VYVANSE- lisdexamfetamine dimesylate capsule
Avera McKennan Hospital
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use VYVANSE safely and effectively. See full prescribing information for VYVANSE.
VYVANSE ® (lisdexamfetamine dimesylate) capsules, for oral use, CII Initial U.S. Approval: 2007 WARNING: ABUSE AND DEPENDENCESee full prescribing information for complete boxed warning.INDICATIONS AND USAGEVYVANSE is a central nervous system (CNS) stimulant indicated for the treatment of ( 1):
Limitation of Use: VYVANSE is not indicated for weight loss. Use of other sympathomimetic drugs for weight loss has been associated with serious cardiovascular adverse events. The safety and effectiveness of VYVANSE for the treatment of obesity have not been established. DOSAGE AND ADMINISTRATIONDOSAGE FORMS AND STRENGTHSCapsules: 10 mg, 20 mg, 30 mg, 40 mg, 50 mg, 60 mg, 70 mg ( 3) CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (incidence ≥5% and at a rate at least twice placebo) in children, adolescents, and/or adults with ADHD were anorexia, anxiety, decreased appetite, decreased weight, diarrhea, dizziness, dry mouth, irritability, insomnia, nausea, upper abdominal pain, and vomiting ( 6.1) Most common adverse reactions (incidence ≥ 5% and at a rate at least twice placebo) in adults with BED were dry mouth, insomnia, decreased appetite, increased heart rate, constipation, feeling jittery, and anxiety ( 6.1) To report SUSPECTED ADVERSE REACTIONS, contact Shire US Inc. at 1-800-828-2088 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch DRUG INTERACTIONSUSE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 12/2015 |
FULL PRESCRIBING INFORMATION
WARNING: ABUSE AND DEPENDENCE
CNS stimulants (amphetamines and methylphenidate-containing products), including VYVANSE, have a high potential for abuse and dependence. Assess the risk of abuse prior to prescribing and monitor for signs of abuse and dependence while on therapy [see Warnings and Precautions (5.1, 5.2), and Drug Abuse and Dependence (9.2, 9.3)].
1 INDICATIONS AND USAGE
VYVANSE ® is indicated for the treatment of:
- Attention Deficit Hyperactivity Disorder (ADHD) [see Clinical Studies (14.1)] Attention Deficit Hyperactivity Disorder (ADHD) [see Clinical Studies (14.1)]
- Moderate to Severe Binge Eating Disorder (BED) [see Clinical Studies (14.2)] . Moderate to Severe Binge Eating Disorder (BED) [see Clinical Studies (14.2)] .
Limitation of Use:
VYVANSE is not indicated or recommended for weight loss. Use of other sympathomimetic drugs for weight loss has been associated with serious cardiovascular adverse events. The safety and effectiveness of VYVANSE for the treatment of obesity have not been established [see Warnings and Precautions (5.2)] .
2 DOSAGE AND ADMINISTRATION
2.1 General Instructions for Use
Take VYVANSE by mouth in the morning with or without food; avoid afternoon doses because of the potential for insomnia. VYVANSE may be administered in one of the following ways:Take VYVANSE by mouth in the morning with or without food; avoid afternoon doses because of the potential for insomnia. VYVANSE may be administered in one of the following ways:
- Swallow VYVANSE capsules whole, orSwallow VYVANSE capsules whole, or
- Open capsules, empty and mix the entire contents with yogurt, water, or orange juice. If the contents of the capsule include any compacted powder, a spoon may be used to break apart the powder. The contents should be mixed until completely dispersed. Consume the entire mixture immediately. It should not be stored. The active ingredient dissolves completely once dispersed; however, a film containing the inactive ingredients may remain in the glass or container once the mixture is consumed. Do not take anything less than one capsule per day, and a single capsule should not be divided. Open capsules, empty and mix the entire contents with yogurt, water, or orange juice. If the contents of the capsule include any compacted powder, a spoon may be used to break apart the powder. The contents should be mixed until completely dispersed. Consume the entire mixture immediately. It should not be stored. The active ingredient dissolves completely once dispersed; however, a film containing the inactive ingredients may remain in the glass or container once the mixture is consumed. Do not take anything less than one capsule per day, and a single capsule should not be divided.
2.2 Dosage for Treatment of ADHD
The recommended starting dose is 30 mg once daily in the morning in patients ages 6 and above. Dosage may be adjusted in increments of 10 mg or 20 mg at approximately weekly intervals up to maximum dose of 70 mg/day. Patients may be maintained on their optimal dose [see Clinical Studies (14.1)] .
2.3 Dosage for Treatment of Moderate to Severe BED
The recommended starting dose is 30 mg/day to be titrated in increments of 20 mg at approximately weekly intervals to achieve the recommended target dose of 50 to 70 mg/day. The maximum dose is 70 mg/day [see Clinical Studies (14.2)] . Discontinue VYVANSE if binge eating does not improve.
2.4 Important Information Prior to Dosing
Prior to treating children, adolescents, and adults with CNS stimulants, assess for the presence of cardiac disease (e.g., a careful history, family history of sudden death or ventricular arrhythmia, and physical exam) [see Warnings and Precautions (5.2)] .
To reduce the abuse of CNS stimulants including VYVANSE, assess the risk of abuse, prior to prescribing. After prescribing, keep careful prescription records, educate patients about abuse, monitor for signs of abuse and overdose, and re-evaluate the need for VYVANSE use [see Warnings and Precautions (5.1), Drug Abuse and Dependence (9.2, 9.3)] .
2.5 Dosage in Patients with Renal Impairment
In patients with severe renal impairment (GFR 15 to < 30 mL/min/1.73 m 2), the maximum dose should not exceed 50 mg/day. In patients with end stage renal disease (ESRD, GFR < 15 mL/min/1.73 m 2), the maximum recommended dose is 30 mg/day [see Use in Specific Populations (8.6)] .
2.6 Dosage Modifications due to Drug Interactions
Agents that alter urinary pH can impact urinary excretion and alter blood levels of amphetamine. Acidifying agents (e.g., ascorbic acid) decrease blood levels, while alkalinizing agents (e.g., sodium bicarbonate) increase blood levels. Adjust VYVANSE dosage accordingly [see Drug Interactions (7.1)] .
3 DOSAGE FORMS AND STRENGTHS
Capsules 10 mg: pink body/pink cap (imprinted with S489 and 10 mg)
Capsules 20 mg: ivory body/ivory cap (imprinted with S489 and 20 mg)
Capsules 30 mg: white body/orange cap (imprinted with S489 and 30 mg)
Capsules 40 mg: white body/blue green cap (imprinted with S489 and 40 mg)
Capsules 50 mg: white body/blue cap (imprinted with S489 and 50 mg)
Capsules 60 mg: aqua blue body/aqua blue cap (imprinted with S489 and 60 mg)
Capsules 70 mg: blue body/orange cap (imprinted with S489 and 70 mg)
4 CONTRAINDICATIONS
VYVANSE is contraindicated in patients with:
- Known hypersensitivity to amphetamine products or other ingredients of VYVANSE. Anaphylactic reactions, Stevens-Johnson Syndrome, angioedema, and urticaria have been observed in postmarketing reports [see Adverse Reactions (6.2)] .
- Concurrent administration of monoamine oxidase inhibitors (MAOI) or administration of VYVANSE within 14 days of the last MAOI dose. Hypertensive crisis can occur [see Drug Interactions (7.2)] .
5 WARNINGS AND PRECAUTIONS
5.1 Potential for Abuse and Dependence
CNS stimulants (amphetamines and methylphenidate-containing products), including VYVANSE, have a high potential for abuse and dependence. Assess the risk of abuse prior to prescribing, and monitor for signs of abuse and dependence while on therapy [see Drug Abuse and Dependence (9.2, 9.3)] .
5.2 Serious Cardiovascular Reactions
Sudden death, stroke and myocardial infarction have been reported in adults with CNS stimulant treatment at recommended doses. Sudden death has been reported in children and adolescents with structural cardiac abnormalities and other serious heart problems taking CNS stimulants at recommended doses for ADHD. Avoid use in patients with known structural cardiac abnormalities, cardiomyopathy, serious heart arrhythmia, coronary artery disease, and other serious heart problems. Further evaluate patients who develop exertional chest pain, unexplained syncope, or arrhythmias during VYVANSE treatment.
5.3 Blood Pressure and Heart Rate Increases
CNS stimulants cause an increase in blood pressure (mean increase about 2-4 mm Hg) and heart rate (mean increase about 3-6 bpm). Monitor all patients for potential tachycardia and hypertension.
5.4 Psychiatric Adverse Reactions
Exacerbation of Pre-existing Psychosis
CNS stimulants may exacerbate symptoms of behavior disturbance and thought disorder in patients with a pre-existing psychotic disorder.
Induction of a Manic Episode in Patients with Bipolar Disorder
CNS stimulants may induce a mixed/manic episode in patients with bipolar disorder. Prior to initiating treatment, screen patients for risk factors for developing a manic episode.
