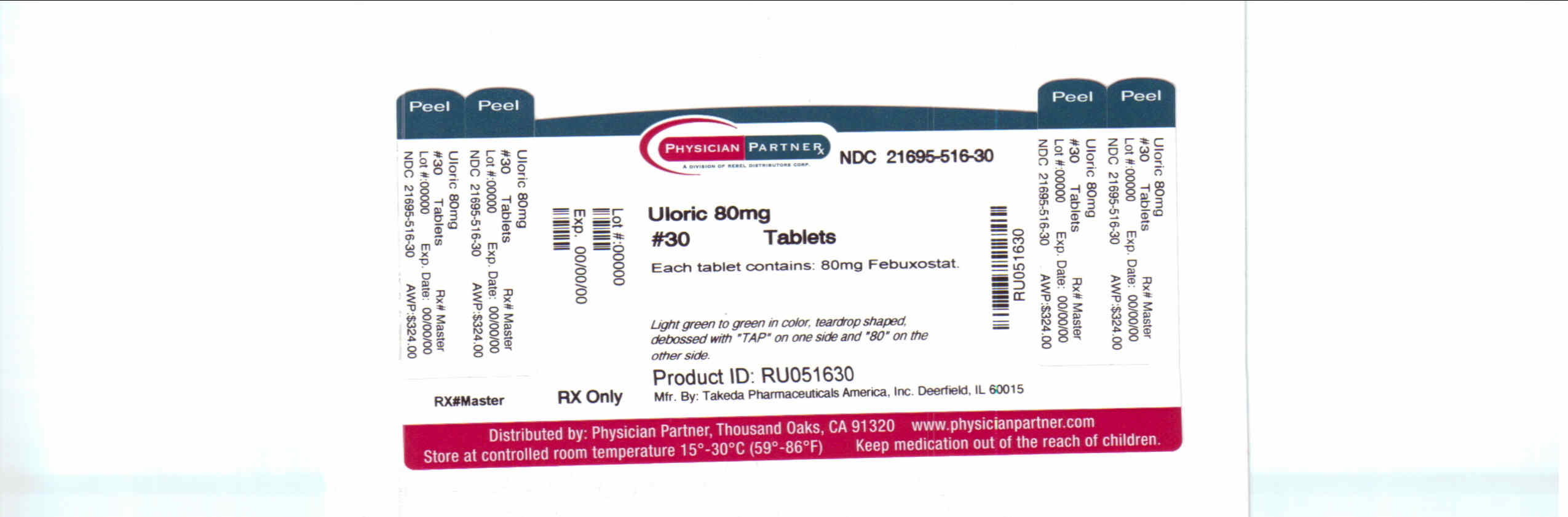
Label: ULORIC- febuxostat tablet

-
Contains inactivated NDC Code(s)
NDC Code(s): 21695-516-30 - Packager: Rebel Distributors Corp
- This is a repackaged label.
- Source NDC Code(s): 64764-677
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated December 27, 2010
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These HIGHLIGHTS do not include all the information needed to use ULORIC safely and effectively. See full prescribing information for ULORIC.
ULORIC (febuxostat) tablet for oral use
Initial U.S. Approval: 2009INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- ULORIC is recommended at 40 mg or 80 mg once daily. The recommended starting dose of ULORIC is 40 mg once daily. For patients who do not achieve a serum uric acid (sUA) less than 6 mg per dL after 2 weeks with 40 mg, ULORIC 80 mg is recommended. (2.1)
- ULORIC can be administered without regard to food or antacid use. (2.1)
- No dose adjustment is necessary when administering ULORIC to patients with mild to moderate renal or hepatic impairment. (2.2)
DOSAGE FORMS AND STRENGTHS
Tablet: 40 mg, 80 mg. (3)
CONTRAINDICATIONS
ULORIC is contraindicated in patients being treated with azathioprine, mercaptopurine, or theophylline. (4)
WARNINGS AND PRECAUTIONS
- Gout Flare: An increase in gout flares is frequently observed during initiation of anti-hyperuricemic agents, including ULORIC. If a gout flare occurs during treatment, ULORIC need not be discontinued. Prophylactic therapy (i.e., non-steroidal anti-inflammatory drug (NSAID) or colchicine upon initiation of treatment) may be beneficial for up to six months. (2.4, 5.1)
- Cardiovascular Events: A higher rate of cardiovascular thromboembolic events was observed in patients treated with ULORIC than allopurinol in clinical trials. Monitor for signs and symptoms of MI and stroke. (5.2)
- Liver Enzyme Elevation: Transaminase elevations have been observed in ULORIC-treated patients. Monitor liver function tests periodically. (5.3)
ADVERSE REACTIONS
Adverse reactions occurring in at least 1% of ULORIC-treated patients, and, at least 0.5% greater than placebo, are liver function abnormalities, nausea, arthralgia, and rash. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals at 1-877-825-3327 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
Concomitant administration of ULORIC with XO substrate drugs, azathioprine, mercaptopurine, or theophylline could increase plasma concentrations of these drugs resulting in severe toxicity. (7)
USE IN SPECIFIC POPULATIONS
- There is insufficient data in patients with severe renal impairment. No studies have been conducted in patients with severe hepatic impairment. Caution should be exercised in these patients. (8.6, 8.7)
- No studies have been conducted in patients with secondary hyperuricemia (including patients being treated for Lesch-Nyhan syndrome or malignant disease, or in organ transplant recipients); therefore, ULORIC is not recommended for use in these patients. (8.8)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2009
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
2.2 Special Populations
2.3 Uric Acid Level
2.4 Gout Flares
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Gout Flare
5.2 Cardiovascular Events
5.3 Liver Enzyme Elevations
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Xanthine Oxidase Substrate Drugs
7.2 Cytotoxic Chemotherapy Drugs
7.3 In Vivo Drug Interaction Studies
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
8.8 Secondary Hyperuricemia
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Management of Hyperuricemia in Gout
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 General Information
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
For treatment of hyperuricemia in patients with gout, ULORIC is recommended at 40 mg or 80 mg once daily.
