FERRLECIT
-
sodium ferric gluconate complex injection
sanofi-aventis U.S. LLC
----------
Ferrlecit®(sodium ferric gluconate complex in sucrose injection)
DESCRIPTION
Ferrlecit® (sodium ferric gluconate complex in sucrose injection) is a stable macromolecular complex with an apparent molecular weight on gel chromatography of 289,000 – 440,000 daltons. The macromolecular complex is negatively charged at alkaline pH and is present in solution with sodium cations. The product has a deep red color indicative of ferric oxide linkages.
The structural formula is considered to be [NaFe2O3(C6H11O7)(C12H22O11)5]n≈200.
Each sterile, single-use ampule of 5 mL of Ferrlecit for intravenous injection contains 62.5 mg (12.5 mg/mL) of elemental iron as the sodium salt of a ferric ion carbohydrate complex in an alkaline aqueous solution with approximately 20% sucrose w/v (195 mg/mL) in water for injection, pH 7.7 – 9.7.
Each mL contains 9 mg of benzyl alcohol as an inactive ingredient.
Therapeutic class: Hematinic
CLINICAL PHARMACOLOGY
Ferrlecit is used to replete the total body content of iron. Iron is critical for normal hemoglobin synthesis to maintain oxygen transport. Additionally, iron is necessary for metabolism and various enzymatic processes.
The total body iron content of an adult ranges from 2 to 4 grams. Approximately 2/3 is in hemoglobin and 1/3 is in reticuloendothelial (RE) storage (bone marrow, spleen, liver) bound to intracellular ferritin. The body highly conserves iron (daily loss of 0.03%) requiring supplementation of about 1 mg/day to replenish losses in healthy, non-menstruating adults. The etiology of iron deficiency in hemodialysis patients is varied and can include blood loss and/or increased iron utilization (e.g., from epoetin therapy). The administration of exogenous epoetin increases red blood cell production and iron utilization. The increased iron utilization and blood losses in the hemodialysis patient may lead to absolute or functional iron deficiency. Iron deficiency is absolute when hematological indicators of iron stores are low. Patients with functional iron deficiency do not meet laboratory criteria for absolute iron deficiency but demonstrate an increase in hemoglobin/ hematocrit or a decrease in epoetin dosage with stable hemoglobin/hematocrit when parenteral iron is administered.
Pharmacokinetics
Multiple sequential single dose intravenous pharmacokinetic studies were performed on 14 healthy iron-deficient volunteers. Entry criteria included hemoglobin ≥10.5 gm/dL and transferrin saturation ≤15% (TSAT) or serum ferritin value ≤20 ng/mL. In the first stage, each subject was randomized 1:1 to undiluted Ferrlecit injection of either 125 mg/hr or 62.5 mg/0.5 hr (2.1 mg/min). Five days after the first stage, each subject was re-randomized 1:1 to undiluted Ferrlecit injection of either 125 mg/7 min or 62.5 mg/4 min (>15.5 mg/min).
Peak drug levels (Cmax) varied significantly by dosage and by rate of administration with the highest Cmax observed in the regimen in which 125 mg was administered in 7 minutes (19.0 mg/L). The initial volume of distribution (VFerr) of 6 L corresponds well to calculated blood volume. VFerr did not vary by dosage or rate of administration. The terminal elimination half-life (λz-HL) for drug bound iron was approximately 1 hour. λz-HL varied by dose but not by rate of administration. The shortest value (0.85 h) occurred in the 62.5 mg/4 min regimen; the longest value (1.45 h) occurred in the 125 mg/7 min regimen. Total clearance of Ferrlecit was 3.02 to 5.35 L/h. There was no significant variation by rate of administration. The AUC for Ferrlecit bound iron varied by dose from 17.5 mg-h/L (62.5 mg) to 35.6 mg-h/L (125 mg). There was no significant variation by rate of administration. Approximately 80% of drug bound iron was delivered to transferrin as a mononuclear ionic iron species within 24 hours of administration in each dosage regimen. Direct movement of iron from Ferrlecit to transferrin was not observed. Mean peak transferrin saturation did not exceed 100% and returned to near baseline by 40 hours after administration of each dosage regimen.