New Psychotic or Manic Symptoms
CNS stimulants, at recommended doses, may cause psychotic or manic symptoms, e.g. hallucinations, delusional thinking, or mania in children and adolescents without a prior history of psychotic illness or mania. If such symptoms occur, consider discontinuing VYVANSE. In a pooled analysis of multiple short-term, placebo-controlled studies of CNS stimulants, psychotic or manic symptoms occurred in 0.1% of CNS stimulant-treated patients compared to 0% in placebo-treated patients.
5.5 Suppression of Growth
CNS stimulants have been associated with weight loss and slowing of growth rate in pediatric patients. Closely monitor growth (weight and height) in pediatric patients treated with CNS stimulants, including VYVANSE. In a 4-week, placebo-controlled trial of VYVANSE in patients ages 6 to 12 years old with ADHD, there was a dose-related decrease in weight in the VYVANSE groups compared to weight gain in the placebo group. Additionally, in studies of another stimulant, there was slowing of the increase in height [see Adverse Reactions (6.1)] .
5.6 Peripheral Vasculopathy, including Raynaud's Phenomenon
Stimulants, including VYVANSE, are associated with peripheral vasculopathy, including Raynaud's phenomenon. Signs and symptoms are usually intermittent and mild; however, very rare sequelae include digital ulceration and/or soft tissue breakdown. Effects of peripheral vasculopathy, including Raynaud's phenomenon, were observed in post-marketing reports at different times and at therapeutic doses in all age groups throughout the course of treatment. Signs and symptoms generally improve after reduction in dose or discontinuation of drug. Careful observation for digital changes is necessary during treatment with stimulants. Further clinical evaluation (e.g., rheumatology referral) may be appropriate for certain patients.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling
- Serious Cardiovascular Reactions [see Warnings and Precautions (5.2)]
- Blood Pressure and Heart Rate Increases [see Warnings and Precautions (5.3)]
- Psychiatric Adverse Reactions [see Warnings and Precautions (5.4)]
- Suppression of Growth [see Warnings and Precautions (5.5)]
- Peripheral Vasculopathy, including Raynaud's phenomenon [see Warnings and Precautions (5.6)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Attention Deficit Hyperactivity Disorder
The safety data in this section is based on data from the 4-week parallel-group controlled clinical studies of VYVANSE in pediatric and adult patients with ADHD [see Clinical Studies (14.1)] .
Adverse Reactions Associated with Discontinuation of Treatment in ADHD Clinical Trials
In the controlled trial in patients ages 6 to 12 years (Study 1), 9% (20/218) of VYVANSE-treated patients discontinued due to adverse reactions compared to 1% (1/72) of placebo-treated patients. The most frequent adverse reactions leading to discontinuation (i.e. leading to discontinuation in at least 1% of VYVANSE-treated patients and at a rate at least twice that of placebo) were ECG voltage criteria for ventricular hypertrophy, tic, vomiting, psychomotor hyperactivity, insomnia, and rash [2 instances for each adverse reaction, i.e., 2/218 (1%)].
In the controlled trial in patients ages 13 to 17 years (Study 4), 4% (10/233) of VYVANSE-treated patients discontinued due to adverse reactions compared to 1% (1/77) of placebo-treated patients. The most frequent adverse reactions leading to discontinuation were irritability (3/233; 1%), decreased appetite (2/233; 1%), and insomnia (2/233; 1%).
In the controlled adult trial (Study 7), 6% (21/358) of VYVANSE-treated patients discontinued due to adverse reactions compared to 2% (1/62) of placebo-treated patients. The most frequent adverse reactions leading to discontinuation (i.e. leading to discontinuation in at least 1% of VYVANSE-treated patients and at a rate at least twice that of placebo) were insomnia (8/358; 2%), tachycardia (3/358; 1%), irritability (2/358; 1%), hypertension (4/358; 1%), headache (2/358; 1%), anxiety (2/358; 1%), and dyspnea (3/358; 1%).
The most common adverse reactions (incidence ≥5% and at a rate at least twice placebo) reported in children, adolescents, and/or adults were anorexia, anxiety, decreased appetite, decreased weight, diarrhea, dizziness, dry mouth, irritability, insomnia, nausea, upper abdominal pain, and vomiting.
Adverse Reactions Occurring at an Incidence of 2% or More Among VYVANSE Treated Patients with ADHD in Clinical Trials
Adverse reactions reported in the controlled trials in pediatric patients ages 6 to 12 years (Study 1), adolescent patients ages 13 to 17 years (Study 4), and adult patients (Study 7) treated with VYVANSE or placebo are presented in Tables 1, 2, and 3 below.
| VYVANSE
(n=218) | Placebo
(n=72) |
|
|---|---|---|
| Decreased Appetite | 39% | 4% |
| Insomnia | 23% | 3% |
| Abdominal Pain Upper | 12% | 6% |
| Irritability | 10% | 0% |
| Vomiting | 9% | 4% |
| Weight Decreased | 9% | 1% |
| Nausea | 6% | 3% |
| Dry Mouth | 5% | 0% |
| Dizziness | 5% | 0% |
| Affect lability | 3% | 0% |
| Rash | 3% | 0% |
| Pyrexia | 2% | 1% |
| Somnolence | 2% | 1% |
| Tic | 2% | 0% |
| VYVANSE
(n=233) | Placebo
(n=77) |
|
|---|---|---|
| Decreased Appetite | 34% | 3% |
| Insomnia | 13% | 4% |
| Weight Decreased | 9% | 0% |
| Dry Mouth | 4% | 1% |
| VYVANSE
(n=358) | Placebo
(n=62) |
|
|---|---|---|
| Decreased Appetite | 27% | 2% |
| Insomnia | 27% | 8% |
| Dry Mouth | 26% | 3% |
| Diarrhea | 7% | 0% |
| Nausea | 7% | 0% |
| Anxiety | 6% | 0% |
| Anorexia | 5% | 0% |
| Feeling Jittery | 4% | 0% |
| Agitation | 3% | 0% |
| Increased Blood Pressure | 3% | 0% |
| Hyperhidrosis | 3% | 0% |
| Restlessness | 3% | 0% |
| Decreased Weight | 3% | 0% |
| Dyspnea | 2% | 0% |
| Increased Heart Rate | 2% | 0% |
| Tremor | 2% | 0% |
In addition, in the adult population erectile dysfunction was observed in 2.6% of males on VYVANSE and 0% on placebo; decreased libido was observed in 1.4% of subjects on VYVANSE and 0% on placebo.
Weight Loss and Slowing Growth Rate in Pediatric Patients with ADHD
In a controlled trial of VYVANSE in children ages 6 to 12 years (Study 1), mean weight loss from baseline after 4 weeks of therapy was -0.9, -1.9, and -2.5 pounds, respectively, for patients receiving 30 mg, 50 mg, and 70 mg of VYVANSE, compared to a 1 pound weight gain for patients receiving placebo. Higher doses were associated with greater weight loss with 4 weeks of treatment. Careful follow-up for weight in children ages 6 to 12 years who received VYVANSE over 12 months suggests that consistently medicated children (i.e. treatment for 7 days per week throughout the year) have a slowing in growth rate, measured by body weight as demonstrated by an age- and sex-normalized mean change from baseline in percentile, of -13.4 over 1 year (average percentiles at baseline and 12 months were 60.9 and 47.2, respectively). In a 4-week controlled trial of VYVANSE in adolescents ages 13 to 17 years, mean weight loss from baseline to endpoint was -2.7, -4.3, and -4.8 lbs., respectively, for patients receiving 30 mg, 50 mg, and 70 mg of VYVANSE, compared to a 2.0 pound weight gain for patients receiving placebo.
Careful follow-up of weight and height in children ages 7 to 10 years who were randomized to either methylphenidate or non-medication treatment groups over 14 months, as well as in naturalistic subgroups of newly methylphenidate-treated and non-medication treated children over 36 months (to the ages of 10 to 13 years), suggests that consistently medicated children (i.e. treatment for 7 days per week throughout the year) have a temporary slowing in growth rate (on average, a total of about 2 cm less growth in height and 2.7 kg less growth in weight over 3 years), without evidence of growth rebound during this period of development. In a controlled trial of amphetamine (d- to l-enantiomer ratio of 3:1) in adolescents, mean weight change from baseline within the initial 4 weeks of therapy was -1.1 pounds and -2.8 pounds, respectively, for patients receiving 10 mg and 20 mg of amphetamine. Higher doses were associated with greater weight loss within the initial 4 weeks of treatment [see Warnings and Precautions (5.5)] .
Weight Loss in Adults with ADHD
In the controlled adult trial (Study 7), mean weight loss after 4 weeks of therapy was 2.8 pounds, 3.1 pounds, and 4.3 pounds, for patients receiving final doses of 30 mg, 50 mg, and 70 mg of VYVANSE, respectively, compared to a mean weight gain of 0.5 pounds for patients receiving placebo.
Binge Eating Disorder
The safety data in this section is based on data from two 12 week parallel group, flexible-dose, placebo-controlled studies in adults with BED [see Clinical Studies 14.2] . Patients with cardiovascular risk factors other than obesity and smoking were excluded.