The recommended starting dose of ULORIC is 40 mg once daily. For patients who do not achieve a serum uric acid (sUA) less than 6 mg per dL after 2 weeks with 40 mg, ULORIC 80 mg is recommended.
ULORIC can be taken without regard to food or antacid use [see Clinical Pharmacology (12.3)].
2.2 Special Populations
No dose adjustment is necessary when administering ULORIC in patients with mild to moderate renal impairment [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)]. The recommended starting dose of ULORIC is 40 mg once daily. For patients who do not achieve a sUA less than 6 mg per dL after 2 weeks with 40 mg, ULORIC 80 mg is recommended.
No dose adjustment is necessary in patients with mild to moderate hepatic impairment [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
2.3 Uric Acid Level
Testing for the target serum uric acid level of less than 6 mg per dL may be performed as early as 2 weeks after initiating ULORIC therapy.
2.4 Gout Flares
Gout flares may occur after initiation of ULORIC due to changing serum uric acid levels resulting in mobilization of urate from tissue deposits. Flare prophylaxis with a non-steroidal anti-inflammatory drug (NSAID) or colchicine is recommended upon initiation of ULORIC. Prophylactic therapy may be beneficial for up to six months [see Clinical Studies (14.1)].
If a gout flare occurs during ULORIC treatment, ULORIC need not be discontinued. The gout flare should be managed concurrently, as appropriate for the individual patient [see Warnings and Precautions (5.1)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
ULORIC is contraindicated in patients being treated with azathioprine, mercaptopurine, or theophylline [see Drug Interactions (7)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Gout Flare
After initiation of ULORIC, an increase in gout flares is frequently observed. This increase is due to reduction in serum uric acid levels resulting in mobilization of urate from tissue deposits.
In order to prevent gout flares when ULORIC is initiated, concurrent prophylactic treatment with an NSAID or colchicine is recommended [see Dosage and Administration (2.4)].
5.2 Cardiovascular Events
In the randomized controlled studies, there was a higher rate of cardiovascular thromboembolic events (cardiovascular deaths, non-fatal myocardial infarctions, and non-fatal strokes) in patients treated with ULORIC [0.74 per 100 P-Y (95% CI 0.36-1.37)] than allopurinol [0.60 per 100 P-Y (95% CI 0.16-1.53)] [see Adverse Reactions (6.1)]. A causal relationship with ULORIC has not been established. Monitor for signs and symptoms of myocardial infarction (MI) and stroke.
5.3 Liver Enzyme Elevations
During randomized controlled studies, transaminase elevations greater than 3 times the upper limit of normal (ULN) were observed (AST: 2%, 2%, and ALT: 3%, 2% in ULORIC and allopurinol-treated patients, respectively). No dose-effect relationship for these transaminase elevations was noted. Laboratory assessment of liver function is recommended at, for example, 2 and 4 months following initiation of ULORIC and periodically thereafter.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 2757 subjects with hyperuricemia and gout were treated with ULORIC 40 mg or 80 mg daily in clinical studies. For ULORIC 40 mg, 559 patients were treated for ≥ 6 months. For ULORIC 80 mg, 1377 subjects were treated for ≥ 6 months, 674 patients were treated for ≥ 1 year and 515 patients were treated for ≥ 2 years.
Most Common Adverse Reactions
In three randomized, controlled clinical studies (Studies 1, 2 and 3), which were 6 to 12 months in duration, the following adverse reactions were reported by the treating physician as related to study drug. Table 1 summarizes adverse reactions reported at a rate of at least 1% in ULORIC treatment groups and at least 0.5% greater than placebo.
Table 1: Adverse Reactions Occurring in ≥ 1% of ULORIC-Treated Patients and at Least 0.5% Greater than Seen in Patients Receiving Placebo in Controlled Studies Adverse Reactions Placebo ULORIC allopurinol* (N=134) 40 mg daily
(N=757)80 mg daily
(N=1279)
(N=1277)- *
- Of the subjects who received allopurinol, 10 received 100 mg, 145 received 200 mg, and 1122 received 300 mg, based on level of renal impairment.
Liver Function Abnormalities 0.7% 6.6% 4.6% 4.2% Nausea 0.7% 1.1% 1.3% 0.8% Arthralgia 0% 1.1% 0.7% 0.7% Rash 0.7% 0.5% 1.6% 1.6% The most common adverse reaction leading to discontinuation from therapy was liver function abnormalities in 1.8% of ULORIC 40 mg, 1.2% of ULORIC 80 mg, and in 0.9% of allopurinol-treated subjects.
In addition to the adverse reactions presented in Table 1, dizziness was reported in more than 1% of ULORIC-treated subjects although not at a rate more than 0.5% greater than placebo.
Less Common Adverse Reactions
In phase 2 and 3 clinical studies the following adverse reactions occurred in less than 1% of subjects and in more than one subject treated with doses ranging from 40 mg to 240 mg of ULORIC. This list also includes adverse reactions (less than 1% of subjects) associated with organ systems from Warnings and Precautions.