Pediatrics: Single dose intravenous pharmacokinetic analyses were performed on 48 iron-deficient pediatric hemodialysis patients. Twenty-two patients received 1.5 mg/kg Ferrlecit and 26 patients received 3.0 mg/kg Ferrlecit (maximum dose 125 mg). The mean Cmax, AUC0–∞ , and terminal elimination half-life values for the 22 patients who received a 1.5 mg/kg dose were 12.9 mg/L, 95.0 mg•hr/L, and 2.0 hours, respectively. The mean Cmax, AUC0–∞ , and terminal elimination half-life values for the 26 patients who received a 3.0 mg/kg dose were 22.8 mg/L, 170.9 mg•hr/L, and 2.5 hours, respectively.
In vitro experiments have shown that less than 1% of the iron species within Ferrlecit can be dialyzed through membranes with pore sizes corresponding to 12,000 to 14,000 daltons over a period of up to 270 minutes. Human studies in renally competent patients suggest the clinical insignificance of urinary excretion.
Drug-drug Interactions: Drug-drug interactions involving Ferrlecit have not been studied. However, like other parenteral iron preparations, Ferrlecit may be expected to reduce the absorption of concomitantly administered oral iron preparations.
CLINICAL STUDIES
Two clinical studies (Studies A and B) were conducted in adults and one clinical study was conducted in pediatric patients (Study C) to assess the efficacy and safety of Ferrlecit.
Study A
Study A was a three-center, randomized, open-label study of the safety and efficacy of two doses of Ferrlecit administered intravenously to iron-deficient hemodialysis patients. The study included both a dose-response concurrent control and an historical control. Enrolled patients received a test dose of Ferrlecit (25 mg of elemental iron) and were then randomly assigned to receive Ferrlecit at cumulative doses of either 500 mg (low dose) or 1000 mg (high dose) of elemental iron. Ferrlecit was given to both dose groups in eight divided doses during sequential dialysis sessions (a period of 16 to 17 days). At each dialysis session, patients in the low-dose group received Ferrlecit 62.5 mg of elemental iron over 30 minutes, and those in the high-dose group received Ferrlecit 125 mg of elemental iron over 60 minutes. The primary endpoint was the change in hemoglobin from baseline to the last available observation through Day 40.
Eligibility for this study included chronic hemodialysis patients with a hemoglobin below 10 g/dL (or hematocrit at or below 32%) and either serum ferritin below 100 ng/mL or transferrin saturation below 18%. Exclusion criteria included significant underlying disease or inflammatory conditions or an epoetin requirement of greater than 10,000 units three times per week. Parenteral iron and red cell transfusion were not allowed for two months before the study. Oral iron and red cell transfusion were not allowed during the study for Ferrlecit-treated patients.
The historical control population consisted of 25 chronic hemodialysis patients who received only oral iron supplementation for 14 months and did not receive red cell transfusion. All patients had stable epoetin doses and hematocrit values for at least two months before initiation of oral iron therapy.
The evaluated population consisted of 39 patients in the low-dose Ferrlecit (sodium ferric gluconate complex in sucrose injection) group (50% female, 50% male; 74% white, 18% black, 5% Hispanic, 3% Asian; mean age 54 years, range 22–83 years), 44 patients in the high-dose Ferrlecit group (50% female, 48% male, 2% unknown; 75% white, 11% black, 5% Hispanic, 7% other, 2% unknown; mean age 56 years, range 20–87 years), and 25 historical control patients (68% female, 32% male; 40% white, 32% black, 20% Hispanic, 4% Asian, 4% unknown; mean age 52 years, range 25–84 years).
The mean baseline hemoglobin and hematocrit were similar between treatment and historical control patients: 9.8 g/dL and 29% and 9.6 g/dL and 29% in low- and high-dose Ferrlecit-treated patients, respectively, and 9.4 g/dL and 29% in historical control patients. Baseline serum transferrin saturation was 20% in the low-dose group, 16% in the high-dose group, and 14% in the historical control. Baseline serum ferritin was 106 ng/mL in the low-dose group, 88 ng/mL in the high-dose group, and 606 ng/mL in the historical control.