Adverse Reactions Associated with Discontinuation of Treatment in BED Clinical Trials
In controlled trials of patients ages 18 to 55 years, 5.1% (19/373) of VYVANSE-treated patients discontinued due to adverse reactions compared to 2.4% (9/372) of placebo-treated patients. No single adverse reaction led to discontinuation in 1% or more of VYVANSE-treated patients.
The most common adverse reactions (incidence ≥5% and at a rate at least twice placebo) reported in adults were dry mouth, insomnia, decreased appetite, increased heart rate, constipation, feeling jittery, and anxiety.
Adverse reactions reported in the pooled controlled trials in adult patients (Study 10 and 11) treated with VYVANSE or placebo are presented in Table 4 below.
| VYVANSE
(N=373) | Placebo
(N=372) |
|
|---|---|---|
| Dry Mouth | 36% | 7% |
| Insomnia * | 20% | 8% |
| Decreased Appetite | 8% | 2% |
| Increased Heart Rate † | 7% | 1% |
| Feeling Jittery | 6% | 1% |
| Constipation | 6% | 1% |
| Anxiety | 5% | 1% |
| Diarrhea | 4% | 2% |
| Decreased Weight | 4% | 0% |
| Hyperhidrosis | 4% | 0% |
| Vomiting | 2% | 1% |
| Gastroenteritis | 2% | 1% |
| Paresthesia | 2% | 1% |
| Pruritis | 2% | 1% |
| Upper Abdominal Pain | 2% | 0% |
| Energy Increased | 2% | 0% |
| Urinary Tract Infection | 2% | 0% |
| Nightmare | 2% | 0% |
| Restlessness | 2% | 0% |
| Oropharyngeal Pain | 2% | 0% |
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of VYVANSE. Because these reactions are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These events are as follows: palpitations, cardiomyopathy, mydriasis, diplopia, difficulties with visual accommodation, blurred vision, eosinophilic hepatitis, anaphylactic reaction, hypersensitivity, dyskinesia, tics, bruxism, depression, dermatillomania, aggression, Stevens-Johnson Syndrome, angioedema, urticaria, seizures, libido changes, frequent or prolonged erections, constipation, and rhabdomyolysis.
7 DRUG INTERACTIONS
7.1 Clinically Important Interactions with VYVANSE
| Concomitant Drug Name or Drug Class | Clinical Rationale | Clinical Recommendation |
|---|---|---|
| Acidifying and Alkalinizing Agents | Ascorbic acid and other agents that acidify urine increase urinary excretion and decrease the half-life of amphetamine. Sodium bicarbonate and other agents that alkalinize urine decrease urinary excretion and extend the half-life of amphetamine. | Adjust the dose accordingly [see Dosage and Administration (2.6)] |
| Concomitant Drug Name or Drug Class | Clinical Rationale | Clinical Recommendation |
|---|---|---|
| Monoamine Oxidase Inhibitors (MAOIs) | Concomitant use of MAOIs and CNS stimulants can cause hypertensive crisis. Potential outcomes include death, stroke, myocardial infarction, aortic dissection, ophthalmological complications, eclampsia, pulmonary edema, and renal failure. | Do not administer VYVANSE concomitantly or within 14 days after discontinuing MAOI treatment [see Contraindications (4)] |
7.2 Drugs Having No Clinically Important Interactions with VYVANSE
From a pharmacokinetic perspective, no dose adjustment of VYVANSE is necessary when VYVANSE is co-administered with guanfacine, venlafaxine, or omeprazole. In addition, no dose adjustment of guanfacine or venlafaxine is needed when VYVANSE is co-administered [see Clinical Pharmacology (12.3)] .
From a pharmacokinetic perspective, no dose adjustment for drugs that are substrates of CYP1A2 (e.g. theophylline, duloxetine, melatonin), CYP2D6 (e.g. atomoxetine, desipramine, venlafaxine), CYP2C19 (e.g. omeprazole, lansoprazole, clobazam), and CYP3A4 (e.g. midazolam, pimozide, simvastatin) is necessary when VYVANSE is co-administered [see Clinical Pharmacology (12.3)] .
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C
Risk Summary
There are no adequate and well-controlled studies with VYVANSE in pregnant women. Adverse pregnancy outcomes, including premature delivery and low birth weight, have been seen in infants born to mothers dependent on amphetamines. Long-term neurochemical and behavioral effects have been reported in animal developmental studies using clinically relevant doses of amphetamine (d- or d,l-). Animal reproduction studies performed with lisdexamfetamine dimesylate in rats and rabbits showed no effects on embryofetal morphological development and survival. VYVANSE should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Clinical Considerations
Amphetamines, such as VYVANSE, cause vasoconstriction and thereby may decrease placental perfusion. Infants born to amphetamine-dependent mothers have an increased risk of premature delivery and low birth weight.
Monitor infants born to mothers taking amphetamines for symptoms of withdrawal such as feeding difficulties, irritability, agitation, and excessive drowsiness.
Human Data
Available data in women using amphetamines during pregnancy do not show a clear increased risk of major congenital malformations. Two case control studies of over a thousand patients in total exposed to amphetamines at different gestational ages did not show an increase in congenital abnormalities.
Animal Data
Lisdexamfetamine dimesylate had no apparent effects on embryofetal morphological development or survival when administered orally to pregnant rats and rabbits throughout the period of organogenesis at doses of up to 40 and 120 mg/kg/day, respectively. These doses are approximately 4 and 27 times, respectively, the maximum recommended human dose of 70 mg/day given to adolescents, on a mg/m 2 body surface area basis.
A number of studies in rodents indicate that prenatal or early postnatal exposure to amphetamine (d- or d,l-) at doses similar to those used clinically can result in long-term neurochemical and behavioral alterations. Reported behavioral effects include learning and memory deficits, altered locomotor activity, and changes in sexual function.
8.3 Nursing Mothers
Amphetamines are excreted into human milk. Long-term neurodevelopmental effects on infants from amphetamine exposure are unknown. Because of the potential for serious adverse reactions in nursing infants, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
ADHD
Safety and effectiveness have been established in pediatric patients with ADHD ages 6 to 17 years [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.1)] . Safety and efficacy in pediatric patients below the age of 6 years have not been established.
Growth Suppression
Growth should be monitored during treatment with stimulants, including VYVANSE, and children who are not growing or gaining weight as expected may need to have their treatment interrupted [see Warnings and Precautions (5.5), Adverse Reactions (6.1)] .
Juvenile Animal Data
Studies conducted in juvenile rats and dogs at clinically relevant doses showed growth suppression that partially or fully reversed in dogs and female rats but not in male rats after a four-week drug-free recovery period.
A study was conducted in which juvenile rats received oral doses of 4, 10, or 40 mg/kg/day of lisdexamfetamine dimesylate from day 7 to day 63 of age. These doses are approximately 0.3, 0.7, and 3 times the maximum recommended human daily dose of 70 mg on a mg/m 2 basis for a child. Dose-related decreases in food consumption, bodyweight gain, and crown-rump length were seen; after a four-week drug-free recovery period, bodyweights and crown-rump lengths had significantly recovered in females but were still substantially reduced in males. Time to vaginal opening was delayed in females at the highest dose, but there were no drug effects on fertility when the animals were mated beginning on day 85 of age.
In a study in which juvenile dogs received lisdexamfetamine dimesylate for 6 months beginning at 10 weeks of age, decreased bodyweight gain was seen at all doses tested (2, 5, and 12 mg/kg/day, which are approximately 0.5, 1, and 3 times the maximum recommended human daily dose on a mg/m 2 basis for a child). This effect partially or fully reversed during a four-week drug-free recovery period.
8.5 Geriatric Use
Clinical studies of VYVANSE did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience and pharmacokinetic data [see Clinical Pharmacology (12.3)] have not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should start at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
Due to reduced clearance in patients with severe renal impairment (GFR 15 to < 30 mL/min/1.73 m 2), the maximum dose should not exceed 50 mg/day. The maximum recommended dose in ESRD (GFR < 15 mL/min/1.73 m 2) patients is 30 mg/day [see Clinical Pharmacology (12.3)] .
Lisdexamfetamine and d-amphetamine are not dialyzable.
8.7 Gender
No dosage adjustment of VYVANSE is necessary on the basis of gender [see Clinical Pharmacology (12.3)] .
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
VYVANSE contains lisdexamfetamine, a prodrug of amphetamine, a Schedule II controlled substance.
9.2 Abuse
CNS stimulants, including VYVANSE, other amphetamines, and methylphenidate-containing products have a high potential for abuse. Abuse is characterized by impaired control over drug use, compulsive use, continued use despite harm, and craving.
Signs and symptoms of CNS stimulant abuse may include increased heart rate, respiratory rate, blood pressure, and/or sweating, dilated pupils, hyperactivity, restlessness, insomnia, decreased appetite, loss of coordination, tremors, flushed skin, vomiting, and/or abdominal pain. Anxiety, psychosis, hostility, aggression, suicidal or homicidal ideation have also been seen. Abusers of CNS stimulants may chew, snort, inject, or use other unapproved routes of administration which can result in overdose and death [see Overdosage (10)] .