Blood and Lymphatic System Disorders: anemia, idiopathic thrombocytopenic purpura, leukocytosis/leukopenia, neutropenia, pancytopenia, splenomegaly, thrombocytopenia.
Cardiac Disorders: angina pectoris, atrial fibrillation/flutter, cardiac murmur, ECG abnormal, palpitations, sinus bradycardia, tachycardia.
Ear and Labyrinth Disorders: deafness, tinnitus, vertigo.
Eye Disorders: vision blurred.
Gastrointestinal Disorders: abdominal distention, abdominal pain, constipation, dry mouth, dyspepsia, flatulence, frequent stools, gastritis, gastroesophageal reflux disease, gastrointestinal discomfort, gingival pain, haematemesis, hyperchlorhydria, hematochezia, mouth ulceration, pancreatitis, peptic ulcer, vomiting.
General Disorders and Administration Site Conditions: asthenia, chest pain/discomfort, edema, fatigue, feeling abnormal, gait disturbance, influenza-like symptoms, mass, pain, thirst.
Hepatobiliary Disorders: cholelithiasis/cholecystitis, hepatic steatosis, hepatitis, hepatomegaly.
Immune System Disorder: hypersensitivity.
Infections and Infestations: herpes zoster.
Procedural Complications: contusion.
Metabolism and Nutrition Disorders: anorexia, appetite decreased/increased, dehydration, diabetes mellitus, hypercholesterolemia, hyperglycemia, hyperlipidemia, hypertriglyceridemia, hypokalemia, weight decreased/increased.
Musculoskeletal and Connective Tissue Disorders: arthritis, joint stiffness, joint swelling, muscle spasms/twitching/tightness/weakness, musculoskeletal pain/stiffness, myalgia.
Nervous System Disorders: altered taste, balance disorder, cerebrovascular accident, Guillain-Barré syndrome, headache, hemiparesis, hypoesthesia, hyposmia, lacunar infarction, lethargy, mental impairment, migraine, paresthesia, somnolence, transient ischemic attack, tremor.
Psychiatric Disorders: agitation, anxiety, depression, insomnia, irritability, libido decreased, nervousness, panic attack, personality change.
Renal and Urinary Disorders: hematuria, nephrolithiasis, pollakiuria, proteinuria, renal failure, renal insufficiency, urgency, incontinence.
Reproductive System and Breast Changes: breast pain, erectile dysfunction, gynecomastia.
Respiratory, Thoracic and Mediastinal Disorders: bronchitis, cough, dyspnea, epistaxis, nasal dryness, paranasal sinus hypersecretion, pharyngeal edema, respiratory tract congestion, sneezing, throat irritation, upper respiratory tract infection.
Skin and Subcutaneous Tissue Disorders: alopecia, angio-edema, dermatitis, dermographism, ecchymosis, eczema, hair color changes, hair growth abnormal, hyperhidrosis, peeling skin, petechiae, photosensitivity, pruritus, purpura, skin discoloration/altered pigmentation, skin lesion, skin odor abnormal, urticaria.
Vascular Disorders: flushing, hot flush, hypertension, hypotension.
Laboratory Parameters: activated partial thromboplastin time prolonged, creatine increased, bicarbonate decreased, sodium increased, EEG abnormal, glucose increased, cholesterol increased, triglycerides increased, amylase increased, potassium increased, TSH increased, platelet count decreased, hematocrit decreased, hemoglobin decreased, MCV increased, RBC decreased, creatinine increased, blood urea increased, BUN/creatinine ratio increased, creatine phosphokinase (CPK) increased, alkaline phosphatase increased, LDH increased, PSA increased, urine output increased/decreased, lymphocyte count decreased, neutrophil count decreased, WBC increased/decreased, coagulation test abnormal, low density lipoprotein (LDL) increased, prothrombin time prolonged, urinary casts, urine positive for white blood cells and protein.
Cardiovascular Safety
Cardiovascular events and deaths were adjudicated to one of the pre-defined endpoints from the Anti-Platelet Trialists' Collaborations (APTC) (cardiovascular death, non-fatal myocardial infarction, and non-fatal stroke) in the randomized controlled and long-term extension studies. In the Phase 3 randomized controlled studies, the incidences of adjudicated APTC events per 100 patient-years of exposure were: Placebo 0 (95% CI 0.00-6.16), ULORIC 40 mg 0 (95% CI 0.00-1.08), ULORIC 80 mg 1.09 (95% CI 0.44-2.24), and allopurinol 0.60 (95% CI 0.16-1.53).
In the long-term extension studies, the incidences of adjudicated APTC events were: ULORIC 80 mg 0.97 (95% CI 0.57-1.56), and allopurinol 0.58 (95% CI 0.02-3.24).
Overall, a higher rate of APTC events was observed in ULORIC than in allopurinol-treated patients. A causal relationship with ULORIC has not been established. Monitor for signs and symptoms of MI and stroke.
-
7 DRUG INTERACTIONS
7.1 Xanthine Oxidase Substrate Drugs
ULORIC is an XO inhibitor. Drug interaction studies of ULORIC with drugs that are metabolized by XO (e.g., theophylline, mercaptopurine, azathioprine) have not been conducted. Inhibition of XO by ULORIC may cause increased plasma concentrations of these drugs leading to toxicity [see Clinical Pharmacology (12.3)]. ULORIC is contraindicated in patients being treated with azathioprine, mercaptopurine, or theophylline [see Contraindications (4)].
7.2 Cytotoxic Chemotherapy Drugs
Drug interaction studies of ULORIC with cytotoxic chemotherapy have not been conducted. No data are available regarding the safety of ULORIC during cytotoxic chemotherapy.