Patients in the high-dose Ferrlecit group achieved significantly higher increases in hemoglobin and hematocrit than either patients in the low-dose Ferrlecit group or patients in the historical control group (oral iron). Patients in the low-dose Ferrlecit group did not achieve significantly higher increases in hemoglobin and hematocrit than patients receiving oral iron. See Table 1.
| Mean Change from Baseline to Two Weeks After Cessation of Therapy | |||
|---|---|---|---|
| Study A | Ferrlecit | Ferrlecit | Historical Control |
| 1000 mg IV | 500 mg IV | Oral Iron | |
| (N=44) | (N=39) | (N=25) | |
| *p<0.01 versus both the 500 mg group and the historical control group. | |||
| Hemoglobin (g/dL) | 1.1* | 0.3 | 0.4 |
| Hematocrit (%) | 3.6* | 1.4 | 0.8 |
| Transferrin Saturation (%) | 8.5 | 2.8 | 6.1 |
| Serum Ferritin (ng/mL) | 199 | 132 | NA |
Study B
Study B was a single-center, non-randomized, open-label, historically-controlled, study of the safety and efficacy of variable, cumulative doses of intravenous Ferrlecit in iron-deficient hemodialysis patients. Ferrlecit administration was identical to Study A. The primary efficacy variable was the change in hemoglobin from baseline to the last available observation through Day 50.
Inclusion and exclusion criteria were identical to those of Study A as was the historical control population. Sixty-three patients were evaluated in this study: 38 in the Ferrlecit-treated group (37% female, 63% male; 95% white, 5% Asian; mean age 56 years, range 22– 84 years) and 25 in the historical control group (68% female, 32% male; 40% white, 32% black, 20% Hispanic, 4% Asian, 4% unknown; mean age 52 years, range 25–84 years).
Ferrlecit-treated patients were considered to have completed the study per protocol if they received at least eight Ferrlecit doses of either 62.5 mg or 125 mg of elemental iron. A total of 14 patients (37%) completed the study per protocol. Twelve (32%) Ferrlecit-treated patients received less than eight doses, and 12 (32%) patients had incomplete information on the sequence of dosing. Not all patients received Ferrlecit at consecutive dialysis sessions and many received oral iron during the study.
| Cumulative Ferrlecit Dose (mg of elemental iron) | 62.5 | 250 | 375 | 562.5 | 625 | 750 | 1000 | 1125 | 1187.5 |
| Patients (#) | 1 | 1 | 2 | 1 | 10 | 4 | 12 | 6 | 1 |
Baseline hemoglobin and hematocrit values were similar between the treatment and control groups, and were 9.1 g/dL and 27.3%, respectively, for Ferrlecit-treated patients. Serum iron studies were also similar between treatment and control groups, with the exception of serum ferritin, which was 606 ng/mL for historical control patients, compared to 77 ng/mL for Ferrlecit-treated patients.
In this patient population, only the Ferrlecit-treated group achieved significant increase in hemoglobin and hematocrit from baseline. This increase was significantly greater than that seen in the historical oral iron treatment group. See Table 2.
| Mean Change from Baseline to One Month After Treatment | ||
|---|---|---|
| a - p<0.05 on group comparison by the ANCOVA method. | ||
| b - p<0.001 from baseline by the paired t-test method. | ||
| Study B | Ferrlecit (N=38) | Oral Iron (N=25) |
| change | change | |
| Hemoglobin (g/dL) | 1.3a,b | 0.4 |
| Hematocrit (%) | 3.8a,b | 0.2 |
| Transferrin Saturation (%) | 6.7b | 1.7 |
| Serum Ferritin (ng/dL) | 73b | -145 |
Study C
Study C was a multicenter, randomized, open-label study of the safety and efficacy of two Ferrlecit dose regimens (1.5 mg/kg or 3.0 mg/kg of elemental iron) administered intravenously to 66 iron-deficient (transferrin saturation < 20% and/or serum ferritin < 100 ng/mL) pediatric hemodialysis patients, 6 to 15 years of age, inclusive who were receiving a stable erythropoietin dosing regimen.
