA. The relapse rates in subjects randomized to receive ribavirin dose reduction or an ESA were 10% (19/196) and 10% (19/197), respectively.
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
VICTRELIS 200 mg capsules are comprised of a red-colored cap with the Merck logo printed in yellow ink, and a yellow-colored body with "314" printed in red ink. The capsules are packaged into a carton with 28 bottles containing 12 capsules (NDC 0085-0314-02).
16.2 Storage and Handling
VICTRELIS Capsules should be refrigerated at 2–8°C (36–46°F) until dispensed. Avoid exposure to excessive heat. For patient use, refrigerated capsules of VICTRELIS can remain stable until the expiration date printed on the label. VICTRELIS can also be stored at room temperature up to 25°C (77°F) for 3 months. Keep container tightly closed.
17 PATIENT COUNSELING INFORMATION
- Advise the patient to read the FDA-approved patient labeling (Medication Guide)
VICTRELIS must be used in combination with peginterferon alfa and ribavirin, and thus all contraindications and warnings for peginterferon alfa and ribavirin also apply. If peginterferon alfa or ribavirin is permanently discontinued, VICTRELIS must also be discontinued [see Dosage and Administration (2.3)].
Pregnancy
Ribavirin must not be used by women who are pregnant or by men whose female partners are pregnant. Ribavirin therapy should not be initiated until a report of a negative pregnancy test has been obtained immediately before starting therapy. Female patients of childbearing potential and male patients with female partners of childbearing potential must be advised of the teratogenic/embryocidal risks of ribavirin and must be instructed to practice effective contraception during therapy and for 6 months post-therapy. Patients should be advised to notify the healthcare provider immediately in the event of a pregnancy [see Contraindications (4) and Warnings and Precautions (5.1)].
Women of childbearing potential and men must use at least two forms of effective contraception during treatment and for at least 6 months after treatment has been stopped; routine monthly pregnancy tests must be performed during this time. One of these reliable forms of contraception can be a combined oral contraceptive product containing at least 1 mg of norethindrone. Oral contraceptives containing lower doses of norethindrone and other forms of hormonal contraception have not been studied or are contraindicated [see Contraindications (4) and Warnings and Precautions (5.1)].
To monitor maternal and fetal outcomes of pregnant women exposed to ribavirin, the Ribavirin Pregnancy Registry has been established. Patients should be encouraged to register by calling 1-800-593-2214.
Anemia
Patients should be informed that anemia may be increased when VICTRELIS is administered with peginterferon alfa and ribavirin [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)]. Patients should be advised that laboratory evaluations are required prior to starting therapy and periodically thereafter [see Warnings and Precautions (5.7)].
Neutropenia
Patients should be informed that neutropenia may be increased when VICTRELIS is administered with peginterferon alfa and ribavirin [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)]. Patients should be advised that laboratory evaluations are required prior to starting therapy and periodically thereafter [see Warnings and Precautions (5.7)].
Pancytopenia
Patients should be informed that serious cases of pancytopenia have been reported during postmarketing when VICTRELIS was administered with peginterferon alfa and ribavirin [see Warnings and Precautions (5.4) and Adverse Reactions (6.2)]. Patients should be advised that laboratory evaluations are required prior to starting therapy and periodically thereafter [see Warnings and Precautions (5.7)].
Hypersensitivity
Patients should be informed that serious acute hypersensitivity reactions have been observed during combination therapy with VICTRELIS, peginterferon alfa, and ribavirin therapy [see Contraindications (4) and Warnings and Precautions (5.5)]. If symptoms of acute hypersensitivity reactions (e.g., itching; hives; swelling of the face, eyes, lips, tongue, or throat; trouble breathing or swallowing) occur, patients should seek medical advice promptly.
Usage Safeguards
Patients should be advised that VICTRELIS must not be used alone due to the high probability of resistance without combination anti-HCV therapies [see Indications and Usage (1)]. See the prescribing information for peginterferon alfa and ribavirin for additional patient counseling information on the use of these drugs in combination with VICTRELIS.
Patients should be informed of the potential for serious drug interactions with VICTRELIS, and that some drugs should not be taken with VICTRELIS [see Contraindications (4), Warnings and Precautions (5.6), Drug Interactions (7), and Clinical Pharmacology (12.3)].
Patients should be advised that the total daily dose of VICTRELIS is packaged into a single bottle containing 12-capsules and the patient should take four capsules three times daily with food.
Missed VICTRELIS Doses
If a patient misses a dose and it is less than 2 hours before the next dose is due, the missed dose should be skipped. If a patient misses a dose and it is 2 or more hours before the next dose is due, the patient should take the missed dose with food and resume the normal dosing schedule.