7.3 In Vivo Drug Interaction Studies
Based on drug interaction studies in healthy subjects, ULORIC does not have clinically significant interactions with colchicine, naproxen, indomethacin, hydrochlorothiazide, warfarin or desipramine [see Clinical Pharmacology (12.3)]. Therefore, ULORIC may be used concomitantly with these medications.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C: There are no adequate and well-controlled studies in pregnant women. ULORIC should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Febuxostat was not teratogenic in rats and rabbits at oral doses up to 48 mg per kg (40 and 51 times the human plasma exposure at 80 mg per day for equal body surface area, respectively) during organogenesis. However, increased neonatal mortality and a reduction in the neonatal body weight gain were observed when pregnant rats were treated with oral doses up to 48 mg per kg (40 times the human plasma exposure at 80 mg per day) during organogenesis and through lactation period.
8.3 Nursing Mothers
Febuxostat is excreted in the milk of rats. It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when ULORIC is administered to a nursing woman.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients under 18 years of age have not been established.
8.5 Geriatric Use
No dose adjustment is necessary in elderly patients. Of the total number of subjects in clinical studies of ULORIC, 16 percent were 65 and over, while 4 percent were 75 and over. Comparing subjects in different age groups, no clinically significant differences in safety or effectiveness were observed but greater sensitivity of some older individuals cannot be ruled out. The Cmax and AUC24 of febuxostat following multiple oral doses of ULORIC in geriatric subjects (≥ 65 years) were similar to those in younger subjects (18-40 years) [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
No dose adjustment is necessary in patients with mild or moderate renal impairment (Clcr 30-89 mL per min). The recommended starting dose of ULORIC is 40 mg once daily. For patients who do not achieve a sUA less than 6 mg per dL after 2 weeks with 40 mg, ULORIC 80 mg is recommended.
There are insufficient data in patients with severe renal impairment (Clcr less than 30 mL per min); therefore, caution should be exercised in these patients [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment is necessary in patients with mild or moderate hepatic impairment (Child-Pugh Class A or B). No studies have been conducted in patients with severe hepatic impairment (Child-Pugh Class C); therefore, caution should be exercised in these patients [see Clinical Pharmacology (12.3)].
8.8 Secondary Hyperuricemia
No studies have been conducted in patients with secondary hyperuricemia (including organ transplant recipients); ULORIC is not recommended for use in patients whom the rate of urate formation is greatly increased (e.g., malignant disease and its treatment, Lesch-Nyhan syndrome). The concentration of xanthine in urine could, in rare cases, rise sufficiently to allow deposition in the urinary tract.
- 10 OVERDOSAGE
-
11 DESCRIPTION
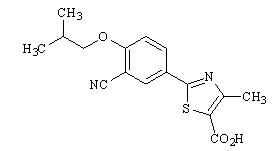
ULORIC (febuxostat) is a xanthine oxidase inhibitor. The active ingredient in ULORIC is 2-[3-cyano-4-(2-methylpropoxy) phenyl]-4-methylthiazole-5-carboxylic acid, with a molecular weight of 316.38. The empirical formula is C16H16N2O3S.
The chemical structure is:
Febuxostat is a non-hygroscopic, white crystalline powder that is freely soluble in dimethylformamide; soluble in dimethylsulfoxide; sparingly soluble in ethanol; slightly soluble in methanol and acetonitrile; and practically insoluble in water. The melting range is 205°C to 208°C.
ULORIC tablets for oral use contain the active ingredient, febuxostat, and are available in two dosage strengths, 40 mg and 80 mg. Inactive ingredients include lactose monohydrate, microcrystalline cellulose, hydroxypropyl cellulose, sodium croscarmellose, silicon dioxide and magnesium stearate. ULORIC tablets are coated with Opadry II, green.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
ULORIC, a xanthine oxidase inhibitor, achieves its therapeutic effect by decreasing serum uric acid. ULORIC is not expected to inhibit other enzymes involved in purine and pyrimidine synthesis and metabolism at therapeutic concentrations.
12.2 Pharmacodynamics
Effect on Uric Acid and Xanthine Concentrations: In healthy subjects, ULORIC resulted in a dose dependent decrease in 24-hour mean serum uric acid concentrations, and an increase in 24-hour mean serum xanthine concentrations. In addition, there was a decrease in the total daily urinary uric acid excretion. Also, there was an increase in total daily urinary xanthine excretion. Percent reduction in 24-hour mean serum uric acid concentrations was between 40% to 55% at the exposure levels of 40 mg and 80 mg daily doses.
12.3 Pharmacokinetics
In healthy subjects, maximum plasma concentrations (Cmax) and AUC of febuxostat increased in a dose proportional manner following single and multiple doses of 10 mg to 120 mg. There is no accumulation when therapeutic doses are administered every 24 hours. Febuxostat has an apparent mean terminal elimination half-life (t1/2) of approximately 5 to 8 hours. Febuxostat pharmacokinetic parameters for patients with hyperuricemia and gout estimated by population pharmacokinetic analyses were similar to those estimated in healthy subjects.
Absorption: The absorption of radiolabeled febuxostat following oral dose administration was estimated to be at least 49% (based on total radioactivity recovered in urine). Maximum plasma concentrations of febuxostat occurred between 1 to 1.5 hours post-dose. After multiple oral 40 mg and 80 mg once daily doses, Cmax is approximately 1.6 ± 0.6 mcg per mL (N=30), and 2.6 ± 1.7 mcg per mL (N=227), respectively. Absolute bioavailability of the febuxostat tablet has not been studied.