Ferrlecit at a dose of 1.5 mg/kg or 3.0 mg/kg (up to a maximum dose of 125 mg of elemental iron) in 25 mL 0.9% sodium chloride was infused intravenously over 1 hour during each hemodialysis session for eight sequential dialysis sessions. Thirty-two patients received the 1.5 mg/kg dosing regimen (47% male, 53% female; 66% Caucasian, 25% Hispanic, and 3% Black, Asian, or Other; mean age 12.3 years). Thirty-four patients received the 3.0 mg/kg dosing regimen (56% male, 44% female; 77% Caucasian, 12% Hispanic, 9% Black, and 3% Other; mean age 12.0 years).
The primary endpoint was the change in hemoglobin concentration from baseline to 2 weeks after last Ferrlecit administration. Patients in both Ferrlecit dose groups had statistically significant changes from baseline in hemoglobin concentrations (Table 3). There was no significant difference between the treatment groups. Statistically significant improvements in hematocrit, transferrin saturation, serum ferritin, and reticulocyte hemoglobin concentrations compared to baseline values were observed 2 weeks after the last Ferrlecit infusion in both the 1.5 mg/kg and 3.0 mg/kg treatment groups (Table 3).
| Study C | Mean Change From Baseline to Two Weeks After Cessation of Therapy in Patients Completing Treatment | |
| 1.5 mg/kg Ferrlecit* | 3.0 mg/kg Ferrlecit | |
| (N=25) | (N=32) | |
| * p < 0.03 versus the baseline values | ||
| Hemoglobin (g/dL) | 0.8* | 0.9* |
| Hamatocrit (%) | 2.6* | 3.0* |
| Transferrin Saturation (%) | 5.5* | 10.5* |
| Serum Ferritin (ng/mL) | 192* | 314* |
| Reticulocyte Hemoglobin Content (pg) | 1.3* | 1.2* |
The increased hemoglobin concentrations were maintained at 4 weeks after the last Ferrlecit infusion in both the 1.5 mg/kg and the 3.0 mg/kg Ferrlecit dose treatment groups.
INDICATIONS AND USAGE
Ferrlecit (sodium ferric gluconate complex in sucrose injection) is indicated for treatment of iron deficiency anemia in adult patients and in pediatric patients age 6 years and older undergoing chronic hemodialysis who are receiving supplemental epoetin therapy.
CONTRAINDICATIONS
-
All anemias not associated with iron deficiency.
-
Hypersensitivity to Ferrlecit or any of its inactive components.
-
Evidence of iron overload.
WARNINGS
Hypersensitivity reactions have been reported with injectable iron products. See PRECAUTIONS.
PRECAUTIONS
General: Iron is not easily eliminated from the body and accumulation can be toxic. Unnecessary therapy with parenteral iron will cause excess storage of iron with consequent possibility of iatrogenic hemosiderosis. Iron overload is particularly apt to occur in patients with hemoglobinopathies and other refractory anemias. Ferrlecit should not be administered to patients with iron overload. See OVERDOSAGE.
Hypersensitivity Reactions: One case of a life-threatening hypersensitivity reaction was observed in 1,097 patients who received a single dose of Ferrlecit in a postmarketing safety study. In the postmarketing spontaneous reporting system, life-threatening hypersensitivity reactions have been reported rarely in patients receiving Ferrlecit. See ADVERSE REACTIONS.
Hypotension: Hypotension associated with lightheadedness, malaise, fatigue, weakness or severe pain in the chest, back, flanks, or groin has been associated with administration of intravenous iron. These hypotensive reactions are not associated with signs of hypersensitivity and have usually resolved within one or two hours. Successful treatment may consist of observation or, if the hypotension causes symptoms, volume expansion. See ADVERSE REACTIONS.
Carcinogenesis, mutagenesis, impairment of fertility: Long term carcinogenicity studies in animals were not performed. Studies to assess the effects of Ferrlecit on fertility were not conducted. Ferrlecit was not mutagenic in the Ames test and the rat micronucleus test. It produced a clastogenic effect in an in vitro chromosomal aberration assay in Chinese hamster ovary cells.
Pregnancy Category B: Ferrlecit was not teratogenic at doses of elemental iron up to 100 mg/kg/day (300 mg/m2/day) in mice and 20 mg/kg/day (120 mg/m2/day) in rats. On a body surface area basis, these doses were 1.3 and 3.24 times the recommended human dose (125 mg/day or 92.5 mg/m2/day) for a person of 50 kg body weight, average height and body surface area of 1.46 m2. There were no adequate and well-controlled studies in pregnant women. Ferrlecit should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers: It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when Ferrlecit is administered to a nursing woman.