Manufactured for:
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ 08889, USA
Manufactured by:
MSD International GmbH (Singapore Branch) Singapore 638414, Singapore
For patent information: www.merck.com/product/patent/home.html
Trademarks depicted herein are the property of their respective owners.
Copyright © 2011-2017 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved.
uspi-mk3034-c-1701r070
MEDICATION GUIDE
VICTRELIS® (vic-TREL-is)
(boceprevir)
capsules
Read this Medication Guide before you start taking VICTRELIS and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or treatment.
VICTRELIS is taken along with peginterferon alfa and ribavirin. You should also read those Medication Guides.
What is the most important information I should know about VICTRELIS?
VICTRELIS, in combination with peginterferon alfa and ribavirin, may cause birth defects or death of your unborn baby. If you are pregnant or your sexual partner is pregnant or plans to become pregnant, do not take these medicines. You or your sexual partner should not become pregnant while taking VICTRELIS, peginterferon alfa, and ribavirin combination therapy and for 6 months after treatment is over.
-
- Females and males must use 2 effective forms of birth control during treatment and for 6 months after treatment with VICTRELIS, peginterferon alfa, and ribavirin combination therapy. Hormonal forms of birth control such as implants, injections, vaginal rings, and some birth control pills may not work during treatment with VICTRELIS. The use of certain types of birth control pills may be acceptable. Talk to your healthcare provider about forms of birth control that may be used during this time.
- Females must have a pregnancy test before starting treatment with VICTRELIS, peginterferon alfa, and ribavirin combination therapy, every month while being treated, and every month for 6 months after treatment with VICTRELIS, peginterferon alfa, and ribavirin combination therapy is over.
- If you or your female sexual partner becomes pregnant while taking VICTRELIS, peginterferon alfa, and ribavirin combination therapy or within 6 months after you stop taking these medicines, tell your healthcare provider right away. You or your healthcare provider should contact the Ribavirin Pregnancy Registry by calling 1-800-593-2214. The Ribavirin Pregnancy Registry collects information about what happens to mothers and their babies if the mother takes ribavirin while she is pregnant.
- Do not take VICTRELIS alone to treat chronic hepatitis C infection. VICTRELIS must be used with peginterferon alfa and ribavirin to treat chronic hepatitis C infection.
What is VICTRELIS?
VICTRELIS is a prescription medicine used with the medicines peginterferon alfa and ribavirin to treat long-lasting (chronic) hepatitis C genotype 1 infection in adults with stable (compensated) liver disease who have not been treated before or who have failed previous treatment.
It is not known if VICTRELIS is safe and effective in children under 18 years of age.
Who should not take VICTRELIS?
See "What is the most important information I should know about VICTRELIS?"
Do not take VICTRELIS if you:
- have had an allergic reaction to boceprevir or any of the ingredients in VICTRELIS. See the end of this Medication Guide for a complete list of ingredients in VICTRELIS.
- take certain medicines. VICTRELIS may cause serious side effects when taken with certain medicines. Read the section "What should I tell my healthcare provider before taking VICTRELIS?"
Talk to your healthcare provider before taking VICTRELIS if you have any of the conditions listed below.
What should I tell my healthcare provider before taking VICTRELIS?
Before and while you take VICTRELIS, tell your healthcare provider if you:
- have certain blood disorders such as low red blood cell count (anemia), a certain type of low white blood cell count (neutropenia) or combination of low platelet, red and white blood cell counts (pancytopenia)
- have liver failure
- have liver problems other than hepatitis C infection
- have had a liver or other organ transplant
- plan to have surgery
- have any other medical condition
- are breastfeeding. It is not known if VICTRELIS passes into breast milk. You and your healthcare provider should decide if you will take VICTRELIS or breastfeed. You should not do both.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
VICTRELIS and other medicines may affect each other. This can cause you to have too much or not enough VICTRELIS or your other medicines in your body, affecting the way VICTRELIS and your other medicines work, or causing side effects that can be serious or life-threatening. Do not start taking a new medicine without telling your healthcare provider or pharmacist.