To reduce the abuse of CNS stimulants, including VYVANSE, assess the risk of abuse prior to prescribing. After prescribing, keep careful prescription records, educate patients and their families about abuse and on proper storage and disposal of CNS stimulants, monitor for signs of abuse while on therapy, and re-evaluate the need for VYVANSE use.
Studies of VYVANSE in Drug Abusers
A randomized, double-blind, placebo-control, cross-over, abuse liability study in 38 patients with a history of drug abuse was conducted with single-doses of 50, 100, or 150 mg of VYVANSE, 40 mg of immediate-release d-amphetamine sulphate (a controlled II substance), and 200 mg of diethylpropion hydrochloride (a controlled IV substance). VYVANSE 100 mg produced significantly less "Drug Liking Effects" as measured by the Drug Rating Questionnaire-Subject score, compared to d-amphetamine 40 mg; and 150 mg of VYVANSE demonstrated similar "Drug-Liking Effects" compared to 40 mg of d-amphetamine and 200 mg of diethylpropion.
Intravenous administration of 50 mg lisdexamfetamine dimesylate to individuals with a history of drug abuse produced positive subjective responses on scales measuring "Drug Liking", "Euphoria", "Amphetamine Effects", and "Benzedrine Effects" that were greater than placebo but less than those produced by an equivalent dose (20 mg) of intravenous d-amphetamine.
9.3 Dependence
Tolerance
Tolerance (a state of adaptation in which exposure to a drug results in a reduction of the drug's desired and/or undesired effects over time) may occur during the chronic therapy of CNS stimulants including VYVANSE.
Dependence
Physical dependence (a state of adaptation manifested by a withdrawal syndrome produced by abrupt cessation, rapid dose reduction, or administration of an antagonist) may occur in patients treated with CNS stimulants including VYVANSE. Withdrawal symptoms after abrupt cessation following prolonged high-dosage administration of CNS stimulants include extreme fatigue and depression.
10 OVERDOSAGE
Consult with a Certified Poison Control Center (1-800-222-1222) for up-to-date guidance and advice for treatment of overdosage. Individual patient response to amphetamines varies widely. Toxic symptoms may occur idiosyncratically at low doses.
Manifestations of amphetamine overdose include restlessness, tremor, hyperreflexia, rapid respiration, confusion, assaultiveness, hallucinations, panic states, hyperpyrexia, and rhabdomyolysis. Fatigue and depression usually follow the central nervous system stimulation. Other reactions include arrhythmias, hypertension or hypotension, circulatory collapse, nausea, vomiting, diarrhea, and abdominal cramps. Fatal poisoning is usually preceded by convulsions and coma.
Lisdexamfetamine and d-amphetamine are not dialyzable.
11 DESCRIPTION
VYVANSE (lisdexamfetamine dimesylate), a CNS stimulant, is a capsule for once-a-day oral administration. The chemical designation for lisdexamfetamine dimesylate is (2S)-2,6-diamino- N-[(1 S)-1-methyl-2-phenylethyl] hexanamide dimethanesulfonate. The molecular formula is C 15H 25N 3O∙(CH 4O 3S) 2, which corresponds to a molecular weight of 455.60. The chemical structure is:
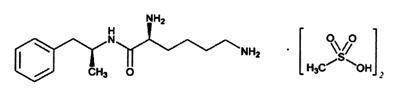
Lisdexamfetamine dimesylate is a white to off-white powder that is soluble in water (792 mg/mL). VYVANSE capsules contain 10 mg, 20 mg, 30 mg, 40 mg, 50 mg, 60 mg, and 70 mg of lisdexamfetamine dimesylate.
Inactive ingredients: microcrystalline cellulose, croscarmellose sodium, and magnesium stearate. The capsule shells contain gelatin, titanium dioxide, and one or more of the following: FD&C Red #3, FD&C Yellow #6, FD&C Blue #1, Black Iron Oxide, and Yellow Iron Oxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lisdexamfetamine is a prodrug of dextroamphetamine. Amphetamines are non-catecholamine sympathomimetic amines with CNS stimulant activity. Amphetamines block the reuptake of norepinephrine and dopamine into the presynaptic neuron and increase the release of these monoamines into the extraneuronal space. The parent drug, lisdexamfetamine, does not bind to the sites responsible for the reuptake of norepinephrine and dopamine in vitro.
12.3 Pharmacokinetics
Pharmacokinetic studies of dextroamphetamine after oral administration of lisdexamfetamine have been conducted in patients ages 6 to 12 years with ADHD and in healthy adult volunteers.
In 18 patients ages 6 to 12 years with ADHD, the T max of dextroamphetamine was approximately 3.5 hours following single-dose oral administration of lisdexamfetamine dimesylate either 30 mg, 50 mg, or 70 mg after an 8-hour overnight fast. The T max of lisdexamfetamine was approximately 1 hour. Linear pharmacokinetics of dextroamphetamine after single-dose oral administration of lisdexamfetamine dimesylate was established over the dose range of 30 mg to 70 mg in children ages 6 to 12 years and over a range of 50 mg to 250 mg in adults. Dextroamphetamine pharmacokinetic parameters following administration of lisdexamfetamine dimesylate in adults exhibited low inter-subject (<25%) and intra-subject (<8%) variability. Safety and efficacy have not been studied above the maximum recommended dose of 70mg.
There is no accumulation of dextroamphetamine AUC at steady state in healthy adults and no accumulation of lisdexamfetamine after once-daily dosing for 7 consecutive days.
Neither food (a high fat meal or yogurt) nor orange juice affect the observed AUC and C max of dextroamphetamine in healthy adults after single-dose oral administration of 70 mg of VYVANSE capsules. Food prolongs T max by approximately 1 hour (from 3.8 hrs at fasted state to 4.7 hrs after a high fat meal or to 4.2 hrs with yogurt). After an 8-hour fast, the AUCs for dextroamphetamine following oral administration of lisdexamfetamine dimesylate in solution and as intact capsules were equivalent.
Weight/Dose normalized AUC and C max were 22% and 12% lower, respectively, in adult females than in males on day 7 following a 70 mg/day dose of lisdexamfetamine dimesylate for 7 days. Weight/Dose normalized AUC and C max values were the same in pediatric patients ages 6 to 12 years following single doses of 30-70 mg.
Metabolism and Excretion
After oral administration, lisdexamfetamine is rapidly absorbed from the gastrointestinal tract. Lisdexamfetamine is converted to dextroamphetamine and l-lysine primarily in blood due to the hydrolytic activity of red blood cells. In vitro data demonstrated that red blood cells have a high capacity for metabolism of lisdexamfetamine; substantial hydrolysis occurred even at low hematocrit levels (33% of normal). Lisdexamfetamine is not metabolized by cytochrome P450 enzymes. Following the oral administration of a 70 mg dose of radiolabeled lisdexamfetamine dimesylate to 6 healthy subjects, approximately 96% of the oral dose radioactivity was recovered in the urine and only 0.3% recovered in the feces over a period of 120 hours. Of the radioactivity recovered in the urine, 42% of the dose was related to amphetamine, 25% to hippuric acid, and 2% to intact lisdexamfetamine. Plasma concentrations of unconverted lisdexamfetamine are low and transient, generally becoming non-quantifiable by 8 hours after administration. The plasma elimination half-life of lisdexamfetamine typically averaged less than one hour in studies of lisdexamfetamine dimesylate in volunteers.
Drug Interaction Studies
|
|
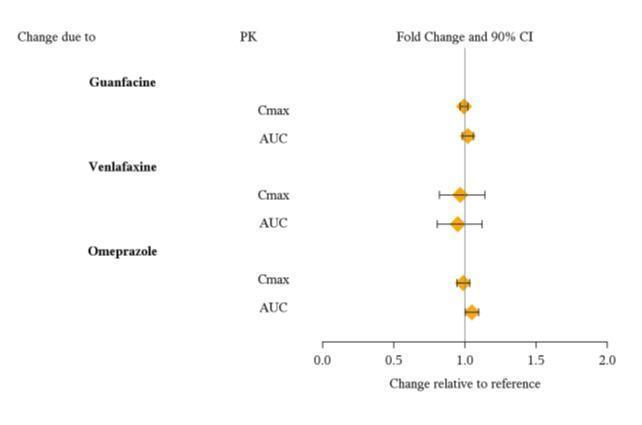
|
|
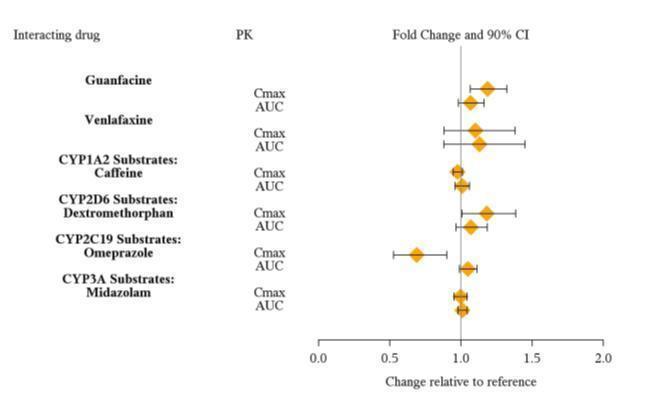
Studies in Specific Populations
Renal Impairment
In a pharmacokinetic study of lisdexamfetamine in subjects with normal and impaired renal function mean d-amphetamine clearance was reduced from 0.7 L/hr/kg in normal subjects to 0.4 L/hr/kg in subjects with severe renal impairment (GFR 15 to <30mL/min/1.73m 2) and 0.3 L/hr/kg in ESRD patients. Dialysis did not significantly affect the clearance of d-amphetamine; the mean clearance of d-amphetamine was 0.3 L/hr/kg for both pre- and post- dialysis [see Use in Specific Populations (8.6)] .