Pediatric Use: Ferrlecit was shown to be safe and effective in pediatric patients ages 6 to 15 years (refer to CLINICAL STUDIES section). Safety and effectiveness in pediatric patients younger than 6 years of age have not been established.
Ferrlecit contains benzyl alcohol and therefore should not be used in neonates.
Geriatric Use: Clinical studies of Ferrlecit did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In particular, 51/159 hemodialysis patients in North American clinical studies were aged 65 years or older. Among these patients, no differences in safety or efficacy as a result of age were identified. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
ADVERSE REACTIONS
Exposure to Ferrlecit has been documented in over 1,400 patients on hemodialysis. This population included in 1,097 Ferrlecit-naïve patients who received a single-dose of Ferrlecit in a placebo-controlled, cross-over, post-marketing safety study. Undiluted Ferrlecit was administered over ten minutes (125 mg of Ferrlecit at 12.5 mg/min). No test dose was used. From a total of 1,498 Ferrlecit-treated patients in medical reports, North American trials, and post-marketing studies, twelve patients (0.8%) experienced serious reactions which precluded further therapy with Ferrlecit.
Hypersensitivity Reactions: See PRECAUTIONS. In the single-dose, post-marketing, safety study one patient experienced a life-threatening hypersensitivity reaction (diaphoresis, nausea, vomiting, severe lower back pain, dyspnea, and wheezing for 20 minutes) following Ferrlecit administration. Among 1,097 patients who received Ferrlecit in this study, there were 9 patients (0.8%) who had an adverse reaction that, in the view of the investigator, precluded further Ferrlecit administration (drug intolerance). These included one life-threatening reaction, six allergic reactions (pruritus x2, facial flushing, chills, dyspnea/chest pain, and rash), and two other reactions (hypotension and nausea). Another 2 patients experienced (0.2%) allergic reactions not deemed to represent drug intolerance (nausea/malaise and nausea/dizziness) following Ferrlecit administration.
Seventy-two (7.0%) of the 1,034 patients who had prior iron dextran exposure had a sensitivity to at least one form of iron dextran (INFeD® or Dexferrum®). The patient who experienced a life-threatening adverse event following Ferrlecit administration during the study had a previous severe anaphylactic reaction to dextran in both forms (INFeD® and Dexferrum®). The incidences of both drug intolerance and suspected allergic events following first dose Ferrlecit administration were 2.8% in patients with prior iron dextran sensitivity compared to 0.8% in patients without prior iron dextran sensitivity.
In this study, 28% of the patients received concomitant angiotensin converting enzyme inhibitor (ACEi) therapy. The incidences of both drug intolerance or suspected allergic events following first dose Ferrlecit administration were 1.6% in patients with concomitant ACEi use compared to 0.7% in patients without concomitant ACEi use. The patient with a life-threatening event was not on ACEi therapy. One patient had facial flushing immediately on Ferrlecit exposure. No hypotension occurred and the event resolved rapidly and spontaneously without intervention other than drug withdrawal.
In multiple dose Studies A and B, no fatal hypersensitivity reactions occurred among the 126 patients who received Ferrlecit. Ferrlecit-associated hypersensitivity events in Study A resulting in premature study discontinuation occurred in three out of a total 88 (3.4%) Ferrlecit-treated patients. The first patient withdrew after the development of pruritus and chest pain following the test dose of Ferrlecit. The second patient, in the high-dose group, experienced nausea, abdominal and flank pain, fatigue and rash following the first dose of Ferrlecit. The third patient, in the low-dose group, experienced a "red blotchy rash" following the first dose of Ferrlecit. Of the 38 patients exposed to Ferrlecit in Study B, none reported hypersensitivity reactions.
Many chronic renal failure patients experience cramps, pain, nausea, rash, flushing, and pruritus.
In the postmarketing spontaneous reporting system, lifethreatening hypersensitivity reactions have been reported rarely in patients receiving Ferrlecit.