Do not take VICTRELIS if you take a medicine that contains:
- alfuzosin hydrochloride (UROXATRAL®)
- anti-seizure medicines:
- carbamazepine (CARBATROL®, EPITOL®, EQUETRO®, TEGRETOL®, TEGRETOL® XR, TERIL™)
- phenobarbital
- phenytoin (DILANTIN®)
- cisapride (PROPULSID®)
- drospirenone-containing birth control medicines, including:
- YAZ®, YASMIN®, ZARAH®, OCELLA®, GIANVI®, BEYAZ®, ANGELIQ®, LORYNA®, SYEDA®, SAFYRAL®
- doxazosin (CARDURA®, CARDURA® XL)
- ergot-containing medicines, including:
- dihydroergotamine mesylate (D.H.E. 45®, MIGRANAL®)
- ergonovine and methylergonovine (ERGOTRATE®, METHERGINE®)
- ergotamine tartrate (CAFERGOT®, MIGERGOT®, ERGOMAR®, ERGOSTAT, MEDIHALER ERGOTAMINE, WIGRAINE, WIGRETTES)
- lovastatin (ADVICOR®, ALTOPREV®, MEVACOR®)
- lurasidone (LATUDA®)
- midazolam, when taken by mouth
- pimozide (ORAP®)
- rifampin (RIFADIN®, RIFAMATE®, RIFATER®, RIMACTANE)
- sildenafil (REVATIO®), when used for treating lung problems
- silodosin (RAPAFLO®)
- simvastatin (SIMCOR®, VYTORIN®, JUVISYNC®, ZOCOR®)
- St. John's Wort (Hypericum perforatum) or products containing St. John's Wort
- tadalafil (ADCIRCA®), when used for treating lung problems
- tamsulosin (FLOMAX®, JALYN®)
- triazolam (HALCION®)
Tell your healthcare provider if you are taking or starting to take any of these medicines:
- atazanavir (REYATAZ®)
- clarithromycin (BIAXIN®, BIAXIN® XL, PREVPAC®)
- darunavir (PREZISTA®)
- dexamethasone
- efavirenz (SUSTIVA®, ATRIPLA®)
- etravirine (INTELENCE®)
- itraconazole (ONMEL®,SPORANOX®)
- ketoconazole (NIZORAL®)
- lopinavir (KALETRA®)
- posaconazole (NOXAFIL®)
- rifabutin (MYCOBUTIN®)
- ritonavir (NORVIR®, KALETRA®)
- voriconazole (VFEND®)
Your healthcare provider may need to monitor your therapy more closely if you take VICTRELIS with the following medicines. Talk to your healthcare provider if you are taking or starting to take a medicine that contains:
- alprazolam (XANAX®)
- amiodarone (CORDARONE®, NEXTERONE®, PACERONE®)
- amlodipine (AMTURNIDE®, NORVASC®, TEKAMLO®)
- atorvastatin (LIPITOR®)
- bepridil (VASCOR)
- bosentan (TRACLEER®)
- budesonide (PULMICORT®, PULMICORT FLEXHALER®, RHINOCORT®, PULMICORT RESPULES®, SYMBICORT®)
- buprenorphine (BUTRANS®, BUPRENEX®, SUBOXONE®, SUBUTEX®)
- cyclosporine (GENGRAF ®, NEORAL®, SANDIMMUNE®)
- desipramine (NORPRAMIN®)
- digoxin (LANOXIN®)
- diltiazem (CARDIZEM®, CARDIZEM® CD, CARDIZEM® LA, CARTIA XT®, DILACOR XR®, DILT-CD, DILTZAC, TIAZAC®, TAZTIA XT®)
- escitalopram (LEXAPRO®)
- felodipine (PLENDIL®)
- fluticasone (VERAMYST®, FLOVENT® HFA, FLOVENT® DISKUS, ADVAIR® HFA, ADVAIR DISKUS®)
- hormonal forms of birth control, including birth control pills, vaginal rings, implants and injections
- hormone replacement therapy
- methadone (METHADOSE®,DOLOPHINE®)
- naloxone
- nifedipine (PROCARDIA®, ADALAT® CC, PROCARDIA XL®, AFEDITAB® CR)
- nicardipine (CARDENE® SR, CARDENE®)
- nisoldipine (SULAR®)
- omeprazole
- prednisone
- oral and IV prednisolone
- pravastatin (PRAVACHOL®)
- propafenone (RYTHMOL®, RYTHMOL® SR)
- quinidine
- raltegravir (ISENTRESS®)
- salmeterol (ADVAIR® HFA, ADVAIR DISKUS®, SEREVENT®)
- sildenafil (VIAGRA®), when used for treating erectile dysfunction
- sirolimus (RAPAMUNE®)
- tacrolimus (PROGRAF®)
- tadalafil (CIALIS®), when used for treating erectile dysfunction
- colchicine (COLCRYS®, Probenecid and Colchicine, COL-Probenecid)
- trazodone (OLEPTRO ®)
- vardenafil (STAXYN®, LEVITRA®), when used for treating erectile dysfunction
- verapamil (CALAN®, CALAN® SR, COVERA-HS®, TARKA®, VERELAN®, VERELAN® PM
- warfarin (COUMADIN®, JANTOVEN®)
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take VICTRELIS?