Following multiple 80 mg once daily doses with a high fat meal, there was a 49% decrease in Cmax and an 18% decrease in AUC, respectively. However, no clinically significant change in the percent decrease in serum uric acid concentration was observed (58% fed vs. 51% fasting). Thus, ULORIC may be taken without regard to food.
Concomitant ingestion of an antacid containing magnesium hydroxide and aluminum hydroxide with an 80 mg single dose of ULORIC has been shown to delay absorption of febuxostat (approximately 1 hour) and to cause a 31% decrease in Cmax and a 15% decrease in AUC∞. As AUC rather than Cmax was related to drug effect, change observed in AUC was not considered clinically significant. Therefore, ULORIC may be taken without regard to antacid use.
Distribution: The mean apparent steady state volume of distribution (Vss/F) of febuxostat was approximately 50 L (CV ~40%). The plasma protein binding of febuxostat is approximately 99.2%, (primarily to albumin), and is constant over the concentration range achieved with 40 mg and 80 mg doses.
Metabolism: Febuxostat is extensively metabolized by both conjugation via uridine diphosphate glucuronosyltransferase (UGT) enzymes including UGT1A1, UGT1A3, UGT1A9, and UGT2B7 and oxidation via cytochrome P450 (CYP) enzymes including CYP1A2, 2C8 and 2C9 and non-P450 enzymes. The relative contribution of each enzyme isoform in the metabolism of febuxostat is not clear. The oxidation of the isobutyl side chain leads to the formation of four pharmacologically active hydroxy metabolites, all of which occur in plasma of humans at a much lower extent than febuxostat.
In urine and feces, acyl glucuronide metabolites of febuxostat (~35% of the dose), and oxidative metabolites, 67M-1 (~10% of the dose), 67M-2 (~11% of the dose), and 67M-4, a secondary metabolite from 67M-1, (~14% of the dose) appeared to be the major metabolites of febuxostat in vivo.
Elimination: Febuxostat is eliminated by both hepatic and renal pathways. Following an 80 mg oral dose of 14C-labeled febuxostat, approximately 49% of the dose was recovered in the urine as unchanged febuxostat (3%), the acyl glucuronide of the drug (30%), its known oxidative metabolites and their conjugates (13%), and other unknown metabolites (3%). In addition to the urinary excretion, approximately 45% of the dose was recovered in the feces as the unchanged febuxostat (12%), the acyl glucuronide of the drug (1%), its known oxidative metabolites and their conjugates (25%), and other unknown metabolites (7%).
The apparent mean terminal elimination half-life (t1/2) of febuxostat was approximately 5 to 8 hours.
Special Populations
Pediatric Use: The pharmacokinetics of ULORIC in patients under the age of 18 years have not been studied.
Geriatric Use: The Cmax and AUC of febuxostat and its metabolites following multiple oral doses of ULORIC in geriatric subjects (≥ 65 years) were similar to those in younger subjects (18-40 years). In addition, the percent decrease in serum uric acid concentration was similar between elderly and younger subjects. No dose adjustment is necessary in geriatric patients [see Use in Specific Populations (8.5)].
Renal Impairment: Following multiple 80 mg doses of ULORIC in healthy subjects with mild (Clcr 50-80 mL per min), moderate (Clcr 30-49 mL per min) or severe renal impairment (Clcr 10-29 mL per min), the Cmax of febuxostat did not change relative to subjects with normal renal function (Clcr greater than 80 mL per min). AUC and half-life of febuxostat increased in subjects with renal impairment in comparison to subjects with normal renal function, but values were similar among three renal impairment groups. Mean febuxostat AUC values were up to 1.8 times higher in subjects with renal impairment compared to those with normal renal function. Mean Cmax and AUC values for 3 active metabolites increased up to 2- and 4-fold, respectively. However, the percent decrease in serum uric acid concentration for subjects with renal impairment was comparable to those with normal renal function (58% in normal renal function group and 55% in the severe renal function group).
No dose adjustment is necessary in patients with mild to moderate renal impairment [see Dosage and Administration (2) and Use in Specific Populations (8.6)]. The recommended starting dose of ULORIC is 40 mg once daily. For patients who do not achieve a sUA less than 6 mg per dL after 2 weeks with 40 mg, ULORIC 80 mg is recommended. There is insufficient data in patients with severe renal impairment; caution should be exercised in those patients [see Use in Specific Populations (8.6)].
ULORIC has not been studied in end stage renal impairment patients who are on dialysis.
Hepatic Impairment: Following multiple 80 mg doses of ULORIC in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment, an average of 20-30% increase was observed for both Cmax and AUC24 (total and unbound) in hepatic impairment groups compared to subjects with normal hepatic function. In addition, the percent decrease in serum uric acid concentration was comparable between different hepatic groups (62% in healthy group, 49% in mild hepatic impairment group, and 48% in moderate hepatic impairment group). No dose adjustment is necessary in patients with mild or moderate hepatic impairment. No studies have been conducted in subjects with severe hepatic impairment (Child-Pugh Class C); caution should be exercised in those patients [see Use in Specific Populations (8.7)].
Gender: Following multiple oral doses of ULORIC, the Cmax and AUC24 of febuxostat were 30% and 14% higher in females than in males, respectively. However, weight-corrected Cmax and AUC were similar between the genders. In addition, the percent decrease in serum uric acid concentrations was similar between genders. No dose adjustment is necessary based on gender.