Hypotension: See PRECAUTIONS. In the single dose safety study, post-administration hypotensive events were observed in 22/1,097 patients (2%) following Ferrlecit administration. Hypotension has also been reported following administration of Ferrlecit in European case reports. Of the 226 renal dialysis patients exposed to Ferrlecit and reported in the literature, 3 (1.3%) patients experienced hypotensive events, which were accompanied by flushing in two. All completely reversed after one hour without sequelae. Transient hypotension may occur during dialysis. Administration of Ferrlecit may augment hypotension caused by dialysis.
Among the 126 patients who received Ferrlecit in Studies A and B, one patient experienced a transient decreased level of consciousness without hypotension. Another patient discontinued treatment prematurely because of dizziness, lightheadedness, diplopia, malaise, and weakness without hypotension that resulted in a 3–4 hour hospitalization for observation following drug administration. The syndrome resolved spontaneously.
Adverse Laboratory Changes: No differences in laboratory findings associated with Ferrlecit (sodium ferric gluconate complex in sucrose injection) were reported in North American clinical trials when normalized against a National Institute of Health database on laboratory findings in 1,100 hemodialysis patients.
Most Frequent Adverse Reactions: In the single-dose, postmarketing safety study, 11% of patients who received Ferrlecit and 9.4% of patients who received placebo reported adverse reactions. The most frequent adverse reactions following Ferrlecit were: hypotension (2%), nausea, vomiting and/or diarrhea (2%), pain (0.7%), hypertension (0.6%), allergic reaction (0.5%), chest pain (0.5%), pruritus (0.5%), and back pain (0.4%). Similar adverse reactions were seen following placebo administration. However, because of the high baseline incidence of adverse events in the hemodialysis patient population, insufficient number of exposed patients, and limitations inherent to the cross-over, single dose study design, no comparison of event rates between Ferrlecit and placebo treatments can be made.
In multiple-dose Studies A and B, the most frequent adverse reactions following Ferrlecit were:
Body as a Whole: injection site reaction (33%), chest pain (10%), pain (10%), asthenia (7%), headache (7%), abdominal pain (6%), fatigue (6%), fever (5%), malaise, infection, abscess, back pain, chills, rigors, arm pain, carcinoma, flu-like syndrome, sepsis.
Nervous System: cramps (25%), dizziness (13%), paresthesias (6%), agitation, somnolence.
Respiratory System: dyspnea (11%), coughing (6%), upper respiratory infections (6%), rhinitis, pneumonia.
Cardiovascular System: hypotension (29%), hypertension (13%), syncope (6%), tachycardia (5%), bradycardia, vasodilatation, angina pectoris, myocardial infarction, pulmonary edema.
Gastrointestinal System: nausea, vomiting and/or diarrhea (35%), anorexia, rectal disorder, dyspepsia, eructation, flatulence, gastrointestinal disorder, melena.
Musculoskeletal System: leg cramps (10%), myalgia, arthralgia.
Skin and Appendages: pruritus (6%), rash, increased sweating.
Genitourinary System: urinary tract infection.
Special Senses: conjunctivitis, abnormal vision, ear disorder.
Metabolic and Nutritional Disorders: hyperkalemia (6%), generalized edema (5%), leg edema, peripheral edema, hypoglycemia, edema, hypervolemia, hypokalemia.
Hematologic System: abnormal erythrocytes (11%), anemia, leukocytosis, lymphadenopathy.
Other Adverse Reactions Observed During Clinical Trials: In the single-dose post-marketing safety study in 1,097 patients receiving Ferrlecit, the following additional events were reported in two or more patients: hypertonia, nervousness, dry mouth, and hemorrhage.
Pediatric Patients: In a clinical trial of 66 iron-deficient pediatric hemodialysis patients, 6 to 15 years of age, inclusive, who were receiving a stable erythropoietin dosing regimen, the most common adverse events, whether or not related to study drug, occurring in ≥5%, regardless of treatment group, were: hypotension (35%), headache (24%), hypertension (23%), tachycardia (17%), vomiting (11%), fever (9%), nausea (9%), abdominal pain (9%), pharyngitis (9%), diarrhea (8%), infection (8%), rhinitis (6%), and thrombosis (6%). More patients in the higher dose group (3.0 mg/kg) than in the lower dose group (1.5 mg/kg) experienced the following adverse events: hypotension (41% vs. 28%), tachycardia (21% vs. 13%), fever (15% vs. 3%), headache (29% vs. 19%), abdominal pain (15% vs. 3%), nausea (12% vs. 6%), vomiting (12% vs. 9%), pharyngitis (12% vs. 6%), and rhinitis (9% vs. 3%).