|
|
|
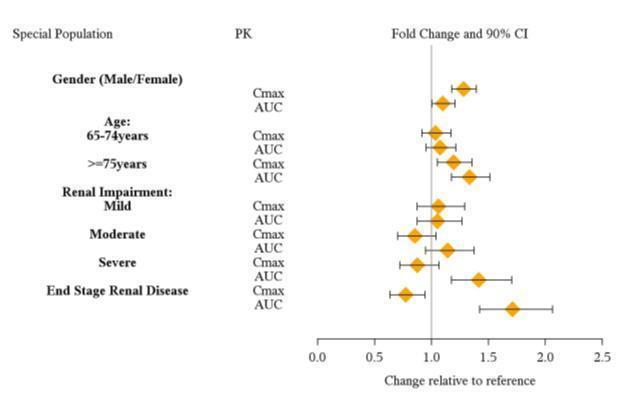
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenesis
Carcinogenicity studies of lisdexamfetamine dimesylate have not been performed. No evidence of carcinogenicity was found in studies in which d-, l-amphetamine (enantiomer ratio of 1:1) was administered to mice and rats in the diet for 2 years at doses of up to 30 mg/kg/day in male mice, 19 mg/kg/day in female mice, and 5 mg/kg/day in male and female rats.
14 CLINICAL STUDIES
Efficacy of VYVANSE in the treatment of ADHD has been established in the following trials:
- Three short-term trials in children (6 to 12 years, Studies 1, 2, 3)
- One short-term trial in adolescents (13 to 17 years, Study 4)
- One short-term trial in children and adolescents (6 to 17 years, Study 5)
- Two short-term trials in adults (18 to 55 years, Studies 7, 8)
- Two randomized withdrawal trials in children and adolescents (6 to 17 years, Study 6), and adults (18 to 55 years, Study 9)
Efficacy of VYVANSE in the treatment of BED has been established in two 12-week trials in adults (18 to 55 years) with moderate to severe BED (Study 10 and 11).
14.1 Attention Deficit Hyperactivity Disorder (ADHD)
Patients Ages 6 to 12 Years Old with ADHD
A double-blind, randomized, placebo-controlled, parallel-group study (Study 1) was conducted in children ages 6 to 12 years (N=290) who met DSM-IV criteria for ADHD (either the combined type or the hyperactive-impulsive type). Patients were randomized to receive final doses of 30 mg, 50 mg, or 70 mg of VYVANSE or placebo once daily in the morning for a total of four weeks of treatment. All patients receiving VYVANSE were initiated on 30 mg for the first week of treatment. Patients assigned to the 50 mg and 70 mg dose groups were titrated by 20 mg per week until they achieved their assigned dose. The primary efficacy outcome was change in Total Score from baseline to endpoint in investigator ratings on the ADHD Rating Scale (ADHD-RS), an 18-item questionnaire with a score range of 0-54 points that measures the core symptoms of ADHD which includes both hyperactive/impulsive and inattentive subscales. Endpoint was defined as the last post-randomization treatment week (i.e. Weeks 1 through 4) for which a valid score was obtained. All VYVANSE dose groups were superior to placebo in the primary efficacy outcome. Mean effects at all doses were similar; however, the highest dose (70 mg/day) was numerically superior to both lower doses (Study 1 in Table 7). The effects were maintained throughout the day based on parent ratings (Conners' Parent Rating Scale) in the morning (approximately 10 am), afternoon (approximately 2 pm), and early evening (approximately 6 pm).
A double-blind, placebo-controlled, randomized, crossover design, analog classroom study (Study 2) was conducted in children ages 6 to 12 years (N=52) who met DSM-IV criteria for ADHD (either the combined type or the hyperactive-impulsive type). Following a 3-week open-label dose optimization with Adderall XR ®, patients were randomly assigned to continue their optimized dose of Adderall XR (10 mg, 20 mg, or 30 mg), VYVANSE (30 mg, 50 mg, or 70 mg), or placebo once daily in the morning for 1 week each treatment. Efficacy assessments were conducted at 1, 2, 3, 4.5, 6, 8, 10, and 12 hours post-dose using the Swanson, Kotkin, Agler, M.Flynn, and Pelham Deportment scores (SKAMP-DS), a 4-item subscale of the SKAMP with scores ranging from 0 to 24 points that measures deportment problems leading to classroom disruptions. A significant difference in patient behavior, based upon the average of investigator ratings on the SKAMP-DS across the 8 assessments were observed between patients when they received VYVANSE compared to patients when they received placebo (Study 2 in Table 7). The drug effect reached statistical significance from hours 2 to 12 post-dose, but was not significant at 1 hour.
A second double-blind, placebo-controlled, randomized, crossover design, analog classroom study (Study 3) was conducted in children ages 6 to 12 years (N=129) who met DSM-IV criteria for ADHD (either the combined type or the hyperactive-impulsive type). Following a 4-week open-label dose optimization with VYVANSE (30 mg, 50 mg, 70 mg), patients were randomly assigned to continue their optimized dose of VYVANSE or placebo once daily in the morning for 1 week each treatment. A significant difference in patient behavior, based upon the average of investigator ratings on the SKAMP-Deportment scores across all 7 assessments conducted at 1.5, 2.5, 5.0, 7.5, 10.0, 12.0, and 13.0 hours post-dose, were observed between patients when they received VYVANSE compared to patients when they received placebo (Study 3 in Table 7, Figure 4).
Patients Ages 13 to 17 Years Old with ADHD
A double-blind, randomized, placebo-controlled, parallel-group study (Study 4) was conducted in adolescents ages 13 to 17 years (N=314) who met DSM-IV criteria for ADHD. In this study, patients were randomized in a 1:1:1:1 ratio to a daily morning dose of VYVANSE (30 mg/day, 50 mg/day or 70 mg/day) or placebo for a total of four weeks of treatment. All patients receiving VYVANSE were initiated on 30 mg for the first week of treatment. Patients assigned to the 50 mg and 70 mg dose groups were titrated by 20 mg per week until they achieved their assigned dose. The primary efficacy outcome was change in Total Score from baseline to endpoint in investigator ratings on the ADHD Rating Scale (ADHD-RS). Endpoint was defined as the last post-randomization treatment week (i.e. Weeks 1 through 4) for which a valid score was obtained. All VYVANSE dose groups were superior to placebo in the primary efficacy outcome (Study 4 in Table 7).
Patients Ages 6 to 17 Years Old: Short-Term Treatment in ADHD
A double-blind, randomized, placebo- and active-controlled parallel-group, dose-optimization study (Study 5) was conducted in children and adolescents ages 6 to 17 years (n=336) who met DSM-IV criteria for ADHD. In this eight-week study, patients were randomized to a daily morning dose of VYVANSE (30, 50 or 70mg/day), an active control, or placebo (1:1:1). The study consisted of a Screening and Washout Period (up to 42 days), a 7-week Double-blind Evaluation Period (consisting of a 4-week Dose-Optimization Period followed by a 3-week Dose-Maintenance Period), and a 1-week Washout and Follow-up Period. During the Dose Optimization Period, subjects were titrated until an optimal dose, based on tolerability and investigator's judgment, was reached. VYVANSE showed significantly greater efficacy than placebo. The placebo-adjusted mean reduction from baseline in the ADHD-RS-IV total score was 18.6. Subjects on VYVANSE also showed greater improvement on the Clinical Global Impression-Improvement (CGI-I) rating scale compared to subjects on placebo (Study 5 in Table 7).