Drug-Drug Interactions
Effect of ULORIC on Other Drugs
Xanthine Oxidase Substrate Drugs-Azathioprine, Mercaptopurine, and Theophylline: Febuxostat is an XO inhibitor. Drug interaction studies of ULORIC with drugs that are metabolized by XO (e.g., theophylline, mercaptopurine, azathioprine) have not been conducted. Inhibition of XO by ULORIC may cause increased plasma concentrations of these drugs leading to toxicity. ULORIC is contraindicated in patients being treated with azathioprine, mercaptopurine, and theophylline [see Contraindications (4) and Drug Interactions (7)].
Azathioprine and mercaptopurine undergo metabolism via three major metabolic pathways, one of which is mediated by XO. Although ULORIC drug interaction studies with azathioprine and mercaptopurine have not been conducted, concomitant administration of allopurinol [a xanthine oxidase inhibitor] with azathioprine or mercaptopurine has been reported to substantially increase plasma concentrations of these drugs. Because ULORIC is a xanthine oxidase inhibitor, it could inhibit the XO-mediated metabolism of azathioprine and mercaptopurine leading to increased plasma concentrations of azathioprine or mercaptopurine that could result in severe toxicity.
Theophylline is a CYP1A2 and XO substrate. Although no ULORIC drug interaction study with theophylline has been conducted, concomitant administration of theophylline with allopurinol, a xanthine oxidase inhibitor at doses ≥ 600 mg per day, has been reported to increase theophylline plasma concentrations. Because ULORIC is a xanthine oxidase inhibitor and theophylline is a low therapeutic index drug, ULORIC could inhibit the XO-mediated metabolism of theophylline leading to increased plasma concentrations of theophylline that could induce severe theophylline toxicity.
P450 Substrate Drugs: In vitro studies have shown that febuxostat does not inhibit P450 enzymes CYP1A2, 2C9, 2C19, 2D6, or 3A4 and it also does not induce CYP1A2, 2B6, 2C9, 2C19, or 3A4 at clinically relevant concentrations. As such, pharmacokinetic interactions between ULORIC and drugs metabolized by these CYP enzymes are unlikely.
Effect of Other Drugs on ULORIC
Febuxostat is metabolized by conjugation and oxidation via multiple metabolizing enzymes. The relative contribution of each enzyme isoform is not clear. Drug interactions between ULORIC and a drug that inhibits or induces one particular enzyme isoform is in general not expected.
In Vivo Drug Interaction Studies
Colchicine: No dose adjustment is necessary for either ULORIC or colchicine when the two drugs are co-administered. Administration of ULORIC (40 mg once daily) with colchicine (0.6 mg twice daily) resulted in an increase of 12% in Cmax and 7% in AUC24 of febuxostat. In addition, administration of colchicine (0.6 mg twice daily) with ULORIC (120 mg daily) resulted in less than 11% change in Cmax or AUC of colchicine for both AM and PM doses. These changes were not considered clinically significant.
Naproxen: No dose adjustment is necessary for ULORIC or naproxen when the two drugs are co-administered. Administration of ULORIC (80 mg once daily) with naproxen (500 mg twice daily) resulted in a 28% increase in Cmax and a 40% increase in AUC of febuxostat. The increases were not considered clinically significant. In addition, there were no significant changes in the Cmax or AUC of naproxen (less than 2%).
Indomethacin: No dose adjustment is necessary for either ULORIC or indomethacin when these two drugs are co-administered. Administration of ULORIC (80 mg once daily) with indomethacin (50 mg twice daily) did not result in any significant changes in Cmax or AUC of febuxostat or indomethacin (less than 7%).
Hydrochlorothiazide: No dose adjustment is necessary for ULORIC when co-administered with hydrochlorothiazide. Administration of ULORIC (80 mg) with hydrochlorothiazide (50 mg) did not result in any clinically significant changes in Cmax or AUC of febuxostat (less than 4%), and serum uric acid concentrations were not substantially affected.
Warfarin: No dose adjustment is necessary for warfarin when co-administered with ULORIC. Administration of ULORIC (80 mg once daily) with warfarin had no effect on the pharmacokinetics of warfarin in healthy subjects. INR and Factor VII activity were also not affected by the co-administration of ULORIC.
Desipramine: Co-administration of drugs that are CYP2D6 substrates (such as desipramine) with ULORIC are not expected to require dose adjustment. Febuxostat was shown to be a weak inhibitor of CYP2D6 in vitro and in vivo. Administration of ULORIC (120 mg once daily) with desipramine (25 mg) resulted in an increase in Cmax (16%) and AUC (22%) of desipramine, which was associated with a 17% decrease in the 2-hydroxydesipramine to desipramine metabolic ratio (based on AUC).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis: Two-year carcinogenicity studies were conducted in F344 rats and B6C3F1 mice. Increased transitional cell papilloma and carcinoma of urinary bladder was observed at 24 mg per kg (25 times the human plasma exposure at maximum recommended human dose of 80 mg per day) and 18.75 mg per kg (12.5 times the human plasma exposure at 80 mg per day) in male rats and female mice, respectively. The urinary bladder neoplasms were secondary to calculus formation in the kidney and urinary bladder.
Mutagenesis: Febuxostat showed a positive mutagenic response in a chromosomal aberration assay in a Chinese hamster lung fibroblast cell line with and without metabolic activation in vitro. Febuxostat was negative in the in vitro Ames assay and chromosomal aberration test in human peripheral lymphocytes, and L5178Y mouse lymphoma cell line, and in vivo tests in mouse micronucleus, rat unscheduled DNA synthesis and rat bone marrow cells.