Postmarketing Surveillance: The following additional adverse reactions have been identified with the use of Ferrlecit from postmarketing spontaneous reports: dysgeusia, hypoesthesia, loss of consciousness, convulsion, skin discoloration, pallor, phlebitis, and shock. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
OVERDOSAGE
Dosages in excess of iron needs may lead to accumulation of iron in iron storage sites and hemosiderosis. Periodic monitoring of laboratory parameters of iron storage may assist in recognition of iron accumulation. Ferrlecit should not be administered in patients with iron overload.
Serum iron levels greater than 300 μg/dL may indicate iron poisoning which is characterized by abdominal pain, diarrhea, or vomiting which progresses to pallor or cyanosis, lassitude, drowsiness, hyperventilation due to acidosis, and cardiovascular collapse. Caution should be exercised in interpreting serum iron levels in the 24 hours following the administration of Ferrlecit since many laboratory assays will falsely overestimate serum or transferrin bound iron by measuring iron still bound to the Ferrlecit complex. Additionally, in the assessment of iron overload, caution should be exercised in interpreting serum ferritin levels in the week following Ferrlecit administration since, in clinical studies, serum ferritin exhibited a nonspecific rise which persisted for five days.
The Ferrlecit iron complex is not dialyzable.
Ferrlecit at elemental iron doses of 125 mg/kg, 78.8 mg/kg, 62.5 mg/kg and 250 mg/kg caused deaths to mice, rats, rabbits, and dogs respectively. The major symptoms of acute toxicity were decreased activity, staggering, ataxia, increases in the respiratory rate, tremor, and convulsions.
Individual doses exceeding 125 mg may be associated with a higher incidence and/or severity of adverse events based on information from postmarketing spontaneous reports. These adverse events included hypotension, nausea, vomiting, abdominal pain, diarrhea, dizziness, dyspnea, urticaria, chest pain, paresthesia, and peripheral swelling. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
DOSAGE AND ADMINISTRATION
The dosage of Ferrlecit is expressed in terms of mg of elemental iron. Each 5 mL sterile, single-use ampule contains 62.5 mg of elemental iron (12.5 mg/mL).
The recommended dosage of Ferrlecit for the repletion treatment of iron deficiency in hemodialysis patients is 10 mL of Ferrlecit (125 mg of elemental iron). Ferrlecit may be diluted in 100 mL of 0.9% sodium chloride administered by intravenous infusion over 1 hour. Ferrlecit may also be administered undiluted as a slow IV injection (at a rate of up to 12.5 mg/min). Most patients will require a minimum cumulative dose of 1.0 gram of elemental iron, administered over eight sessions at sequential dialysis treatments, to achieve a favorable hemoglobin or hematocrit response. Patients may continue to require therapy with intravenous iron at the lowest dose necessary to maintain target levels of hemoglobin, hematocrit, and laboratory parameters of iron storage within acceptable limits. Ferrlecit has been administered at sequential dialysis sessions by infusion or by slow IV injection during the dialysis session itself.
Data from Ferrlecit postmarketing spontaneous reports indicate that individual doses exceeding 125 mg may be associated with a higher incidence and/or severity of adverse events. See OVERDOSAGE.
Pediatric Dosage: The recommended pediatric dosage of Ferrlecit for the repletion treatment of iron deficiency in hemodialysis patients is 0.12 mL/kg Ferrlecit (1.5 mg/kg of elemental iron) diluted in 25 mL 0.9% sodium chloride and administered by intravenous infusion over 1 hour at eight sequential dialysis sessions. The maximum dosage should not exceed 125 mg per dose.
Note: Do not mix Ferrlecit with other medications, or add to parenteral nutrition solutions for intravenous infusion. The compatibility of Ferrlecit with intravenous infusion vehicles other than 0.9% sodium chloride has not been evaluated. Parenteral drug products should be inspected visually for particulate matter and discoloration before administration, whenever the solution and container permit.
