ALENDRONATE SODIUM- alendronate sodium tablet
Blu Pharmaceuticals, LLC
----------
These highlights do not include all the information needed to use alendronate sodium tablets safely and effectively. See full prescribing information for alendronate sodium tablets USP.
Alendronate Sodium Tablets USP for oral use
Initial U.S. Approval: 1995
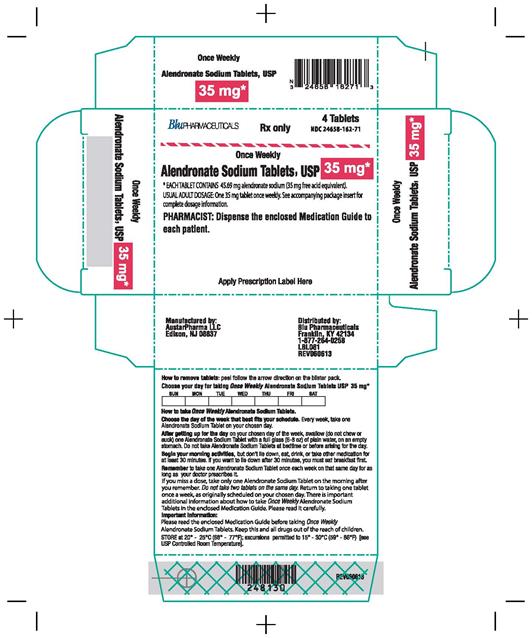

| ALENDRONATE SODIUM
alendronate sodium tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ALENDRONATE SODIUM
alendronate sodium tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Blu Pharmaceuticals, LLC (784445061) |
Revised: 5/2016
Document Id: f181186f-2659-429e-9f77-78e579ef5b25
Set id: a6dd3f4c-2a7d-4e3d-9b91-78e9f9261742
Version: 14
Effective Time: 20160517
Blu Pharmaceuticals, LLC