13.2 Animal Toxicology and/or Pharmacology
A 12-month toxicity study in beagle dogs showed deposition of xanthine crystals and calculi in kidneys at 15 mg per kg (approximately 4 times the human plasma exposure at 80 mg per day). A similar effect of calculus formation was noted in rats in a six-month study due to deposition of xanthine crystals at 48 mg per kg (approximately 35 times the human plasma exposure at 80 mg per day).
-
14 CLINICAL STUDIES
A serum uric acid level of less than 6 mg per dL is the goal of anti-hyperuricemic therapy and has been established as appropriate for the treatment of gout.
14.1 Management of Hyperuricemia in Gout
The efficacy of ULORIC was demonstrated in three randomized, double-blind, controlled trials in patients with hyperuricemia and gout. Hyperuricemia was defined as a baseline serum uric acid level ≥ 8 mg per dL.
Study 1 randomized patients to: ULORIC 40 mg daily, ULORIC 80 mg daily, or allopurinol (300 mg daily for patients with estimated creatinine clearance (Clcr) ≥ 60 mL per min or 200 mg daily for patients with estimated Clcr ≥ 30 mL per min and ≤ 59 mL per min). The duration of Study 1 was 6 months.
Study 2 randomized patients to: placebo, ULORIC 80 mg daily, ULORIC 120 mg daily, ULORIC 240 mg daily or allopurinol (300 mg daily for patients with a baseline serum creatinine ≤ 1.5 mg per dL or 100 mg daily for patients with a baseline serum creatinine greater than 1.5 mg per dL and ≤ 2 mg per dL). The duration of Study 2 was 6 months.
Study 3, a 1-year study, randomized patients to: ULORIC 80 mg daily, ULORIC 120 mg daily, or allopurinol 300 mg daily. Subjects who completed Study 2 and Study 3 were eligible to enroll in a phase 3 long-term extension study in which subjects received treatment with ULORIC for over three years.
In all three studies, subjects received naproxen 250 mg twice daily or colchicine 0.6 mg once or twice daily for gout flare prophylaxis. In Study 1 the duration of prophylaxis was 6 months; in Study 2 and Study 3 the duration of prophylaxis was 8 weeks.
The efficacy of ULORIC was also evaluated in a 4 week dose ranging study which randomized patients to: placebo, ULORIC 40 mg daily, ULORIC 80 mg daily, or ULORIC 120 mg daily. Subjects who completed this study were eligible to enroll in a long-term extension study in which subjects received treatment with ULORIC for up to five years.
Patients in these studies were representative of the patient population for which ULORIC use is intended. Table 2 summarizes the demographics and baseline characteristics for the subjects enrolled in the studies.
Table 2: Patient Demographics and Baseline Characteristics in Study 1, Study 2 and Study 3 Male 95% Race: Caucasian 80% African American 10% Ethnicity: Hispanic or Latino 7% Alcohol User 67% Mild to Moderate Renal Insufficiency
[percent with estimated Clcr less than 90 mL per min]59% History of Hypertension 49% History of Hyperlipidemia 38% BMI ≥ 30 kg per m2 63% Mean BMI 33 kg per m2 Baseline sUA ≥ 10 mg per dL 36% Mean baseline sUA 9.7 mg per dL Experienced a gout flare in previous year 85% Serum Uric Acid Level less than 6 mg per dL at Final Visit: ULORIC 80 mg was superior to allopurinol in lowering serum uric acid to less than 6 mg per dL at the final visit. ULORIC 40 mg daily, although not superior to allopurinol, was effective in lowering serum uric acid to less than 6 mg per dL at the final visit (Table 3).
Table 3: Proportion of Patients with Serum Uric Acid Levels Less Than 6 mg per dL at Final Visit Difference in Proportion
(95% CI)Study* ULORIC
40 mg dailyULORIC
80 mg dailyallopurinol Placebo ULORIC 40 mg
vs
allopurinolULORIC 80 mg
vs
allopurinol- *
- Randomization was balanced between treatment groups, except in Study 2 in which twice as many patients were randomized to each of the active treatment groups compared to placebo.
Study 1
(6 months)
(N=2268)45% 67% 42% 3%
(-2%, 8%)25%
(20%, 30%)Study 2
(6 months)
(N=643)72% 39% 1% 33%
(26%, 42%)Study 3
(12 months)
(N=491)74% 36% 38%
(30%, 46%)In 76% of ULORIC 80 mg patients, reduction in serum uric acid levels to less than 6 mg per dL was noted by the Week 2 visit. Average serum uric acid levels were maintained at 6 mg per dL or below throughout treatment in 83% of these patients.
In all treatment groups, fewer subjects with higher baseline serum urate levels (≥ 10 mg per dL) and/or tophi achieved the goal of lowering serum uric acid to less than 6 mg per dL at the final visit; however, a higher proportion achieved a serum uric acid less than 6 mg per dL with ULORIC 80 mg than with ULORIC 40 mg or allopurinol.
Study 1 evaluated efficacy in patients with mild to moderate renal impairment (i.e., baseline estimated Clcr less than 90 mL per minute). The results in this sub-group of patients are shown in Table 4.
Table 4: Proportion of Patients with Serum Uric Acid Levels Less Than 6 mg per dL in Patients with Mild or Moderate Renal Impairment at Final Visit Difference in Proportion
(95% CI)ULORIC
40 mg daily
(N=479)ULORIC
80 mg daily
(N=503)allopurinol*
300 mg daily
(N=501)ULORIC 40 mg vs allopurinol ULORIC 80 mg vs allopurinol - *
- Allopurinol patients (n=145) with estimated Clcr ≥ 30 mL per min and Clcr ≤ 59 mL per min were dosed at 200 mg daily.