If diluted in saline, use immediately after dilution.
HOW SUPPLIED
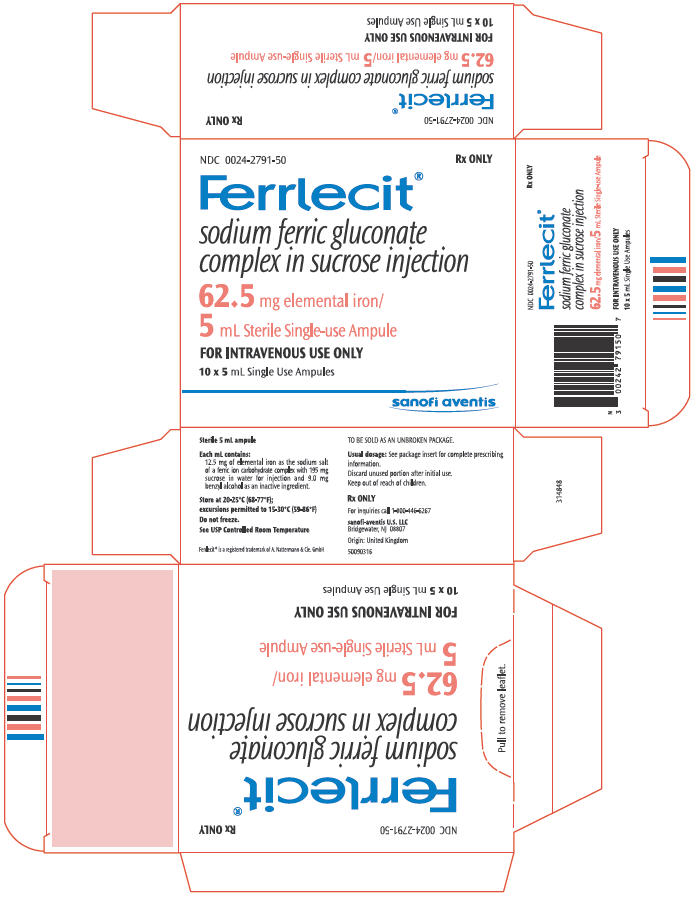
NDC 0024-2791-50
Ferrlecit is supplied in colorless glass ampules. Each sterile, single-use ampule contains 62.5 mg of elemental iron in 5 mL for intravenous use, packaged in cartons of 10 ampules.
Store at 20 – 25°C (68 – 77°F); excursions permitted to 15 – 30°C (59 – 86°F). Do not freeze. See USP Controlled Room Temperature.
Keep out of the reach of children.
Revised: January 2010
sanofi-aventis U.S. LLC
Bridgewater, NJ 08807
©2010 sanofi-aventis U.S. LLC
PRINCIPAL DISPLAY PANEL - 5 mL Ampule
Rx Only
NDC 0024-2791-50
INTRAVENOUS
USE ONLY
Ferrlecit®
sodium ferric gluconate complex in sucrose injection
62.5 mg elemental iron/
5 mL Sterile Single-use Ampule
Each mL contains:
12.5 mg elemental iron
195 mg sucrose in water
for injection and
9.0 mg benzyl alcohol
For inquiries call
1-800-446-6267
sanofi-aventis U.S. LLC
Bridgewater, NJ 08807
Origin: United Kingdom
See package insert
for complete
prescribing
information.
Store at 20-25°C
(68-77°F);
excursions permitted
to 15-30°C (59-86°F)
50090313
sanofi aventis

PRINCIPAL DISPLAY PANEL - 10 Ampule Carton
NDC 0024-2791-50
Rx ONLY
Ferrlecit®
sodium ferric gluconate
complex in sucrose injection
62.5 mg elemental iron/
5 mL Sterile Single-use Ampule
FOR INTRAVENOUS USE ONLY
10 × 5 mL Single Use Ampules
sanofi aventis
| FERRLECIT
sodium ferric gluconate complex injection |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020955 | 02/18/1999 | |
| Labeler - sanofi-aventis U.S. LLC (824676584) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Aventis Pharma Ltd. | 737163787 | MANUFACTURE | |