Patients Ages 6 to 17 Years Old: Maintenance Treatment in ADHD
Maintenance of Efficacy Study (Study 6) - A double-blind, placebo-controlled, randomized withdrawal study was conducted in children and adolescents ages 6 to 17 (N=276) who met the diagnosis of ADHD (DSM-IV criteria). A total of 276 patients were enrolled into the study, 236 patients participated in Study 5 and 40 subjects directly enrolled. Subjects were treated with open-label VYVANSE for at least 26 weeks prior to being assessed for entry into the randomized withdrawal period. Eligible patients had to demonstrate treatment response as defined by CGI-S <3 and Total Score on the ADHD-RS ≤22. Patients that maintained treatment response for 2 weeks at the end of the open label treatment period were eligible to be randomized to ongoing treatment with the same dose of VYVANSE (N=78) or switched to placebo (N=79) during the double-blind phase. Patients were observed for relapse (treatment failure) during the 6 week double blind phase. A significantly lower proportion of treatment failures occurred among VYVANSE subjects (15.8%) compared to placebo (67.5%) at endpoint of the randomized withdrawal period. The endpoint measurement was defined as the last post-randomization treatment week at which a valid ADHD-RS Total Score and CGI-S were observed. Treatment failure was defined as a ≥50% increase (worsening) in the ADHD-RS Total Score and a ≥2-point increase in the CGI-S score compared to scores at entry into the double-blind randomized withdrawal phase. Subjects who withdrew from the randomized withdrawal period and who did not provide efficacy data at their last on-treatment visit were classified as treatment failures (Study 6, Figure 5).
Adults: Short-Term Treatment in ADHD
A double-blind, randomized, placebo-controlled, parallel-group study (Study 7) was conducted in adults ages 18 to 55 (N=420) who met DSM-IV criteria for ADHD. In this study, patients were randomized to receive final doses of 30 mg, 50 mg, or 70 mg of VYVANSE or placebo for a total of four weeks of treatment. All patients receiving VYVANSE were initiated on 30 mg for the first week of treatment. Patients assigned to the 50 mg and 70 mg dose groups were titrated by 20 mg per week until they achieved their assigned dose. The primary efficacy outcome was change in Total Score from baseline to endpoint in investigator ratings on the ADHD Rating Scale (ADHD-RS). Endpoint was defined as the last post-randomization treatment week (i.e. Weeks 1 through 4) for which a valid score was obtained. All VYVANSE dose groups were superior to placebo in the primary efficacy outcome (Study 7 in Table 7).
The second study was a multi-center, randomized, double-blind, placebo-controlled, cross-over, modified analog classroom study (Study 8) of VYVANSE to simulate a workplace environment in 142 adults ages 18 to 55 who met DSM-IV-TR criteria for ADHD. There was a 4-week open-label, dose optimization phase with VYVANSE (30 mg/day, 50 mg/day, or 70 mg/day in the morning). Patients were then randomized to one of two treatment sequences: 1) VYVANSE (optimized dose) followed by placebo, each for one week, or 2) placebo followed by VYVANSE, each for one week. Efficacy assessments occurred at the end of each week, using the Permanent Product Measure of Performance (PERMP), a skill-adjusted math test that measures attention in ADHD. PERMP total score results from the sum of the number of math problems attempted plus the number of math problems answered correctly. VYVANSE treatment, compared to placebo, resulted in a statistically significant improvement in attention across all post-dose time points, as measured by average PERMP total scores over the course of one assessment day, as well as at each time point measured. The PERMP assessments were administered at pre-dose (-0.5 hours) and at 2, 4, 8, 10, 12, and 14 hours post-dose (Study 8 in Table 7, Figure 6).
Adults: Maintenance Treatment in ADHD
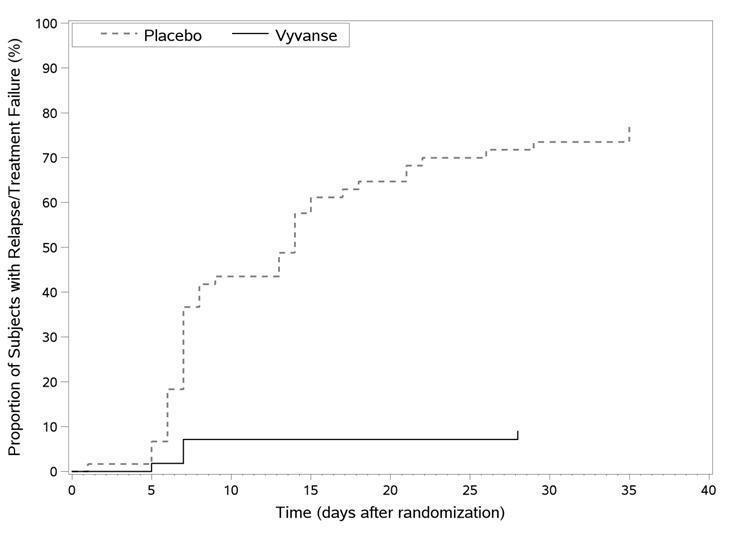
A double-blind, placebo-controlled, randomized withdrawal design study (Study 9) was conducted in adults ages 18 to 55 (N=123) who had a documented diagnosis of ADHD or met DSM-IV criteria for ADHD. At study entry, patients must have had documentation of treatment with VYVANSE for a minimum of 6 months and had to demonstrate treatment response as defined by Clinical Global Impression Severity (CGI-S) ≤3 and Total Score on the ADHD-RS <22. ADHD-RS Total Score is a measure of core symptoms of ADHD. The CGI-S score assesses the clinician's impression of the patient's current illness state and ranges from 1 (not at all ill) to 7 (extremely ill). Patients that maintained treatment response at week 3 of the open label treatment phase (N=116) were eligible to be randomized to ongoing treatment with the same dose of VYVANSE (N=56) or switched to placebo (N=60) during the double-blind phase. Patients were observed for relapse (treatment failure) during the 6-week double-blind phase. The efficacy endpoint was the proportion of patients with treatment failure during the double-blind phase. Treatment failure was defined as a ≥50% increase (worsening) in the ADHD-RS Total Score and ≥2-point increase in the CGI-S score compared to scores at entry into the double-blind phase. Maintenance of efficacy for patients treated with VYVANSE was demonstrated by the significantly lower proportion of patients with treatment failure (9%) compared to patients receiving placebo (75%) at endpoint during the double-blind phase (Study 9, Figure 7).
| Study Number (Age range) | Primary Endpoint | Treatment Group | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference * (95% CI) |
|---|---|---|---|---|---|
| SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: confidence interval. | |||||
|
|||||
| Study 1
(6 - 12 years) | ADHD-RS-IV | VYVANSE (30 mg/day) † | 43.2 (6.7) | -21.8 (1.6) | -15.6 (-19.9, -11.2) |
| VYVANSE (50 mg/day) † | 43.3 (6.7) | -23.4 (1.6) | -17.2 (-21.5, -12.9) | ||
| VYVANSE (70 mg/day) † | 45.1(6.8) | -26.7 (1.5) | -20.5 (-24.8, -16.2) | ||
| Placebo | 42.4 (7.1) | -6.2 (1.6) | -- | ||
| Study 2
(6 - 12 years) | Average SKAMP-DS | VYVANSE (30, 50 or 70 mg/day) † | -- ‡ | 0.8 (0.1) § | -0.9 (-1.1, -0.7) |
| Placebo | -- ‡ | 1.7 (0.1) § | -- | ||
| Study 3
(6 - 12 years) | Average SKAMP-DS | VYVANSE (30, 50 or 70 mg/day) † | 0.9 (1.0) ¶ | 0.7 (0.1) § | -0.7 (-0.9, -0.6) |
| Placebo | 0.7 (0.9) ¶ | 1.4 (0.1) § | -- | ||
| Study 4
(13 - 17 years) | ADHD-RS-IV | VYVANSE (30 mg/day) † | 38.3 (6.7) | -18.3 (1.2) | -5.5 (-9.0, -2.0) |