- Take VICTRELIS exactly as your healthcare provider tells you to take it.
- Your healthcare provider will tell you how much to take and when to take it.
- Take VICTRELIS with food (a meal or light snack).
- VICTRELIS is packaged into single daily-use bottles. Each bottle has your entire day's worth of medicine. Make sure you are taking the correct amount of medicine each time.
- If you miss a dose of VICTRELIS and it is less than 2 hours before the next dose, the missed dose should be skipped.
- If you miss a dose of VICTRELIS and it is 2 or more hours before the next dose, take the missed dose with food. Take your next dose at your normal time and continue the normal dosing schedule.
- Do not double the next dose. If you have questions about what to do, call your healthcare provider.
- Your healthcare provider should do blood tests before you start treatment, at weeks 2, 4, 8, 12 and 24, and at other times as needed during treatment, to see how well the medicines are working and to check for side effects.
- If you take too much VICTRELIS, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of VICTRELIS?
VICTRELIS may cause serious side effects, including:
See "What is the most important information I should know about VICTRELIS?"
Serious allergic reactions. Serious allergic reactions can happen and may become severe requiring treatment in a hospital. Tell your healthcare provider right away if you have any of these symptoms:
- itching
- hives
- swelling of your face, eyes, lips, tongue, or throat
- trouble breathing or swallowing
Blood problems. VICTRELIS can affect your bone marrow and cause low red blood cell, low white blood cell, and low platelet counts. In some people, these blood counts may fall to dangerously low levels. If your blood cell counts become very low, you can get anemia, infections, or bleed easily.
The most common side effects of VICTRELIS in combination with peginterferon alfa and ribavirin include:
- tiredness
- nausea
- headache
- change in taste
Additionally, while the medicine has been on the market, serious skin reactions, including blistering or peeling of the skin, and infections of the blood and pneumonia have been reported. Tell your healthcare provider about any side effect that bothers you or that does not go away.
These are not all the possible side effects of VICTRELIS. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store VICTRELIS?
- Store VICTRELIS capsules in a refrigerator at 36 to 46°F (2 to 8°C). Safely throw away refrigerated VICTRELIS after the expiration date.
- VICTRELIS capsules may also be stored at room temperature up to 77°F (25°C) for 3 months.
- Keep VICTRELIS in a tightly closed container and away from heat.
Keep VICTRELIS and all medicines out of the reach of children.
General information about the safe and effective use of VICTRELIS.
It is not known if treatment with VICTRELIS will prevent you from infecting another person with the hepatitis C virus during your treatment. Talk with your healthcare provider about ways to prevent spreading the hepatitis C virus.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide.
Do not use VICTRELIS for a condition for which it was not prescribed. Do not give VICTRELIS to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about VICTRELIS. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about VICTRELIS that is written for health professionals.
For more information, go to www.victrelis.com or call 1-877-888-4231.
What are the ingredients in VICTRELIS?
Active ingredients: boceprevir
Inactive ingredients: sodium lauryl sulfate, microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, pre-gelatinized starch, and magnesium stearate.
Red capsule shell: gelatin, titanium dioxide, D&C Yellow #10, FD&C Blue #1, FD&C Red #40.
Yellow capsule shell: gelatin, titanium dioxide, D&C Yellow #10, FD&C Red #40, FD&C Yellow #6.
Red printing ink: shellac, red iron oxide. Yellow printing ink: shellac, titanium dioxide, povidone, D&C Yellow #10 Aluminum Lake.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Manufactured for:
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ 08889, USA
Manufactured by:
MSD International GmbH (Singapore Branch) Singapore 638414, Singapore
Revised: 01/2017
For patent information: www.merck.com/product/patent/home.html
Trademarks depicted herein are the property of their respective owners.
Copyright © 2011-2017 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved.
usmg-mk3034-c-1701r039
PRINCIPAL DISPLAY PANEL - 200 mg Capsule Carton
NDC 0085-0314-02
Victrelis®
(boceprevir) Capsules
REFRIGERATE
200 mg
Usual Dosage: Four capsules by mouth three times daily
(every 7 to 9 hours) with food. See package insert.
ATTENTION PHARMACIST: Dispense the enclosed Medication Guide to each patient.
Each capsule contains 200 mg boceprevir.
Rx only
28 bottles x 12 capsules
| VICTRELIS
boceprevir capsule |
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
| Labeler - Merck Sharp & Dohme Corp. (001317601) |