50% 72% 42% 7%
(1%, 14%)29%
(23%, 35%) - 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
[see FDA-Approved Patient Labeling (17.2)]
17.1 General Information
Patients should be advised of the potential benefits and risks of ULORIC. Patients should be informed about the potential for gout flares, elevated liver enzymes and adverse cardiovascular events after initiation of ULORIC therapy.
Concomitant prophylaxis with an NSAID or colchicine for gout flares should be considered.
Patients should be instructed to inform their healthcare professional if they develop a rash, chest pain, shortness of breath or neurologic symptoms suggesting a stroke. Patients should be instructed to inform their healthcare professional of any other medications they are currently taking with ULORIC, including over-the-counter medications.
-
PATIENT PACKAGE INSERT
Patient Information
ULORIC® (Ū – 'lor – ik)
(febuxostat) tabletsRead the Patient Information that comes with ULORIC before you start taking it and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or your treatment.
What is ULORIC?
ULORIC is a prescription medicine called a xanthine oxidase (XO) inhibitor, used to lower blood uric acid levels in adults with gout.
It is not known if ULORIC is safe and effective in children under 18 years of age.
Who should not take ULORIC?
Do not take ULORIC if you:
- take Azathioprine (Azasan®, Imuran®)
- take Mercaptopurine (Purinethol®)
- take Theophylline (Theo-24®, Elixophyllin®, Theochron®, Theolair®, Uniphyl®)
It is not known if ULORIC is safe and effective in children under 18 years of age.
What should I tell my healthcare provider before taking ULORIC?
Before taking ULORIC tell your healthcare provider about all of your medical conditions, including if you:
- have liver or kidney problems
- have a history of heart disease or stroke
- are pregnant or plan to become pregnant. It is not known if ULORIC will harm your unborn baby. Talk with your healthcare provider if you are pregnant or plan to become pregnant.
- are breast-feeding or plan to breast-feed. It is not known if ULORIC passes into your breast milk. You and your healthcare provider should decide if you should take ULORIC while breast-feeding.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. ULORIC may affect the way other medicines work, and other medicines may affect how ULORIC works.
Know the medicines you take. Keep a list of them and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take ULORIC?
- Take ULORIC exactly as your healthcare provider tells you to take it.
- ULORIC can be taken with or without food.
- ULORIC can be taken with antacids.
- Your gout may flare up when you start taking ULORIC, do not stop taking your ULORIC even if you have a flare. Your healthcare provider may give you other medicines to help prevent your gout flares.
- Your healthcare provider may do certain tests while you take ULORIC.
What are the possible side effects of ULORIC?
Heart problems. A small number of heart attacks, strokes and heart-related deaths were seen in clinical studies. It is not certain that ULORIC caused these events.
The most common side effects of ULORIC include:
- liver problems
- nausea
- gout flares
- joint pain
- rash
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of ULORIC. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to the FDA at 1-800-FDA-1088.
How should I store ULORIC?
Store ULORIC between 59°F - 86°F (15°C - 30°C).
Keep ULORIC out of the light.
Keep ULORIC and all medicines out of the reach of children.
General information about the safe and effective use of ULORIC
Medicines are sometimes prescribed for purposes other than those listed in a patient information leaflet. Do not use ULORIC for a condition for which it was not prescribed. Do not give ULORIC to other people, even if they have the same symptoms that you have. It may harm them.
This patient information leaflet summarizes the most important information about ULORIC. If you would like more information about ULORIC talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about ULORIC that is written for health professionals. For more information go to www.uloric.com, or call 1-877-825-3327.
What are the ingredients in ULORIC?
Active Ingredient: febuxostat
Inactive ingredients include: lactose monohydrate, microcrystalline cellulose, hydroxypropyl cellulose, sodium croscarmellose, silicon dioxide, magnesium stearate, and Opadry II, green
Distributed by
Takeda Pharmaceuticals America, Inc.
Deerfield, IL 60015U.S. Patent Nos. - 6,225,474; 7,361,676; 5,614,520.
ULORIC® is a registered trademark of Teijin Pharma Limited and used under license by Takeda Pharmaceuticals America, Inc.
All other trademark names are the property of their respective owners
©2009 Takeda Pharmaceuticals America, Inc.
PI1114 R1
February 2009Repackaged by Rebel Distributors Corp
Thousand Oaks, CA 91320
- MEDICATION GUIDE
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
ULORIC
febuxostat tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:21695-516(NDC:64764-677) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength febuxostat (UNII: 101V0R1N2E) (febuxostat - UNII:101V0R1N2E) febuxostat 80 mg Inactive Ingredients Ingredient Name Strength lactose monohydrate (UNII: EWQ57Q8I5X) cellulose, microcrystalline (UNII: OP1R32D61U) hydroxypropyl cellulose (UNII: RFW2ET671P) croscarmellose sodium (UNII: M28OL1HH48) silicon dioxide (UNII: ETJ7Z6XBU4) magnesium stearate (UNII: 70097M6I30) Product Characteristics Color GREEN (light green to green) Score no score Shape TEAR (teardrop shaped) Size 14mm Flavor Imprint Code TAP;80 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:21695-516-30 30 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021856 02/13/2009 Labeler - Rebel Distributors Corp (118802834) Establishment Name Address ID/FEI Business Operations Rebel Distributors Corp 118802834 RELABEL, REPACK