| VYVANSE (50 mg/day) † | 37.3 (6.3) | -21.1 (1.3) | -8.3 (-11.8, -4.8) | ||
| VYVANSE (70 mg/day) † | 37.0 (7.3) | -20.7 (1.3) | -7.9 (-11.4, -4.5) | ||
| Placebo | 38.5 (7.1) | -12.8 (1.2) | -- | ||
| Study 5
(6 - 17 years) | ADHD-RS-IV | VYVANSE (30, 50 or 70 mg/day) † | 40.7 (7.3) | -24.3 (1.2) | -18.6 (-21.5, -15.7) |
| Placebo | 41.0 (7.1) | -5.7 (1.1) | -- | ||
| Study 7
(18 - 55 years) | ADHD-RS-IV | VYVANSE (30 mg/day) † | 40.5 (6.2) | -16.2 (1.1) | -8.0 (-11.5, -4.6) |
| VYVANSE (50 mg/day) † | 40.8 (7.3) | -17.4 (1.0) | -9.2 (-12.6, -5.7) | ||
| VYVANSE (70 mg/day) † | 41.0 (6.0) | -18.6 (1.0) | -10.4 (-13.9, -6.9) | ||
| Placebo | 39.4 (6.4) | -8.2 (1.4) | -- | ||
| Study 8
(18 - 55 years) | Average PERMP | VYVANSE (30, 50 or 70 mg/day) † | 260.1 (86.2) ¶ | 312.9 (8.6) § | 23.4 (15.6, 31.2) |
| Placebo | 261.4 (75.0) ¶ | 289.5 (8.6) § | -- | ||
Figure 4 LS Mean SKAMP Deportment Subscale Score by Treatment and Time-point for Children Ages 6 to 12 with ADHD after 1 Week of Double Blind Treatment (Study 3)
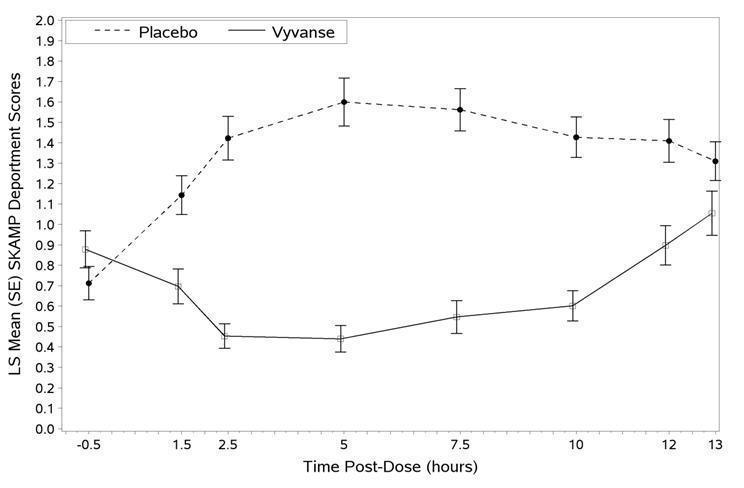
Higher score on the SKAMP-Deportment scale indicates more severe symptoms
Figure 5 Kaplan-Meier Estimation of Proportion of Patients with Treatment Failure for Children and Adolescent Ages 6-17 (Study 6)
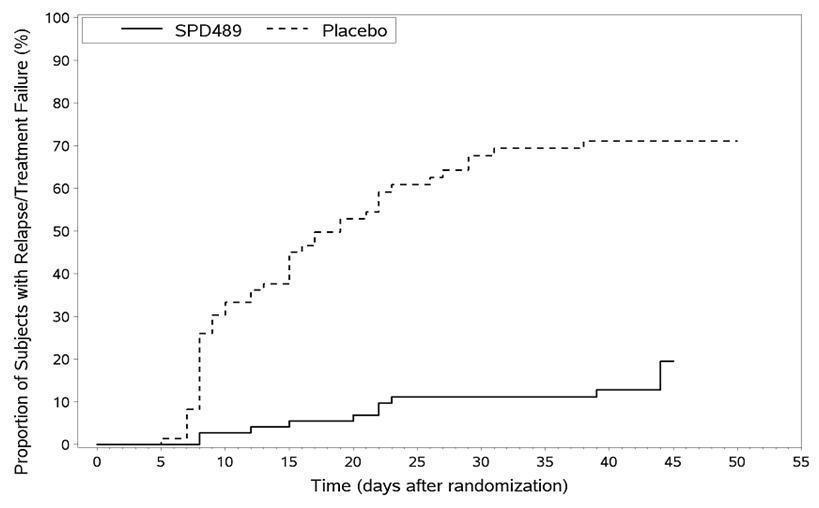
Figure 6 LS Mean (SE) PERMP Total Score by Treatment and Time-point for Adults Ages 18 to 55 with ADHD after 1 Week of Double Blind Treatment (Study 8)
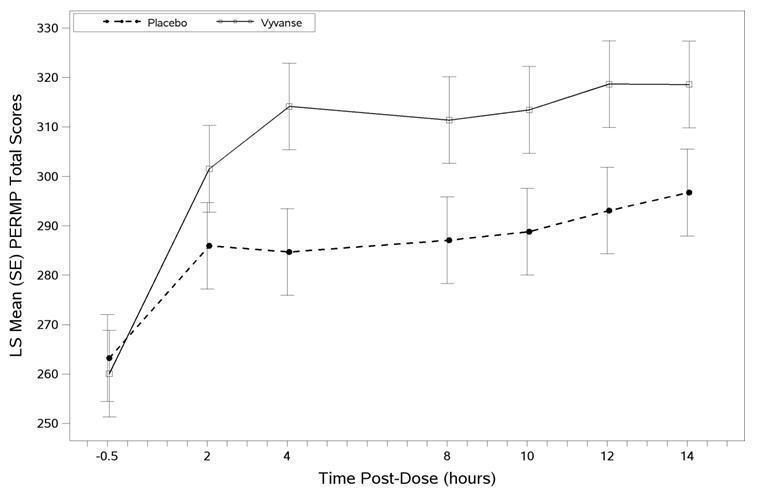
Higher score on the PERMP scale indicates less severe symptoms.
Figure 7 Kaplan-Meier Estimation of Time to Treatment Failure in Adults with ADHD (Study 9)
14.2 Binge Eating Disorder (BED)
A phase 2 study evaluated the efficacy of VYVANSE 30, 50 and 70 mg/day compared to placebo in reducing the number of binge days/week in adults with at least moderate to severe BED. This randomized, double-blind, parallel-group, placebo-controlled, forced-dose titration study consisted of an 11-week double-blind treatment period (3 weeks of forced-dose titration followed by 8 weeks of dose maintenance). VYVANSE 30 mg/day was not statistically different from placebo on the primary endpoint. The 50 and 70 mg/day doses were statistically superior to placebo on the primary endpoint.
The efficacy of VYVANSE in the treatment of BED was demonstrated in two 12-week randomized, double-blind, multi-center, parallel-group, placebo-controlled, dose-optimization studies in adults aged 18-55 years (Study 10: N=374, Study 11: N=350) with moderate to severe BED. A diagnosis of BED was confirmed using DSM-IV criteria for BED. Severity of BED was determined based on having at least 3 binge days per week for 2 weeks prior to the baseline visit and on having a Clinical Global Impression Severity (CGI-S) score of ≥4 at the baseline visit. For both studies, a binge day was defined as a day with at least 1 binge episode, as determined from the subject's daily binge diary.
Both 12-week studies consisted of a 4-week dose-optimization period and an 8-week dose-maintenance period. During dose-optimization, subjects assigned to VYVANSE began treatment at the titration dose of 30 mg/day and, after 1 week of treatment, were subsequently titrated to 50mg/day. Additional increases to 70 mg/day were made as tolerated and clinically indicated. Following the dose-optimization period, subjects continued on their optimized dose for the duration of the dose-maintenance period.
The primary efficacy outcome for the two studies was defined as the change from baseline at Week 12 in the number of binge days per week. Baseline is defined as the weekly average of the number of binge days per week for the 14 days prior to the baseline visit. Subjects from both studies on VYVANSE had a statistically significantly greater reduction from baseline in mean number of binge days per week at Week 12. In addition, subjects on VYVANSE showed greater improvement as compared to placebo across key secondary outcomes with higher proportion of subjects rated improved on the CGI-I rating scale, higher proportion of subjects with 4-week binge cessation, and greater reduction in the Yale-Brown Obsessive Compulsive Scale Modified for Binge Eating (Y-BOCS-BE) total score.
| Study Number | Treatment Group | Primary Efficacy Measure: Binge Days per Week at Week 12 | ||
|---|---|---|---|---|
| Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference *(95% CI) | ||
| SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: confidence interval. | ||||
| Study 10 | VYVANSE (50 or 70 mg/day) † | 4.79 (1.27) | -3.87 (0.12) | -1.35 (-1.70, -1.01) |
| Placebo | 4.60 (1.21) | -2.51 (0.13) | -- | |
| Study 11 | VYVANSE (50 or 70 mg/day) † | 4.66 (1.27) | -3.92 (0.14) | -1.66 (-2.04, -1.28) |
| Placebo | 4.82 (1.42) | -2.26 (0.14) | -- | |
Examination of population subgroups based on age (there were no patients over 65), gender, and race did not reveal any clear evidence of differential responsiveness in the treatment of BED.
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
VYVANSE capsules 70 mg: blue body/orange cap (imprinted with S489 and 70 mg), bottles of 100, NDC 59417-107-10
NDC 69189-0137-1 single dose pack with 1 capsule as repackaged by Avera McKennan Hospital
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Controlled Substance Status/High Potential for Abuse and Dependence
Advise patients that VYVANSE is a controlled substance and it can be abused and lead to dependence and not to give VYVANSE to anyone else [see Drug Abuse and Dependence (9.1, 9.2, and 9.3)] . Advise patients to store VYVANSE in a safe place, preferably locked, to prevent abuse. Advise patients to dispose of remaining, unused, or expired VYVANSE by a medicine take-back program.
Serious Cardiovascular Risks
Advise patients that there is a potential serious cardiovascular risk including sudden death, myocardial infarction, stroke, and hypertension with VYVANSE use. Instruct patients to contact a healthcare provider immediately if they develop symptoms such as exertional chest pain, unexplained syncope, or other symptoms suggestive of cardiac disease [see Warnings and Precautions (5.2)] .
Hypertension and Tachycardia
Instruct patients that VYVANSE can cause elevations of their blood pressure and pulse rate and they should be monitored for such effects.
Psychiatric Risks
Advise patients that VYVANSE at recommended doses may cause psychotic or manic symptoms even in patients without prior history of psychotic symptoms or mania [see Warnings and Precautions (5.4)] .
Suppression of Growth
Advise patients that VYVANSE may cause slowing of growth including weight loss [see Warnings and Precautions (5.5)] .
Impairment in Ability to Operate Machinery or Vehicles
Advise patients that VYVANSE may impair their ability to engage in potentially dangerous activities such as operating machinery or vehicles. Instruct patients to find out how VYVANSE will affect them before engaging in potentially dangerous activities [see Adverse Reactions (6.1, 6.2)] .
Circulation problems in fingers and toes [Peripheral vasculopathy, including Raynaud's phenomenon]
Instruct patients beginning treatment with VYVANSE about the risk of peripheral vasculopathy, including Raynaud's Phenomenon, and associated signs and symptoms: fingers or toes may feel numb, cool, painful, and/or may change from pale, to blue, to red. Instruct patients to report to their physician any new numbness, pain, skin color change, or sensitivity to temperature in fingers or toes. Instruct patients to call their physician immediately with any signs of unexplained wounds appearing on fingers or toes while taking VYVANSE. Further clinical evaluation (e.g. rheumatology referral) may be appropriate for certain patients [see Warnings and Precautions (5.6)] .
Manufactured for: Shire US Inc., Wayne, PA 19087
Made in USA
For more information call 1-800-828-2088
VYVANSE ® is a trademark of Shire LLC
©2015 Shire US Inc.
US Pat No. 7,105,486 and US Pat No. 7,223,735
MEDICATION GUIDE
VYVANSE
® [Vī' - văns]
(lisdexamfetamine dimesylate) CII
Capsules
Read the Medication Guide that comes with VYVANSE before you start taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your doctor about your medical condition or your treatment.
What is the most important information I should know about VYVANSE?
VYVANSE is a federally controlled substance (CII) because it can be abused or lead to dependence. Keep VYVANSE in a safe place to prevent misuse and abuse. Selling or giving away VYVANSE may harm others, and is against the law.
Tell your doctor if you have ever abused or been dependent on alcohol, prescription medicines or street drugs.
VYVANSE is a stimulant medicine. Some people have had the following problems when taking stimulant medicines such as VYVANSE:
1. Heart-related problems including:
- sudden death in people who have heart problems or heart defects
- sudden death, stroke and heart attack in adults
- increased blood pressure and heart rate
Tell your doctor if you have any heart problems, heart defects, high blood pressure, or a family history of these problems.
Your doctor should check you carefully for heart problems before starting VYVANSE.
Your doctor should check your blood pressure and heart rate regularly during treatment with VYVANSE.
Call your doctor right away if you have any signs of heart problems such as chest pain, shortness of breath, or fainting while taking VYVANSE.
- Mental (psychiatric) problems including:
- In Children, Teenagers, and Adults:
-
- new or worse behavior and thought problems
- new or worse bipolar illness
- In Children and Teenagers
-
-
new psychotic symptoms such as:
- hearing voices
- believing things that are not true
- being suspicious
- new manic symptoms
-
new psychotic symptoms such as:
Tell your doctor about any mental problems you have or if you have a family history of suicide, bipolar illness, or depression.
Call your doctor right away if you have any new or worsening mental symptoms or problems while taking VYVANSE, especially:
- seeing or hearing things that are not real
- believing things that are not real
- being suspicious
3. Circulation problems in fingers and toes [Peripheral vasculopathy, including Raynaud's phenomenon]:
- Fingers or toes may feel numb, cool, painful
- Fingers or toes may change color from pale, to blue, to red
Tell your doctor if you have numbness, pain, skin color change, or sensitivity to temperature in your fingers or toes.
Call your doctor right away if you have any signs of unexplained wounds appearing on fingers or toes while taking VYVANSE.
What Is VYVANSE?
VYVANSE is a central nervous system stimulant prescription medicine used to treat:
- Attention-Deficit/Hyperactivity Disorder (ADHD). VYVANSE may help increase attention and decrease impulsiveness and hyperactivity in patients with ADHD.
- Binge Eating Disorder (BED). VYVANSE may help reduce the number of binge eating days in patients with BED.
VYVANSE is not for weight loss. It is not known if VYVANSE is safe and effective for the treatment of obesity.
It is not known if VYVANSE is safe and effective in children with ADHD under 6 years of age or in patients with BED under 18 years of age.
Who should not take VYVANSE?
Do not take VYVANSE if you:
- are taking or have taken within the past 14 days an anti-depression medicine called a monoamine oxidase inhibitor or MAOI.
- are sensitive to, allergic to, or had a reaction to other stimulant medicines.
What should I tell my doctor before taking VYVANSE?
Before you take VYVANSE, tell your doctor if you have or if there is a family history of:
- heart problems, heart defects, high blood pressure
- mental problems including psychosis, mania, bipolar illness, or depression
- circulation problems in fingers and toes
Tell your doctor if:
- you have any kidney problems. Your doctor may lower your dose.
- you are pregnant or plan to become pregnant. It is not known if VYVANSE will harm your unborn baby.
- you are breastfeeding or plan to breastfeed. VYVANSE passes into breast milk. Discuss with your doctor before you breastfeed while you are taking VYVANSE.
Tell your doctor about all of the medicines that you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
VYVANSE can affect the way other medicines work, and other medicines may affect how VYVANSE works. Using VYVANSE with other medicines can cause serious side effects.
Especially tell your doctor if you take anti-depression medicines including MAOIs.
Ask your doctor or pharmacist for a list of these medicines if you are not sure.
Know the medicines that you take. Keep a list of them to show your doctor and pharmacist when you get a new medicine.
Do not start any new medicine while taking VYVANSE without talking to your doctor first.
How should I take VYVANSE?
- Take VYVANSE exactly as your doctor tells you to take it.
- Your doctor may change your dose until it is right for you.
- Take VYVANSE 1 time each day in the morning.
- VYVANSE can be taken with or without food.
- VYVANSE capsules may be swallowed whole.
- If you have trouble swallowing capsules, open your VYVANSE capsule and pour all the powder into yogurt, water, or orange juice.
- Use all of the VYVANSE powder from the capsule so you get all of the medicine.
- Using a spoon, break apart any powder that is stuck together. Stir the VYVANSE powder and yogurt, water or orange juice until they are completely mixed together.
- Eat all the yogurt or drink all the water or orange juice right away after it has been mixed with VYVANSE. Do not store the yogurt, water, or orange juice after it has been mixed with VYVANSE. It is normal to see a filmy coating on the inside of your glass or container after you eat or drink all the VYVANSE.
- Your doctor may sometimes stop VYVANSE treatment for a while to check your ADHD or your BED symptoms.
- Your doctor may do regular checks of your heart, and blood pressure while taking VYVANSE.
- Children should have their height and weight checked often while taking VYVANSE. VYVANSE treatment may be stopped if a problem is found during these check-ups.
- If you take too much VYVANSE, call your doctor or poison control center right away, or get to the nearest hospital emergency room.
What should I avoid while taking VYVANSE?
Do not drive, operate machinery, or do other dangerous activities until you know how VYVANSE affects you.
What are possible side effects of VYVANSE?
VYVANSE may cause serious side effects, including:
- See " What is the most important information I should know about VYVANSE?"
- slowing of growth (height and weight) in children
| The most common side effects of VYVANSE in ADHD include: | |
|
|
The most common side effects of VYVANSE in BED include:
- dry mouth
- trouble sleeping
- decreased appetite
- increased heart rate
- constipation
- feeling jittery
- anxiety
Talk to your doctor if you have any side effects that bother you or do not go away.
These are not all the possible side effects of VYVANSE. For more information ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store VYVANSE?
- Store VYVANSE at room temperature, 68°F to 77°F (20°C to 25°C).
- Protect VYVANSE from light.
- Store VYVANSE in a safe place, like a locked cabinet.
- Do not throw away unused VYVANSE in your household trash as it may harm other people or animals. Ask your doctor or pharmacist about a medicine take-back program in your community.
Keep VYVANSE and all medicines out of the reach of children.
General information about the safe and effective use of VYVANSE.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use VYVANSE for a condition for which it was not prescribed. Do not give VYVANSE to other people, even if they have the same condition. It may harm them.
This Medication Guide summarizes the most important information about VYVANSE. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about VYVANSE that is written for healthcare professionals.
For more information about VYVANSE, go to www.vyvanse.com or call 1-800-828-2088.
What are the ingredients in VYVANSE?
Active Ingredient: lisdexamfetamine dimesylate
Inactive Ingredients: microcrystalline cellulose, croscarmellose sodium, and magnesium stearate. The capsule shells (imprinted with S489) contain gelatin, titanium dioxide, and one or more of the following: FD&C Red #3, FD&C Yellow #6, FD&C Blue #1, Black Iron Oxide, and Yellow Iron Oxide.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Manufactured for: Shire US Inc., Wayne, PA 19087.
© 2015 Shire US Inc.
Revised April 2015

| VYVANSE
lisdexamfetamine dimesylate capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Avera McKennan Hospital (068647668) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Avera McKennan Hospital | 068647668 | relabel(69189-0137) , repack(69189-0137) | |